

Теплофизика высоких температур, 2020, T. 58, № 2, стр. 202-207
Термодинамические функции ArO и ArO+
М. А. Мальцев 1, 2, *, И. В. Морозов 1, 2, Е. Л. Осина 1
1 ФГБУН Объединенный институт высоких температур РАН
Москва, Россия
2 Московский физико-технический институт
Москва, Россия
* E-mail: maksim.malcev@phystech.edu
Поступила в редакцию 23.07.2019
После доработки 23.07.2019
Принята к публикации 22.10.2019
Аннотация
С использованием потенциалов межатомного взаимодействия рассчитаны колебательно-вращательные статистические суммы и термодинамические функции ArO и ArO+ в интервале температур 298.15–10 000 К. Получены значения энтальпий образования ${{\Delta }_{f}}H$ (ArO, 298.15 K) = 243 ± 5 кДж моль–1 и ${{\Delta }_{f}}H$ (ArO+, 298.15 K) = 1508 ± 5 кДж моль–1. Результаты расчетов внесены в базу данных ИВТАНТЕРМО.
ВВЕДЕНИЕ
Данная работа является продолжением исследований термодинамических свойств двухатомных молекул с атомом аргона (аргидов), опубликованных в [1–3]. Аргиды представляют большой интерес в масс-спектрометрии с индуктивно связанной плазмой (ИСП) [4], так как перекрывают сигналы исследуемых соединений. Изучение свойств оксидов инертных газов, в том числе и аргона, мотивировано также их возможным применением в эксимерных лазерах, широко распространенных в глазной хирургии и полупроводниковом производстве.
Целью настоящей работы являются: во-первых, расчет термодинамических функций молекулы ArO и иона ArO+ на основе современных данных о потенциалах межатомного взаимодействия, так как данные о термодинамических свойствах этих соединений в настоящее время отсутствуют в литературе; во-вторых, получение величин энтальпий образования с использованием принятых значений энергии диссоциации и рассчитанных в данной работе термодинамических функций ArO и ArO+.
Для расчета термодинамических функций использовалась методика, описанная в работах [1, 2]. Эта методика основана на численном решении стационарного уравнения Шредингера для заданного межатомного потенциала взаимодействия и получения полного колебательно-вращательного спектра с последующим вычислением колебательно-вращательной статистической суммы. При этом рассматривается основное и несколько возбужденных состояний молекулы, вносящих наиболее существенный вклад в статистическую сумму в выбранном интервале температур (298.15–10 000 К). Таким образом, значительное внимание в работе уделено выбору и оценке достоверности потенциальных кривых, полученных другими авторами. Там, где это возможно, результаты сопоставлены с расчетом термодинамических функций по молекулярным постоянным.
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ ОКСИДА АРГОНА
Первые экспериментальные данные о потенциале взаимодействия Ar и атома кислорода в основном электронном состоянии $^{3}\Pi $ были получены из экспериментов по упругому рассеянию, выполненных в [5–7]. В [8] разработан новый подход в изучении тонкой структуры системы Ar + O, основанный на измерении дифференциальных сечений для межмультиплетных переходов при атом-атомных столкновениях. Авторы интерпретировали полученные результаты, используя данные ab initio-расчетов методом RHF (Restricted Hartree Fock). Удалось установить значительную чувствительность результатов измерений к особенностям потенциалов взаимодействия, что позволило отказаться от эмпирического потенциала [7] в пользу несколько модифицированного ab initio-потенциала, который давал дифференциальные и интегральные сечения в соответствии с экспериментом. В работах [9, 10] молекулы ArO были изучены методом фотоэлектронной спектроскопии, позволяющим определять сродство к электрону, энергии возбужденных электронных состояний и колебательные частоты соответствующих нейтральных молекул. Из-за недостаточно высокого экспериментального разрешения в [9, 10] наблюдаемые фотоэлектронные переходы были сильно перекрыты, образуя широкую полосу, состоящую из двух пиков, разрешить которую авторам не удалось.
Оксид аргона был объектом нескольких теоретических исследований [8, 11–14]. Основная цель [11] состояла в разработке теоретической модели для моделирования фотоэлектронных спектров с помощью современных ab initio-расчетов потенциалов взаимодействия. Расчеты нерелятивистских кривых потенциальной энергии ArO и ArO– для низших электронных состояний симметрии $\Pi $ и $\Sigma $ выполнены с использованием теории возмущений UMP4. Элементы матрицы спин-орбитального расщепления рассчитаны с использованием модели AIM (atom-in-molecule). Полученные смоделированные фотоэлектронные спектры согласовывались с экспериментальными спектрами [9, 10]. Теоретические расчеты более высокого уровня RCCSD(T) выполнены в работах [12, 13].
Фотоэлектронные спектры высокого разрешения ArO–, полученные с использованием новой методики SEVI (slow electron velocity-map imaging), приведены в работе [14]. В отличие от предыдущих фотоэлектронных спектров [9, 10] авторы [14] получили спектр с хорошо разрешенными фотоэлектронными переходами. В [14] также выполнены квантово-химические расчеты высокого уровня RCCSD(T), которые позволили разработать новые потенциалы межатомного взаимодействия для ArO– и ArO и использовать их для моделирования SEVI-спектра. Параметры потенциалов, полученных в [14] для молекулы ArO, приведены в табл. 1. Кроме молекулярных постоянных авторы [14] привели рассчитанные ими потенциальные кривые, не предоставив тем не менее численных значений энергий при различных межъядерных расстояниях.
Таблица 1.
Параметры ab initio нерелятивистских потенциалов [14] с учетом спин-орбитального расщепления
| Состояние | ${{r}_{e}}$, Å | ${{D}_{e}}$, см–1 | ${{T}_{0}}$, см–1 | ${{D}_{0}}$, см–1 | ${{\omega }_{e}}$, см–1 | ${{\omega }_{e}}{{x}_{e}}$, см–1 |
|---|---|---|---|---|---|---|
| X2 | 3.347 | 87.8 | 0 | 68.6 | 38.5 | 4.34 |
| I1 | 3.559 | 61.3 | 21 | 47.6 | 28.5 | 3.35 |
| I0 | 3.659 | 55.6 | 26.6 | 42 | 26.9 | 3.30 |
| II1 | 3.647 | 58.2 | 184.2 | 42.7 | 30.7 | 4.23 |
| II0 | 3.347 | 87.8 | 158.3 | 68.6 | 38.5 | 4.34 |
| III0 | 3.569 | 63.9 | 248.3 | 47.3 | 30.7 | 3.83 |
В настоящей работе в качестве исходных данных для потенциальных кривых основного и пяти низколежащих возбужденных электронных состояний использовались результаты квантово-химических расчетов [14], полученные путем оцифровки представленных в статье графиков. Для численного решения уравнения Шредингера и нахождения колебательно-вращательного спектра необходимо представить эти данные в виде гладкой аналитической кривой. Для этого оцифрованные данные [14] аппроксимировались потенциалом EMO (Extended Morse oscillator) [15] с использованием энергии диссоциации и равновесного межатомного расстояния, указанных в табл. 1. Потенциал EMO имеет вид
(1)
$\begin{gathered} V(r) = {{D}_{e}}\mathop {\left[ {1 - {{e}^{{\beta \left( r \right)\left( {r - {{r}_{e}}} \right)}}}} \right]}\nolimits^2 ,\,\,\,\,\beta \left( r \right) = \sum\limits_{i = 0}^{{{N}_{\beta }}} {{{\beta }_{i}}\mathop {\left[ {y_{q}^{{{\text{ref}}}}\left( r \right)} \right]}\nolimits^i } , \\ y_{q}^{{{\text{ref}}}}\left( r \right) = \frac{{{{r}^{q}} - r_{{{\text{ref}}}}^{q}}}{{{{r}^{q}} + r_{{{\text{ref}}}}^{q}}}, \\ \end{gathered} $Рис. 1.
Потенциальные кривые основного и пяти низколежащих возбужденных электронных состояний ArO: 1 – основное электронное состояние X2; возбужденные электронные состояния: 2 – I1, 3 – I0, 4 – II1, 5 – II0, 6 – III0 (обозначения как в [14]).
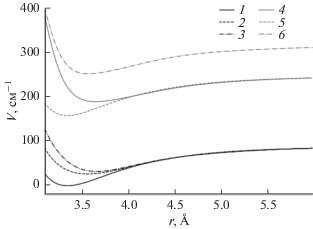
Отличительной особенностью EMO-потенциала является то, что он более корректно описывает отталкивательную зону потенциальной кривой молекулы по сравнению с потенциалом Морзе. Чтобы определить влияние этих особенностей, проведены аналогичные расчеты с использованием потенциала Морзе, построенного по молекулярным постоянным из [14]. Разница в значениях для приведенной энергии Гиббса составила 1.17–1.24 Дж К–1 моль–1 в температурном интервале 298.15–10 000 К. Такое малое различие показывает, что в данном случае вид потенциала слабо влияет на величины рассчитанных термодинамических функций. Однако при расчете термодинамических функций по молекулярным постоянным без использования потенциалов межатомного взаимодействия напрямую наблюдалось более значительное отклонение.
Для сравнения рассчитаны термодинамические функции по молекулярным постоянным из [14] по методике [16]. Разница в приведенной энергии Гиббса не превышала 5 Дж К–1 моль–1 и слабо менялась при изменении температуры в диапазоне 298.15–10 000 К. Это свидетельствует о том, что основное различие в значениях термодинамических функций вносит вращательная составляющая статистической суммы. Такая разница связана с методами экстраполяции энергий вращательных уровней, которые используются при расчете по молекулярным постоянным. При нахождении максимального вращательного квантового числа в расчетах по молекулярным постоянным не учитываются случаи, когда потенциальная кривая имеет минимум, но при этом связанные состояния отсутствуют. Например, по этой причине при расчете термодинамических функций по молекулярным постоянным в статистическую сумму для основного состояния входит больше вращательных уровней энергии (${{J}_{{{\text{max}}}}}$ = 47) по сравнению с расчетом, основанным на потенциале межатомного взаимодействия (${{J}_{{{\text{max}}}}}$ = 28).
Результаты расчетов термодинамических функций с использованием потенциала (1) для основного и пяти низколежащих электронных состояний в температурном диапазоне 298.15–10 000 К представлены в табл. 2. Высоколежащие электронные состояния (15 000 см–1 и выше) не приняты во внимание из-за малого вклада в электронную статистическую сумму. На основе полученных результатов рассчитаны энтальпии образования оксида аргона:
Таблица 2.
Термодинамические функции ArO
| $T$, | $C_{p}^{^\circ }(T)$, | ${{\Phi }^{^\circ }}(T)$, | ${{S}^{^\circ }}(T)$, | ${{H}^{^\circ }}(T) - {{H}^{^\circ }}(0)$, |
|---|---|---|---|---|
| К | Дж К–1 моль–1 | кДж моль–1 | ||
| 298.15 | 22.007 | 213.28 | 238.219 | 7.436 |
| 300 | 21.995 | 213.434 | 238.355 | 7.476 |
| 400 | 21.537 | 220.486 | 244.612 | 9.65 |
| 500 | 21.293 | 225.808 | 249.389 | 11.791 |
| 600 | 21.149 | 230.071 | 253.257 | 13.912 |
| 700 | 21.059 | 233.621 | 256.51 | 16.022 |
| 800 | 20.998 | 236.662 | 259.318 | 18.125 |
| 900 | 20.955 | 239.319 | 261.789 | 20.222 |
| 1000 | 20.924 | 241.679 | 263.995 | 22.316 |
| 1500 | 20.849 | 250.624 | 272.462 | 32.756 |
| 2000 | 20.822 | 256.869 | 278.455 | 43.173 |
| 2500 | 20.809 | 261.668 | 283.1 | 53.58 |
| 3000 | 20.802 | 265.566 | 286.893 | 63.983 |
| 4000 | 20.795 | 271.681 | 292.877 | 84.781 |
| 5000 | 20.792 | 276.402 | 297.517 | 105.574 |
| 6000 | 20.79 | 280.246 | 301.307 | 126.365 |
| 7000 | 20.789 | 283.49 | 304.512 | 147.155 |
| 8000 | 20.788 | 286.295 | 307.288 | 167.943 |
| 9000 | 20.788 | 288.766 | 309.736 | 188.731 |
| 10 000 | 20.788 | 290.975 | 311.927 | 209.519 |
Использованное для расчета значение энергии диссоциации ${{D}_{0}}$ составляло 71.8 см–1, а данные об энтальпиях образования Ar и O взяты из [16]. Указанные погрешности принятой энтальпии образования складываются из погрешностей энергии диссоциации и погрешности расчета инкремента энтальпии.
Температурный диапазон 298.15–10 000 К разбит на несколько меньших температурных интервалов, и на каждом интервале рассчитанные термодинамические функции аппроксимированы полиномом
(2)
$\begin{gathered} \Phi ^\circ (T) = {{\phi }_{1}} + {{\phi }_{2}}lnX + {{\phi }_{3}}{{X}^{{ - 2}}} + \\ + \,\,{{\phi }_{4}}{{X}^{{ - 1}}} + {{\phi }_{5}}X + {{\phi }_{6}}{{X}^{2}} + {{\phi }_{7}}{{X}^{3}}, \\ \end{gathered} $Таблица 3.
Коэффициенты полинома (2) для ArO и ArO+
| Интервал температур, K | ${{\phi }_{1}}$ | ${{\phi }_{2}}$ | ${{\phi }_{3}}$ | ${{\phi }_{4}}$ | ${{\phi }_{5}}$ | ${{\phi }_{6}}$ | ${{\phi }_{7}}$ |
|---|---|---|---|---|---|---|---|
| ArO | |||||||
| 298.15–3198 | 291.9035 | 21.1134 | 0.0004 | –0.1461 | –2.2687 | 3.4654 | –2.5384 |
| 3198–10 000 | 289.6044 | 19.1525 | 0.0223 | –0.5994 | 2.9052 | –1.2177 | 0.2581 |
| ArO+ | |||||||
| 298.15–1044 | 339.1083 | 39.2818 | –0.0006 | 0.2085 | –57.116 | 509.9801 | –1471.4015 |
| 1044–3154 | 446.8524 | 92.5407 | –0.0607 | 4.5252 | –267.8273 | 255.733 | –124.2619 |
| 3154–7956 | 314.1998 | 6.7299 | 0.3483 | –8.4695 | 25.3336 | –1.5092 | –1.5531 |
| 7956–10 000 | 337.8213 | 10.9033 | 2.2331 | –17.3788 | 0 | 8.5815 | –2.9002 |
Колебательно-вращательная статистическая сумма рассчитывалась также в работе [18]. Авторы использовали только основное электронное состояние с энергией диссоциации 61.4 см–1 без учета спин-орбитального расщепления. Принятое значение энергии диссоциации сильно отличается от величины, полученной в эксперименте [7] (84 см–1) и в более поздних расчетах [14] (87.8 см–1). Также при определении уровней колебательно-вращательной энергии авторы не рассматривали квазисвязанные состояния и состояния, которые лежат выше диссоционного предела, что привело к нахождению меньшего числа уровней. В результате авторы получили 57 колебательно-вращательных уровней, в то время как в настоящих расчетах использовано 83 энергетических уровня только для основного состояния.
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ ПОЛОЖИТЕЛЬНОГО ИОНА ОКСИДА АРГОНА
Основным состоянием иона ArO+ является состояние $^{4}{{\Sigma }^{ - }}.$ Экспериментальное исследование рассеяния ионов кислорода в основном состоянии $^{4}S$ на атомах аргона выполнено в работе [19]. Авторами получена эмпирическая зависимость потенциальной энергии взаимодействия от межатомного расстояния, а также значения равновесного межатомного расстояния и энергии диссоциации, которые представлены в табл. 4. Первые теоретические расчеты методом POL-CI для основного и для ряда возбужденных состояний проведены в работе [20]. Были получены два возбужденных связывающих электронных состояния (${{a}^{2}}\Pi $ и ${{b}^{2}}{{\Sigma }^{ - }}$), оба из которых имеют диссоционный предел, соответствующий распаду молекулы на атом кислорода в состоянии $^{3}P$ и положительный ион аргона в состоянии $^{2}P.$ Энергия диссоциации возбужденного состояния ${{a}^{2}}\Pi $ существенно больше (14 518 см–1), чем для основного состояния (5404 см–1), а потенциальная кривая пересекает кривую основного состояния при межатомном расстоянии, меньшем, чем равновесное расстояние для $^{4}{{\Sigma }^{ - }}.$ Второе возбужденное состояние ${{b}^{2}}{{\Sigma }^{ - }}$ имеет энергию диссоциации (4517 см–1), близкую к величине диссоциации основного состояния, однако в дальнейших работах по изучению электронной структуры ArO+ это электронное состояние не было обнаружено, поэтому здесь оно также исключено из рассмотрения.
Расчеты методами MP4(SDTQ)/6-311G(2df,2pd), MP2/6-31G(d,p) выполнены в [21]. Кроме основного состояния $^{4}{{\Sigma }^{ - }}$ и первого возбужденного состояния $^{2}\Pi $ авторы рассчитали энергию диссоциации электронного состояния с симметрией $^{2}\Pi ,$ которое приводит к диссоциации молекулы на атом Ar в состоянии $^{1}S$ и иона кислорода в состоянии $^{2}D.$ Потенциальные кривые основного и низколежащих электронных состояний приведены на рис. 2.
Рис. 2.
Диабатические потенциальные кривые низколежащих электронных состояний ArO+: 1 – потенциальная кривая, которая соответствует реакции ArO+(X4Σ) → Ar(1S) + O+($^{4}S$); 2 – ArO+(2П) → → Ar+(2P) + O($^{3}P$); 3 – ArO+${{(}^{2}}\Pi )$ → Ar($^{1}S$) + O+${{(}^{2}}D).$

Наиболее поздним исследованием электронной структуры ArO+ является работа [22] по определению подвижности ионов кислорода O+ в аргоне. Авторы выполнили квантово-химические расчеты методом RCCSD(T)/daug-cc-pV$\infty $Z для основного $^{4}{{\Sigma }^{ - }}$ и первого возбужденного $^{2}\Pi $ состояний. Полученные результаты показали хорошее согласие не только с данными по подвижности иона кислорода, но также лучшее согласие с экспериментальными данными по энергии диссоциации и равновесному межатомному расстоянию для основного состояния $^{4}{{\Sigma }^{ - }}$ [19, 20], чем расчеты [21] (табл. 4).
Также авторы [22] более детально изучили пересечение потенциальных кривых и показали, что этого пересечения удается избежать, если учитывать спин-орбитальное расщепление (расчет был проведен методом CASSCF). В результате было показано, что основное состояние имеет два минимума, а величина спин-орбитального расщепления вблизи точки пересечения потенциальных кривых состояний $^{4}{{\Sigma }^{ - }}$ и $^{2}\Pi $ составляет всего 240 см–1, что мало по сравнению с энергией диссоциации в этих электронных состояниях. Однако использование адиабатических (с учетом спин-орбитального расщепления) потенциальных кривых приводило к большому отклонению рассчитанных величин от полученных в эксперименте [22].
Для того чтобы определить, какое влияние спин-орбитальное расщепление оказывает на термодинамические функции, в настоящей работе рассчитаны термодинамические функции с учетом расщепления и в его отсутствии для основного состояния. Кривая $^{4}{{\Sigma }^{ - }}$ без учета спин-орбитального расщепления была аппроксимирована к EMO-потенциалу (1) с энергией диссоциации и равновесным расстоянием из [22]. Адиабатическая кривая, полученная с учетом спин-орбитального взаимодействия и имеющая два локальных минимума, проинтерполирована с использованием сплайнов. Расчеты показали, что максимальная разница для приведенной энергии Гиббса составляет 0.06 Дж К–1 моль–1 при температуре 10 000 К. Указанные различия являются малыми, что позволяет рассчитывать термодинамические функции исходя из диабатических (без учета спин-орбитального взаимодействия) кривых, практически не теряя в точности расчета.
Для итоговых расчетов термодинамических функций иона ArO+ взята потенциальная диабатическая кривая основного состояния из [22], так как расчет выполнен наиболее точным квантово-химическим методом. Данные о первом возбужденном $^{2}\Pi $-состоянии, которое диссоциирует на ион аргона и атом кислорода, взято из [21], так как различие с [22] для данного состояния по равновесному расстоянию незначительно. При этом в [22] отсутствуют данные о потенциальной кривой вблизи диссоциационного предела, которые являются важными для численного интегрирования уравнения Шредингера. Данные о втором $^{2}\Pi $-состоянии, продуктами распада которого являются атом аргона и ион кислорода, несмотря на низкую точность расчета, взяты из [21], так как другой информации об этом состоянии в литературе не найдено. Результаты расчетов термодинамических функций и коэффициенты аппроксимирующих полиномов (2) представлены в табл. 5 и 3. На основе полученных результатов об энергии основного колебательного уровня и принятых энергий диссоциации рассчитаны следущие энергии реакций:
для реакции ArO+ = Ar+ + O,
для реакции ArO+ = Ar + O+,
а также энтальпии образования:
Таблица 5.
Термодинамические функции ArO+
| $T$ | $C_{p}^{^\circ }(T)$ | ${{\Phi }^{^\circ }}(T)$ | $S^\circ (T)$ | $H^\circ (T) - H^\circ (0)$ |
|---|---|---|---|---|
| К | Дж К–1 моль–1 | кДж моль–1 | ||
| 298.15 | 36.739 | 206.131 | 238.896 | 9.769 |
| 300 | 36.763 | 206.334 | 239.123 | 9.837 |
| 400 | 37.739 | 215.93 | 249.843 | 13.566 |
| 500 | 38.494 | 223.592 | 258.347 | 17.378 |
| 600 | 39.248 | 229.99 | 265.431 | 21.265 |
| 700 | 39.979 | 235.499 | 271.537 | 25.227 |
| 800 | 40.537 | 240.347 | 276.915 | 29.255 |
| 900 | 40.777 | 244.681 | 281.707 | 33.323 |
| 1000 | 40.637 | 248.602 | 286 | 37.397 |
| 1500 | 36.296 | 263.925 | 301.778 | 56.779 |
| 2000 | 31.799 | 274.697 | 311.557 | 73.722 |
| 2500 | 29.316 | 282.783 | 318.352 | 88.924 |
| 3000 | 28.317 | 289.163 | 323.591 | 103.284 |
| 4000 | 28.678 | 298.835 | 331.735 | 131.602 |
| 5000 | 30.263 | 306.089 | 338.297 | 161.036 |
| 6000 | 31.811 | 311.939 | 343.957 | 192.107 |
| 7000 | 32.766 | 316.877 | 348.942 | 224.454 |
| 8000 | 33.03 | 321.166 | 353.341 | 257.406 |
| 9000 | 32.739 | 324.961 | 357.219 | 290.328 |
| 10 000 | 32.09 | 328.361 | 360.637 | 322.765 |
Для расчета энтальпий образования были использованы данные об энтальпиях образования Ar и O+ из [16]. Указанные погрешности энтальпий образования, как и в случае ArO, складываются из погрешностей принятых энергий диссоциации и погрешности расчета инкремента энтальпии.
ЗАКЛЮЧЕНИЕ
В работе впервые получены термодинамические функции ArO и ArO+ и энтальпии их образования. На примере молекулы ArO проведено сравнение методов расчета термодинамических функций по молекулярным постоянным и по потенциалу межатомного взаимодействия. Показано влияние выбора вида потенциальной кривой межатомного взаимодействия на рассчитываемые термодинамические функции. Для иона ArO+ выполнено сравнение результатов расчетов термодинамических функций с использованием диабатических и адиабатических потенциалов межатомного взаимодействия для основного и низколежащих возбужденных состояний. Результаты расчетов представлены в форме аппроксимационных полиномов, используемых в базе данных ИВТАНТЕРМО.
Список литературы
Maltsev M.A., Kulikov A.N., Morozov I.V. Thermodynamic Properties of Vanadium and Cobalt Argide Ions, VAr+ and CoAr+ // J. Phys.: Conf. Ser. 2016. V. 774. P. 012023.
Мальцев М.А., Морозов И.В., Осина Е.Л. Термодинамические свойства димеров аргона ${\text{Ar}}_{2}^{ + }$ и Ar2 // ТВТ. 2019. Т. 57. № 1. С. 42.
Мальцев М.А., Морозов И.В., Осина Е.Л. Термодинамические свойства ArH+ и ArH // ТВТ. 2019. Т. 57. № 3. С. 367.
Черноножкин С.М., Сапрыкин А.И. Применение лазерной абляции для анализа твердых образцов методом масс-спектрометрии с индуктивно-связанной плазмой // Масс-спектрометрия. 2012. Т. 9. С. 157.
Aquilanti V., Liuti G., Pirani F. et al. Absolute Total Elastic Cross Sections for Collisions of Oxygen Atoms with the Rare Gases at Thermal Energies // J. Chem. Phys. 1976. V. 65. P. 4751.
Aquilanti V., Luzzatti E., Pirani F. et al. Scattering of Magnetically Selected O($^{3}P$) Atoms: Characterization of the Low Lying States of the Heavy Rare Gas Oxides // J. Chem. Phys. 1980. V. 73. P. 1181.
Aquilanti V., Candori R., Pirani F. Molecular Beam Studies of Weak Interactions for Open-shell Systems: The Ground and Lowest Excited States of Rare Gas Oxides // J. Chem. Phys. 1988. V. 89. P. 6157.
Ma Z., Liu K., Harding L.B. Differential Cross Sections for Fine Structure Transitions in O($^{3}{{P}_{2}}$) + Ar Collisions // J. Chem. Phys. 1994. V. 100. P. 8026.
Arnold S.T., Hendricks J.H., Bowen K.H. Photoelectron Spectroscopy of the Solvated Anion Clusters ${{{\text{O}}}^{ - }}{{\left( {{\text{Ar}}} \right)}_{{n = 1 - 26.34}}}{\text{:}}$ Energetics and Structure // J. Chem. Phys. 1995. V. 102. P. 39.
DeClercq H.L., Hendricks J.H., Bowen K.H. Low Lying Electronic States of Rare Gas Oxide Anions: Photoelectron Spectroscopy of Complexes of O– with Ar, Kr, Xe, and N2 // J. Chem. Phys. 2002. V. 117. P. 2619.
Buchachenko A.A., Jakowski J., Chalasinski G. et al. Ab initio Based Study of the ArO– Photoelectron Spectra: Selectivity of Spin Orbit Transitions // J. Chem. Phys. 2000. V. 112. № 13. P. 5852.
Krems R.V., Buchachenko A.A., Szczsniak M.M. et al. Dynamics of O($^{3}{{P}_{j}}$) + Rg Collisions on ab initio and Scattering Potentials // J. Chem. Phys. 2002. V. 116. P. 1457.
Veihland L.A., Webb R., Lee E.P.F. et al. Accurate Potential Energy Curves for HeO–, NeO–, and ArO–: Spectroscopy and Transport Coefficients // J. Chem. Phys. 2005. V. 122. P. 114302.
Garand E, Buchachenko A.A., Yacovitch T.I. et al. Study of ArO– and ArO via Slow Photoelectron Velocity-map Imaging Spectroscopy and ab initio Calculations // J. Phys. Chem. 2009. V. 113. P. 4631.
Lee E.G., Seto J.Y., Hirao T. et al. FTIR Emission Spectra, Molecular Constants, and Potential Curve of Ground State GeO // J. Mol. Spectrosc. 1999. V. 194. № 2. P. 197.
Gurvich L.V., Veyts I.V., Alcock C.B. Thermodynamic Properties of Individual Substances, V. 1. Hemisphere Publishing Corp., 1989. P. 664.
Belov G.V., Aristova N.M., Morozov I.V. et al. On Approximation of the Heat Capacity of Substances in the Gasous State // J. Math. Chem. 2017. V. 55. P. 1683.
Prudente F.V., Riganelli A., Varandas A.J.C. Calculation of the Rovibrational Partition Function Using Classical Methods with Quantum Corrections // J. Chem. Phys. 2001. V. 105. P. 5272.
Ding A., Karlau J., Weise J. The Potential of Ar–O+(4${{\Sigma }^{ - }}$) // Chem. Phys. Lett. 1976. V. 45. № 1. P. 92.
Guest M.F., Ding A., Karlau J. et al. Potential Interactions between O+ and Rare Gases // Mol. Phys. 1979. V. 38. № 5. P. 1427.
Frenking G., Koch W, Cremer D. et al. Neon and Argon Bonding in First-row Cations NeX+ and ArX+ (X = = Li–Ne) // J. Phys. Chem. 1989. V. 93. № 9. P. 3410.
Danailov D.V., Viehland L.A., Johnsen R. et al. Transport of O+ Through Argon Gas // J. Chem. Phys. 2008. V. 128. P. 134 302.
Дополнительные материалы отсутствуют.
Инструменты
Теплофизика высоких температур