

Теплофизика высоких температур, 2019, T. 57, № 3, стр. 404-407
Анализ стабильности малых металлических кластеров при конденсации паров металла
А. Г. Воронцов 1, *, А. Е. Коренченко 1, 2, Б. Р. Гельчинский 2
1 Южно-Уральский государственный университет (НИУ)
г. Челябинск, Россия
2 Институт металлургии УрО РАН,
г. Екатеринбург, Россия
* E-mail: vorontsovag@susu.ru
Поступила в редакцию 24.04.2018
После доработки 14.05.2018
Принята к публикации 10.10.2018
Аннотация
Выполнен статистический анализ результатов молекулярно-динамических расчетов конденсации паров металла (Cu или Ti) в среде инертного газа (Ar). Конденсация протекает с образованием малых частиц, состоящих из атомов металла, – кластеров. В качестве ключевой характеристики для описания состояния металлического кластера выбрана его внутренняя энергия, представляющая собой сумму кинетической энергии атомов в системе центра масс кластера и их потенциальной энергии. Показано, что значение внутренней энергии позволяет предсказать длительность существования кластера от образования до распада, т.е. описывает способность кластера к росту. Представлена временнáя эволюция функций распределения кластеров по значениям внутренней энергии.
ВВЕДЕНИЕ
Один из распространенных методов получения металлических порошков основан на испарении металла или сплава с последующей конденсацией [1]. Для оптимизации технологических процессов производства требуется математическая модель, описывающая процессы конденсации с количественной точностью, однако она до сих пор не построена. Аналитические методы [2] не позволяют достичь количественной точности, так как плохо описывают неравновесный процесс нуклеации. Для применения кинетических или статистических моделей [3, 4] требуется надежное определение вероятностей протекания различных процессов, описывающих столкновения в парогазовой смеси, содержащей кластеры малых размеров. Данные вероятности не поддаются прямому экспериментальному определению, поэтому эффективным методом исследования начального этапа конденсации стало атомистическое молекулярно-динамическое (МД) моделирование.
Замечено, что должное внимание не уделяется вопросу обратного влияния конденсации на параметры среды [5, 6]. Образование кластера приводит к локальному снятию пересыщения, что может на несколько порядков менять скорость нуклеации. В [7] показано, что причиной изменения скорости нуклеации является неравновесность малых кластеров с окружающей парогазовой смесью. Внутренняя “температура” кластера обычно выше температуры парогазовой смеси, т.е. необходимо рассматривать среднюю внутреннюю энергию кластеров как дополнительный параметр, определяющий скорость нуклеации. Моделирование серии столкновений отдельного кластера и атома [8] показало, что для железа коэффициент аккомодации атома к кластеру существенно зависит от температуры кластера и температуры потока налетающих атомов. Вышесказанное позволяет заключить, что необходимо дальнейшее исследование механизмов протекания начального этапа конденсации.
Цель данной работы состоит в нахождении условий, определяющих стабильность металлических кластеров малых размеров в процессе их роста при помощи МД-моделирования. В работе рассмотрены кластеры меди и титана в среде буферного газа – аргона. Результаты предполагается использовать в макроскопической модели газофазной нуклеации для прогнозирования распределения наночастиц по размерам в опытном реакторе.
СХЕМА МД-ЭКСПЕРИМЕНТА
Исследование формирования наночастиц из пересыщенных паров металла проводилось методом молекулярной динамики, реализованном в пакете LAMMPS [9]. Система состояла из атомов буферного газа (Аr) и атомов металла: меди или титана. Взаимодействие атомов металла описывалось потенциалами погруженного атома [10] для Cu и [11] для Ti. Потенциал меди хорошо (с ошибкой менее 25%) описывает энергию и длину связи двухатомной молекулы Cu2, поверхностную и объемную энергию конденсированной фазы [7], поэтому можно ожидать, что он также приемлемо описывает малые кластеры меди. Применимость потенциала титана для описания наночастиц детально не изучена, однако параметры двухатомной молекулы (межатомное расстояние, энергия взаимодействия) отличаются от экспериментальных не более чем на 30%.
При изучении столкновений совместный анализ траекторий выполняется для атомов, расстояние между которыми не превышает некоторой фиксированной величины Δ. Такая группа атомов в данной работе называется кластером. Значение Δ выбиралось в соответствии с правилом, изложенным в [12]: Δ – это наименьшее расстояние, при котором атомы не испытывают отталкивания при сближении, т.е. они пролетают мимо практически без отклонения или с небольшими отклонениями за счет сил притяжения. Определено, что для меди Δ = 4.95 Å и для титана – 5.9 Å. Радиус отсечки потенциалов составлял 10 Å.
Шаг интегрирования равен 3 фс. Тепловое перемещение отдельного атома за это время не превышает 0.02 Å, что составляет в среднем менее 0.005Δ. Малая величина перемещения на каждом шаге гарантирует, что за это время может произойти либо объединение двух частиц, либо распад кластера на две частицы. Это подтверждено тем, что на всем протяжении моделирования с таким временны́м шагом ни разу не наблюдались одновременное (за один шаг) тройное столкновение или распад кластера на три фрагмента.
В работе анализировались время жизни каждого кластера τ и значение его энергии в расчете на один атом в системе центра масс ε. Под “жизнью” кластера подразумевается пребывание его в определенном состоянии, т.е. сохранение в нем количества атомов. При добавлении или отделении атома отсчет времени начинается заново. Также для каждого кластера известна его история, т.е. является он результатом распада (“осколком” другого кластера) или же появился в результате столкновения кластеров/атомов.
Атомы помещались в кубическую ячейку с периодическими граничными условиями. Количество атомов металла взято сравнительно небольшим (5000, 10 000, 25 000), но это компенсировалось несколькими независимыми запусками для различных конфигураций атомов. В результате получалось необходимое количество статистического материала. Рассматривались разные соотношения между количеством атомов аргона и атомов металла. Температура аргона выбиралась в интервале 300 ≤ ТAr ≤ 1500 К, размеры ячейки подбирались так, чтобы плотность пара металла находилась в диапазоне 0.1 ≤ ρ ≤ 0.3 кг/м3. Парциальное давление металлического пара составляло 3 × 103 < p < 5 × 103 Па, что вполне реалистично и наблюдается в установках для газофазного синтеза с испарителем [1]. Термостатирование системы происходило только для атомов инертного газа. В начальный момент времени температуры аргона и атомарного металлического пара были одинаковы. Эволюция атомной системы рассматривалась в течение времен порядка 500 нс.
ВНУТРЕННЯЯ ЭНЕРГИЯ КЛАСТЕРОВ РАЗНОГО РАЗМЕРА
Как отмечено выше, одним из ключевых параметров, определяющих эволюцию кластера в пространстве размеров, является ε. На рис. 1 показаны функции распределения (ФР) димеров по энергии ε для Ti2 и Сu2 в несколько моментов времени после начала нуклеации. Расчеты выполнены для TAr = 300 и 1500 К, плотность паров металла равна 0.2 кг/м3. Для построения графиков подсчитывалось количество кластеров в системе, энергии которых приходятся на интервал [ε; ε + Δε], Δε = 0.01 эВ/атом, и затем полученная зависимость нормировалась так, чтобы площадь под кривой была равна единице. Погрешности значений ФР получены с помощью статистического анализа результатов расчетов четырех различных начальных конфигураций для каждого значения TAr.
Рис. 1.
Временнáя эволюция функций распределения димеров меди Cu2 (а) и титана Ti2 (б) по энергиям: 1 – 1500 К, 2 – 300 К.

ФР димеров меди и титана демонстрируют сходное поведение. Отметим основные закономерности этих зависимостей. Во-первых, в начале процесса нуклеации большинство димеров в ячейке имеет внутреннюю энергию вблизи ε = = 0 эВ/атом. Во-вторых, с течением времени появляется еще один максимум кривой функции распределения, приходящийся на ε ~ –1.17 эВ/атом для Ti2 и ε ~ –1.22 эВ/атом для Cu2. Эти энергии равны половине энергии связи соответствующих двухатомных молекул для использующихся потенциалов. Два максимума кривых распределения разделены участком энергий, которыми обладает очень мало кластеров. Координаты максимумов не изменяются с течением времени, но зависят от температуры: при повышении температуры каждый максимум сдвигается вправо, становится шире и ниже. В-третьих, левый максимум ФР с течением времени растет, правый – уменьшается. По истечении ~24 нс на ФР остается только левый максимум.
Схожие закономерности наблюдаются у ФР кластеров больших размеров (см., например, рис. 2), однако энергии, на которые приходятся максимумы, становятся ближе. Так, для Cu2 расстояние между максимумами составляет примерно 1.2 эВ/атом, для Cu3 – 0.71, Cu4 – 0.57, Cu8 – 0.28 и Cu12 – 0.22.
Рис. 2.
Зависимость вероятности появления долгоживущего кластера от энергии и ФР для кластеров меди (а) и титана (б) различного размера: 1 – вероятность, полученная с помощью МД моделирования; 2 – ФР для кластеров, образовавшихся в реакции объединения; 3 – для кластеров, появившихся в результате распада.
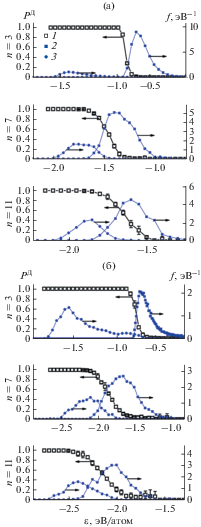
При анализе траекторий обнаружилось, что кластеры, дающие вклад в правый и левый максимумы ФР, имеют различное происхождение: правый максимум образован кластерами, возникшими в результате столкновения, а в левый максимум входят частицы, образовавшиеся после распада больших частиц. Причиной увеличения левого пика со временем является увеличение количества крупных частиц, которые при распаде могут давать кластеры с низкой энергией. Причина уменьшения правого пика – уменьшение со временем атомов и малых кластеров, которые при столкновении могут образовать больший кластер. Такая особенность временнóй эволюции ФР связана с тем, что рассматриваются системы с постоянным числом частиц. В реальных системах происходит постоянное восполнение числа атомов, поэтому исчезновения правого пика не произойдет. Область энергий между двумя пиками ФР, для которых наблюдается очень малое число кластеров, соответствует димерам, перешедшим из правого пика после потери энергии из-за столкновения с другим атомом металла или аргона. В этой области димеры не задерживаются, и после одного или нескольких столкновений переходят в левый пик ФР, где остаются, пока не образуют тример.
ВНУТРЕННЯЯ ЭНЕРГИЯ И ВРЕМЯ ЖИЗНИ КЛАСТЕРОВ
Результат взаимодействия атома и кластера рассматривается как новый кластер, который может оказаться стабильным, а может случиться так, что атом и кластер разлетятся сразу. В качестве критерия стабильности выбрано время существования нового кластера. Кластеры с большим временем жизни назовем “долгоживущими”. Количественное определение границы для долгоживущих кластеров не является строгим, однако удобно считать долгоживущим кластер, время жизни которого больше среднего времени между столкновениями атом–атом в системе. Такой кластер имеет возможность дальнейшего роста, т.е. за время существования он должен испытать несколько последующих столкновений с атомами или кластерами.
На рис. 2 показаны графики зависимости вероятности появления долгоживущего кластера P Д(ε) от энергии для тримеров, 7- и 11-атомных кластеров металла. Вероятность определялась отношением количества долгоживущих кластеров с энергиями в интервале [ε; ε + Δε] к общему числу кластеров с такой же энергией. Время накопления статистики в одной модели составило 100 нс. Данные для графиков получены статистической обработкой значений вероятностей для 26 независимых моделей. При построении графиков на рис. 2 взаимодействия с атомом Ar не учитывались как распад. Функции распределения нормированы так, что в сумме площади под кривыми дают единицу, поэтому их сумма представляет собой общую ФР. Отметим, что уже для кластера с k = 7 максимумы общей ФР не разрешены. Кластеры каждого размера существуют в определенном диапазоне энергии, например, возможные значения энергии составляют –1.17 и 0.7 эВ для Ti2 (см. рис. 1б), –1.9 и –0.1 эВ для Ti3, –2.6 и –0.9 эВ для 7-атомных кластеров. С увеличением количества атомов область возможных энергий атомов в кластере сужается. Сопоставление графиков P Д(ε) и ФР выявило следующие особенности. Во-первых, вероятность появления долгоживущего кластера не зависит от предыстории кластера, от температуры аргона и от плотности паров металла, а определяется только его внутренней энергией. Эта вероятность стремится к единице на левой границе возможных значений энергии для кластеров данного размера и к нулю – на правой. Во-вторых, погрешности определения вероятности определяются объемом выборки, а не зависимостью вероятности от температуры, плотности паров металла и соотношения между количеством атомов аргона и металла. При значениях энергии, при которых можно набрать выборку достаточного объема (100 и более испытаний), погрешности не превышают 5%. В-третьих, у кластеров, содержащих более семи атомов, максимумы на ФР различить не удается, а расстояния между пиками можно узнать, только если по отдельности строить функции распределения для кластеров, полученных в результате объединения и распада, как это показано на рис. 2. Таким образом, доля долгоживущих кластеров любого размера не зависит от температуры и плотности паров металла, а определяется только внутренней энергией кластера.
ЗАКЛЮЧЕНИЕ
В работе выполнен статистический анализ результатов МД-моделирования ранней стадии нуклеации в пересыщенном металлическом паре и описан механизм получения долгоживущих димеров, тримеров и т.д. в результате распада крупных кластеров. Показано, что внутренняя энергия кластера в расчете на один атом является хорошим параметром для построения численной модели нуклеации, так как для определенного материала только она влияет на вероятность получения долгоживущих кластеров PД(ε). Найдены универсальные зависимости PД(ε) для кластеров меди и титана малых размеров. Таким образом, кроме традиционной эволюции кластеров в пространстве размеров, необходимо дополнительно рассматривать эволюцию кластеров в пространстве энергий.
Работа выполнена при поддержке Правительства РФ (Постановление № 211 от 16.03.2013 г.), соглашение № 02.A03.21.0011, а также при частичной поддержке ИМЕТ УрО РАН в рамках выполнения госзадания (тема № 0396-2019-0004).
Список литературы
Фришберг И.В., Кватер Л.И., Кузьмин Б.П., Грибовский С.В. Газофазный метод получения порошков. М.: Наука, 1978. 223 с.
Becker R., Döring W. The Kinetic Treatment of Nuclear Formation in Supersaturated Vapors // Ann. Phys. 1935. V. 24. P. 719.
Зельдович Я.Б. К теории образования новой фазы. Кавитация // ЖЭТФ. 1942. Т. 12. С. 525.
Френкель Я.И. Статистическая теория конденсации (ассоциации) и полимеризации // ЖЭТФ. 1939. Т. 9. № 2. С. 199.
Фисенко С.П. Микроструктура поля пересыщения при гомогенной нуклеации в парогазовой смеси // ЖТФ. 2013. Т. 83. № 5. С. 35.
Вараксин А.Ю. Влияние частиц на турбулентность несущего потока газа // ТВТ. 2015. Т. 53. № 3. С. 441.
Воронцов А.Г., Гельчинский Б.Р., Коренченко А.Е. Кинетика и энергетические состояния нанокластеров в начальной стадии процесса гомогенной конденсации при высоких степенях пересыщения // ЖЭТФ. 2012. Т. 142. № 11. С. 897.
Lenev D.Yu., Norman G.E. Sticking Coefficient for Fe Atoms Interacting with Iron Cluster // J. Phys.: Conf. Ser. 2018. V. 946. P. 012111.
Plimpton S. Fast Parallel Algorithms for Short-range Molecular Dynamics // J. Comp. Phys. 1995. V. 117. P. 1.
Foiles S.M., Baskes M.I., Daw M.S. Embedded-atom-method functions for the FCC Metals Cu, Ag, Au, Ni, Pd, Pt, and their Alloys // Phys. Rev. B. 1986. V. 33. P. 7983.
Zhou X.W., Johnson R.A., Wadley H.N.G. Misfit-energy-increasing Dislocations in Vapor-deposited CoFe/NiFe Multilayers // Phys. Rev. B. 2004. V. 69. P. 144 113.
Korenchenko A.E., Vorontsov A.G., Gel’chinskii B.R., Sannikov G.P. Statistical Analysis of Dimer Formation in Supersaturated Metal Vapor Based on Molecular Dynamics Simulation // Phys. A. 2018. V. 496. P. 147.
Дополнительные материалы отсутствуют.
Инструменты
Теплофизика высоких температур