

Теплофизика высоких температур, 2020, T. 58, № 6, стр. 915-950
Проектирование материалов атомной энергетики: первопринципные расчеты и методы искусственного интеллекта
И. А. Абрикосов 1, Э. Е. Сон 2, *, Б. О. Мухамедов 1, А. В. Хван 1
1 Национальный исследовательский технологический университет “МИСиС”
Москва, Россия
2 Объединенный институт высоких температур РАН
Москва, Россия
* E-mail: son.eduard@gmail.com
Поступила в редакцию 10.03.2020
После доработки 20.08.2020
Принята к публикации 14.10.2020
Аннотация
Обзор содержит описание cовременных методов и результатов проектирования новых материалов атомной энергетики. Первая часть, представленная в данном обзоре, содержит описание первопринципных квантовых методов расчета свойств материалов, описаны строгие и приближенные квантовые методы (метод функционала плотности) для возможности предсказания свойств материалов на основе суперкомпьютерных расчетов и распространенные пакеты численного моделирования для высокопроизводительных кластеров, а также базы данных первопринципных расчетов, существующие в мире. С накоплением большого количества трудоемких квантовых расчетов все более эффективными становятся методы искусственного интеллекта, в частности методы машинного обучения, результаты использования которого для проектирования материалов представлены в обзоре. Приведены результаты квантового моделирования бинарных систем железа, хрома и никеля. Вторая часть обзора посвящена квантовому моделированию жидких и плазменных состояний вещества в реакторах, а также экспериментальным исследованиям материалов атомной энергетики. Третья часть обзора посвящена неравновесной плазме, образующейся в реакторах вследствие вторичных процессов при торможении быстрых частиц.
ОГЛАВЛЕНИЕ
Введение
1. Квантовые методы моделирования свойств материалов атомной энергетики
1.1. Теоретические основы квантово-механических расчетов
1.2. Теория функционала плотности
1.2.1. Обменно-корреляционный функционал
1.2.2. Приближение локальной плотности
1.2.3. Приближение обобщенного градиента
1.2.4. Методы решения уравнений Кона–Шэма
1.2.5. Метод псевдопотенциалов
1.2.6. Метод функций Грина. Теория многократного рассеяния
2. Основные программные пакеты, используемые для квантового моделирования материалов
2.1. VASP
2.2. Quantum ESPRESSO
2.3. ABINIT
2.4. WIEN2k
2.5. EMTO
3. Базы данных результатов первопринципных расчетов
3.1. Materials Project
3.2. Лаборатория открытия новых материалов (лаборатория NOMAD)
3.3. AFLOW (Automatic flow)
3.4. База данных квантовых материалов (OQMD)
3.5. Использование результатов первопринципных расчетов в создании новых материалов
4. Методы машинного обучения в создании новых материалов
4.1. Категории методов машинного обучения
4.1.1. Метод регуляризации Тихонова
4.1.2. Деревья принятия решений
4.1.3. Нейронные сети
4.2. Примеры применения машинных методов в материаловедении
4.3. Адаптивный дизайн материалов
4.4. Дескрипторы квантовых структур металлов
4.5. Предсказание критических температур машинными методами
5. Термодинамические свойства бинарных сплавов
5.1. Диаграммы состояния бинарных систем Fe–Cr
5.1.1. Фазовые равновесия
5.1.2. Термодинамические свойства
5.2. Диаграммы состояния бинарных систем Fe–Ni
5.2.1. Фазовые равновесия
5.2.2. Термодинамические свойства
5.3. Диаграммы состояния бинарных систем Сr–Ni
5.3.1. Фазовые равновесия
5.3.2. Термодинамические свойства
Заключение
Список литературы
ВВЕДЕНИЕ
В настоящее время происходит рост числа доступных материалов, связанный с увеличением информационной составляющей в их разработке. Экспоненциальный рост вычислительных мощностей и плотности хранения данных в сочетании с прогрессом в области компьютерных наук (e‑science) произвел информационную революцию. Методы управления данными в настоящее время нашли использование в технологиях поиска информации, науках о жизни, экономике, социальных сетях и т.д. Недавно была сформулирована инновационная идея интеграции методов разработки материалов с технологиями управления данными, так называемая информатика материалов или геном материалов. Ее реализация принесет изменение основной парадигмы в материаловедении, заменив традиционный метод проб и ошибок на высокопродуктивный научно-обоснованный подход. В ряде стран уже запущены программы с сильной государственной поддержкой в этом направлении (например, Центр проектирования материалов для функциональной электроники в США). В 2016 г. три Центра превосходства для исследования управления большими массивами данных в материаловедении начали свою деятельность в Европе: MAX, NOMAD и ECAM. Очень важно, чтобы Россия заняла ведущую роль в этой области. С этой целью в данной работе ставятся задачи и приводится обзор результатов, необходимых для проектирования материалов атомной энергетики. Для обеспечения безопасности действующих атомных станций возникает потребность в создании материалов, обладающих свойствами, которые удовлетворяли бы требованиям стандартов, долговременной эксплуатации и надежности. После определения необходимых свойств материалов на основе существующих интерактивных (постоянно дополняемых и корректируемых) баз данных может быть произведен анализ кандидатов на новые материалы. При этом производятся первопринципные расчеты и определяется молекулярная структура нового материала. После этого определяются технологии получения этих материалов и, например, на основе аддитивных технологий создается новый материал. Полученный материал должен пройти полный цикл испытаний и сертифицикацию. После этого он готов для внедрения в организациях, проектирующих и создающих новые реакторы. Хотя это непростой путь, включающий использование интерактивных баз данных, первопринципные квантовые расчеты, верификацию, валидацию, разработку технологий и экспериментальные исследования, по-видимому, это наиболее эффективный путь, который может привести к успеху в области нового атомного машиностроения.
Целью настоящего обзора является дать представление о возможности научно обоснованной разработки новых материалов на основе управления большими массивами данных, предлагаемой революционным прогрессом теории моделирования, вычислительных мощностей, информатики, статистики и визуализации. Для решения поставленных задач необходимо производить, сохранять и классифицировать большие массивы данных о материалах на основе компьютерного моделирования и экспериментов, а также использовать широкий круг литературных источников и доступных баз данных. Исследования больших массивов данных с использованием эффективных алгоритмов машинного обучения позволит открыть новые материалы и явления с многообещающими технологическими применениями, распознать новые соотношения “параметр–свойства” и “параметр–параметр”. Разрабатываемый подход будет иметь достаточный потенциал, чтобы значительно снизить (на порядок и более) стоимость, риски и время открытия и коммерциализации новых материалов. Он позволит получать качественно новые идеи, недоступные для обычных методов исследования. Действительно, эффективность перехода от данных к знаниям зависит от адекватности и правильности методов абстракции. Примерами успешных абстракций являются Периодическая таблица элементов Менделеева или так называемые диаграммы Эшби, графически отображающие соотношения между двумя или большим числом свойств материалов или их классов. Подобные диаграммы широко используются учеными-материаловедами и инженерами. Однако использование подходов, разработанных для больших массивов данных в материаловедении, до последнего времени ограничивалось чрезмерной трудоемкостью экспериментального получения самих данных. С использованием компьютерного моделирования эффективность получения данных о свойствах материалов значительно возрастает. При этом задача перехода от данных к знаниям с использованием традиционных эмпирических методов становится трудноразрешимой. С использованием новых революционных подходов можно выявить научный контекст ключевых концепций в материаловедении и использовать его для моделирования и прогнозирования свойств и поведения материалов при различных условиях и в массе приложений.
За последние пять лет научными группами, включающими авторов настоящего обзора, выполнены многочисленные проекты в таких сферах, как моделирование свойств и разработка материалов из первых принципов, экспериментальное определение термодинамических свойств, критическая оценка фазовых диаграмм и термодинамических данных, построение и управление базами данных и расчеты из них для промышленности, кинетическое моделирование процессов и экспериментальных исследований, синтез и характеристика новых материалов. Основная цель исследований заключалась в разработке нового поколения теоретических инструментов, обладающих достаточной предсказательной силой для теоретически обоснованной разработки материалов.
1. КВАНТОВЫЕ МЕТОДЫ МОДЕЛИРОВАНИЯ СВОЙСТВ МАТЕРИАЛОВ АТОМНОЙ ЭНЕРГЕТИКИ
1.1. Теоретические основы квантово-механических расчетов
Одна из важных задач современного материаловедения – необходимость создавать сплавы с определенными технологическими свойствами. Для достижения этой цели необходимо иметь глубокое понимание фундаментальных механизмов, лежащих в основе поведения материалов.
В середине 1960-х годов Кон и Шэм сформулировали теорию функционала плотности (ТФП) [1, 2], которая не только дала возможность теоретического изучения электронной структуры, но и привела к возможности предсказывать свойства материалов с использованием компьютерного моделирования без каких-либо экспериментальных или феноменологических параметров с точностью, сопоставимой с экспериментом. Поэтому подобные расчеты принято называть первопринципными или ab initio. При этом современная практика показала, что эффективность определения свойств материалов на основе квантово-механических расчетов с использованием ТФП очень высока и компьютерное моделирование стало новым инструментом исследования свойств материалов, дополняющим эксперимент и существенно снижающим затраты и время, необходимые на проведение исследований. В 1998 г. Уолтер Кон был удостоен Нобелевской премии за эту теорию. В настоящее время в рамках ТФП могут быть успешно решены многие практические проблемы материаловедения. Более того, за прошедший с момента открытия Кона период были разработаны различные пакеты компьютерных программ для расчетов ab initio. Важно, что данные методы расчета используются для моделирования не только в физике, но и в химии, геофизике, биофизике, металлургии и в других областях знаний. Приведем обзор теоретических основ и основных используемых подходов для первопринципного исследования и создания новых материалов.
1.2. Теория функционала плотности
Описание электронной структуры твердых тел можно получить исходя из законов квантовой механики. Вся информация о системе содержится в многочастичной волновой функции электронов. Волновая функция $\Psi $ является решением уравнения Шредингера, но из-за огромного числа степеней свободы это уравнение не может быть решено точно. Поэтому для решения задач, связанных с описанием электронной структуры кристаллов, используются дополнительные приближения, подробно описанные в работе [3], взятой за основу.
Во-первых, в большинстве случаев используется приближение Борна–Оппенгеймера (или адиабатическое приближение). Это приближение позволяет рассматривать движение ядер и электронов отдельно, то есть можно считать, что электроны движутся в поле неподвижных ядер. Обоснованность этого приближения определяется тем, что скорости движения электронов примерно на два порядка выше, чем скорости ядер, поэтому соответствующее электронное равновесие будет устанавливаться при любой, даже неравновесной, конфигурации ядер. В этом случае электронная и ядерная подсистемы не обмениваются энергией, поэтому это приближение называется адиабатическим.
С учетом приближения Борна–Оппенгеймера уравнение Шредингера в общем виде выглядит следующим образом:
Теория функционала электронной плотности (ТФП), основанная на трудах Хоэнберга–Кона [1] и Кона–Шэма [2], была предложена для описания многоэлектронной системы. Основная идея ТФП состоит в том, чтобы решить многочастичное уравнение Шредингера, заменив полную электронную волновую функцию гораздо более простой электронной плотностью основного состояния в качестве фундаментальной переменной. Согласно этой теории, все свойства электронной структуры системы в невырожденном основном состоянии полностью определяются ее электронной плотностью. Таким образом, многоэлектронная волновая функция 3N переменных заменяется электронной плотностью с тремя переменными, что существенно упрощает задачу. Главным преимуществом ТФП является возможность изучения невзаимодействующих друг с другом электронов в эффективном потенциале, т.е. вместо системы с большим числом электронов во внешнем потенциале ядер можно рассматривать электронную плотность в некотором внешнем эффективном потенциале.
ТФП строится на двух основных теоремах Хоэнберга и Кона [1, 2]: 1) электронная плотность основного состояния $n\left( r \right)$ однозначно определяет внешний потенциал системы ${{\upsilon }_{e}}\left( r \right)$ с точностью до произвольной константы. Электронная плотность может быть использована как базовая функция, однозначно характеризующая систему; 2) для некоторого вполне определенного внешнего потенциала ${{\upsilon }_{e}}\left( r \right)$ минимум функционала полной энергии ${{E}_{e}}\left[ n \right]$ достигается для электронной плотности основного состояния $n\left( r \right)$.
Основное состояние электронной системы для газа взаимодействующих частиц во внешнем потенциале ${{\upsilon }_{e}}\left( r \right)$ можно описать функционалом полной энергии:
Согласно вариационному принципу, равновесному значению электронной плотности соответствует минимум функционала ${{E}_{e}}\left[ n \right]$, равный полной энергии электронной системы. Универсальный функционал можно представить в виде
(2)
$F\left[ n \right] = {{T}_{0}}\left[ n \right] + {{E}_{H}}\left[ n \right] + {{E}_{{xc}}}\left[ n \right],$Используя вариационный метод, Кон и Шэм получили систему одноэлектронных уравнений:
(3)
$\begin{gathered} H\psi = \left( { - {{\nabla }^{2}} + {{\upsilon }_{{{\text{eff}}}}}} \right)\psi = E\psi , \\ n\left( r \right) = \sum\limits_{occ} {\psi {\text{*}}\psi } , \\ {{\upsilon }_{{{\text{eff}}}}} = {{\upsilon }_{H}}\left( {n\left( r \right)} \right) + {{\upsilon }_{{xc}}}\left( {n\left( r \right)} \right) + {{\upsilon }_{{ei}}}\left( {n\left( r \right)} \right). \\ \end{gathered} $Решение уравнений Кона–Шэма проводится самосогласованно следующими этапами.
1. Для некоторой затравочной плотности вычисляется эффективный потенциал.
2. Рассчитывается волновая функция, соответствующая эффективному потенциалу.
3. Определяется новая электронная плотность.
4. Вычисляется новый потенциал, соответствующий новой плотности.
5. Если достигнута необходимая точность вычисления энергии, расчет прекращается, в противном случае он повторяется до достижения сходимости.
Для магнитных систем, исследуемых в настоящем проекте, в описании системы необходимо учитывать скалярную электронную плотность $n\left( r \right)$ и плотность намагничивания $m\left( r \right)$. Как вариант, можно использовать $2 \times 2$ матрицу спин-разрешенной плотности ${{n}^{{\alpha \beta }}}\left( r \right)$, где $\alpha $ и $\beta $ – индексы спина, которые могут принимать два значения: спин-вверх $ \uparrow $ и спин-вниз $ \downarrow $. Связь между параметрами $n\left( r \right)$, ${{n}^{{\alpha \beta }}}\left( r \right)$ и вектором $m\left( r \right)$ описывается следующими уравнениями:
1.2.1. Обменно-корреляционный функционал. Обменно-корреляционный функционал ${{E}_{{xc}}}$ точно не определен. Основные используемые в современной практике расчетов электронной структуры приближения: приближение локальной плотности (для магнитных систем приближение локальной спиновой плотности) и приближение обобщенного градиента (ПОГ).
1.2.2. Приближение локальной плотности. Для описания обменно-корреляционных эффектов Кон и Шэм предложили приближение, в котором действительная электронная плотность заменяется плотностью однородного электронного газа, т.е. плотность обменно-корреляционной энергии в данной точке должна быть равна плотности обменно-корреляционной энергии однородного электронного газа с такой же электронной плотностью. Электронная плотность может быть рассчитана и параметризована в удобном для использования виде. Это приближение получило название приближение локальной плотности (ПЛП). Согласно этому приближению, обменно-корреляционный функционал может быть представлен в виде
Это приближение позволяет достаточно хорошо описывать объемные и поверхностные свойства $4d$ и $5d$ переходных металлов [3]. Но, например, для $3d$ переходных металлов результаты, полученные в рамках ПЛП, могут достаточно сильно отклоняться от экспериментальных значений. Для атомов и молекул метод ПЛП занижает обменную энергию примерно на 10%, тогда как корреляционные энергии могут отличаться на 100%. Для большинства систем эффект обменного взаимодействия существенно (на порядок) больше корреляционных взаимодействий, поэтому итоговый результат не столь значительно (обычно порядка 7%) отличается от реальных значений [3].
1.2.3. Приближение обобщенного градиента. Приближение обобщенного градиента [4] позволяет частично разрешить проблемы, возникающие при использовании ПЛП. Это приближение учитывает в функционале энергии зависимость от градиента электронной плотности и в общем виде выглядит как
Для приближения обобщенного градиента существует ряд подходящих параметризаций, позволяющих увеличить точность расчетов. Наиболее часто встречающиеся – это параметризации PW91 [4] и PBE [5].
Использование приближения ОГП позволило значительно повысить точность описания свойств твердых тел в рамках теории функционала плотности, однако не все проблемы удалось решить. Поэтому продолжается поиск приближений для более точного описания обменно-корреляционного функционала.
1.2.4. Методы решения уравнений Кона–Шэма. Для эффективного численного решения уравнений Кона–Шэма был разработан ряд методов. Например, методы, использующие полноэлектронный потенциал, являются оптимальными для наиболее точного описания локальной электронной плотности или ее градиента в твердых телах [6–8]. Также эти методы хорошо подходят для расчетов физических свойств упорядоченных структур и дефектов в этих системах. Однако применение этих методов очень ресурсозатратно и они имеют ряд ограничений, обусловленных различными численными приближениями. Необходимая точность решений уравнений Кона–Шэма определяется исследуемыми характеристиками материала. Для описания свойств статически неподвижной кристаллической структуры не требуется высокой точности, тогда как для оценки энергии дефектов кристаллической структуры точность полученных результатов очень важна. Поэтому для каждой задачи необходимо подобрать параметры расчетов исходя из баланса точности и эффективности вычислений. Часто вместо полнозарядовых потенциалов используется сочетание замороженного остова и псевдопотенциала. В этом случае можно исключить из итераций глубоко лежащие коровские состояния и учитывать лишь валентные электроны. В междоузельных областях сохраняется полнопотенциальное описание, тогда как кулоновский потенциал вблизи ядра заменяется на гладкий псевдопотенциал с непрерывной сшивкой на границе областей. Полученные таким образом физические свойства практически не отличаются от тех, что были получены полноэлектронными методами, но скорость вычислений существенно выросла. Рассмотрим наиболее часто встречающиеся методы.
1.2.5. Метод псевдопотенциалов. Волновые функции электронов в атоме можно разделить на две области: коровская (вблизи ядра) область с осциллирующей волновой функцией и валентная область с гладкой волновой функцией [9] (рис. 1). Так как глубоко лежащие коровские электроны практически не участвуют в образовании химической связи, то основной задачей метода псевдопотенциала является замена осциллирующей части волновой функции на гладкую функцию, которая тем не менее не меняет свойства системы. Псевдопотенциал должен быть гладким, но его форма также не должна влиять на точность расчета физических свойств.
Рис. 1.
Схематическое изображение метода псевдопотенциала в соответствии с [9]: (а) реальный потенциал заменяется на специально построенный псевдопотенциал (1), (б) осциллирующая волновая функция вблизи ядра аппроксимируется гладкой функцией, совпадающей с исходной волновой функцией за пределами радиуса обрезания (2).
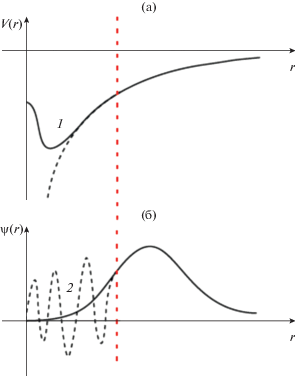
Основной характеристикой метода псевдопотенциалов является энергия обрезания ${{E}_{{{\text{cut}}}}}$ – энергия, определяющая максимальную кинетическую энергию плоской волны, которая будет входить в базисный набор плоских волн. Таким образом, если энергия обрезания равна ${{E}_{{{\text{cut}}}}}$, то все волновые векторы $k$ используемых в качестве базиса плоских волн вместе с базисом обратного пространства $G$ должны удовлетворять следующему условию:
гдеПри увеличении энергии обрезания растет число используемых плоских волн, улучшается точность вычислений, общее время расчета также возрастает, поэтому в каждом случае необходимо проводить исследование влияния энергии обрезания на свойства системы и выбирать соответствующую граничную энергию ${{E}_{{{\text{cut}}}}}$. Более того, существенного увеличения точности моделирования с использованием метода псевдопотенциала удалось добиться в методе проекторов присоединенных волн, который будет подробно описан ниже.
1.2.6. Метод функций Грина. Теория многократного рассеяния. Метод Корринги, Кона и Ростокера (ККР) [10, 11], основанный на теории многократного рассеяния [12, 13], был разработан для решения уравнений Кона–Шэма. Основная идея метода ККР заключается в использовании формализма функций Грина. Основной принцип заключается в определении функции Грина $G$, связанной с гамильтонианом Кона–Шэма ${{H}^{{{\text{KS}}}}}$:
Рис. 2.
Схема формализма многократного рассеяния в соответствии с [14]: волна выходит из узла i и несколько раз рассеивается, прежде чем достигнет узла j; влияние всех рассеяний между i и j описывается оператором пути рассеяния τij, рассеяние на одном узле k описывается t-матрицей t k, связанной с потенциалом υk на узле k.
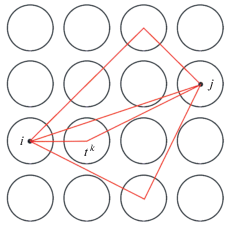
Плотность электронных состояний, как спектральная характеристика системы, может быть получена из мнимой части функции Грина. Зарядовая плотность $n\left( r \right)$ соответствует условию
2. ОСНОВНЫЕ ПРОГРАММНЫЕ ПАКЕТЫ, ИСПОЛЬЗУЕМЫЕ ДЛЯ КВАНТОВОГО МОДЕЛИРОВАНИЯ МАТЕРИАЛОВ
Расчеты с использованием теории функционала плотности (ТФП) являются ярким примером области, которая зависит от разработки и правильного использования сложного программного обеспечения. Базируясь на законах квантовой механики, ТФП описывает структуру и свойства молекул и твердых тел в атомном масштабе. За прошедшие годы многие академические группы разработали реализации ТФП в компьютерных кодах, и некоторые из них были приняты большими сообществами пользователей. Также существуют коммерческие продукты в этой области. В настоящее время ежегодно публикуется более 15 000 статей, в которых используются различные программные пакеты, базирующиеся на ТФП. Более того, расчеты ТФП используются в настоящее время для создания больших баз данных и в многомасштабных вычислениях, в которых они являются одним из звеньев цепочки моделирования.
За последние годы программные пакеты ТФП значительно изменились. Переход от небольших и персонализированных кодов к широко распространенным универсальным пакетам способствовал значительному увеличению точности расчетов. Путем сравнения результатов расчетов для разных кодов с помощью погрешности D было показано, что ошибки в определении уравнения состояния сравнимы с соответствующими ошибками между различными высокоточными экспериментами. Это показывает достоверность результатов ТФП-расчетов и подтверждает, что правильно сходящиеся вычисления дают надежные прогнозы.
Рассмотрим наиболее часто используемые программные пакеты.
2.1. VASP
Vienna Ab initio Simulation Package (VASP) – компьютерная программа для моделирования материалов на атомном уровне [15, 16]. VASP вычисляет приближенное решение уравнения Шредингера для многих тел как в рамках теории функционала плотности (ТФП) (решения уравнений Кона–Шэма), так и в приближении Хартри–Фока (HF) (решения уравнений Рутана). Также в этом пакете реализованы гибридные функционалы, которые смешивают подход Хартри–Фока с теорией функционала плотности. Кроме того, в VASP доступны методы функций Грина (квазичастицы GW и ACFDT-RPA). В VASP основные величины, такие как одноэлектронные орбитали, плотность электронного заряда и локальный потенциал, выражаются в базисах плоских волн. Взаимодействия между электронами и ионами описываются с использованием ультрамягких псевдопотенциалов, или методом проектора присоединенных плоских волн.
Чтобы определить основное состояние, VASP использует эффективные методы диагонализации итерационных матриц, такие как метод минимизации невязки с прямым обращением итеративного подпространства (RMM-DIIS) или блокированные алгоритмы Дэвидсона. Они связаны с высокоэффективными схемами смешивания плотности Бройдена и Пулая для ускорения цикла самосогласования.
2.2. Quantum ESPRESSO
Quantum ESPRESSO (QE) – интегрированный набор компьютерных кодов с открытым исходным кодом для расчетов электронной структуры и моделирования материалов на наноуровне [17]. Он основан на теории функционала плотности, плоских волнах и псевдопотенциалах.
Расчеты в QE проводятся в базисе плоских волн с использованием сепарабельных сохраняющих норму (Гаманн–Шлютер–Чинг, Труллер–Мартинс и др. типов) или ультрамягких псевдопотенциалов, а также метода PAW. QE позволяет проводить расчеты с использованием обменно-корреляционных функционалов различного вида: от приближения локальной плотности (ПЛП), до обобщенно-градиентного приближения (ОГП). В QE существует обширная библиотека псевдопотенциалов различных атомов. Кроме того, псевдопотенциалы с необходимыми параметрами могут быть получены с использованием входящей в состав пакета программы ld1.
Ключевые возможности программного пакета QE:
• решение уравнений Кона–Шэма и определение полной энергии многоэлектронных систем,
• расчет распределения электронной плотности,
• вычисление полной и парциальных плотностей состояний,
• расчет атомных зарядов по схеме Левдина,
• расчет межатомных сил, тензора напряжений и структурная оптимизация,
• исследование эволюции многоатомных систем методом молекулярной динамики,
• определение энергетических барьеров и путей реакций,
• расчет эффективных зарядов Борна и диэлектрической проницаемости,
• расчет фононных частот и векторов поляризации в рамках теории возмущения для функционала плотности (DFPT), вычисление силовых постоянных кристалла,
• расчет коэффициентов электрон-фононного взаимодействия для металлов,
• расчет макроскопической поляризации методом фазы Берри.
2.3. ABINIT
ABINIT – это программный комплекс для расчета оптических, механических, вибрационных и термодинамических свойств материалов [18].
ABINIT базируется на теории функционала плотности, решая уравнения Кона–Шэма путем разложения в базисный набор плоских волн и используя метод самосогласованного сопряженного градиента для определения минимума энергии. Вычислительная эффективность достигается за счет использования быстрых преобразований Фурье и псевдопотенциалов для описания остовных электронов. В качестве альтернативы стандартным псевдопотенциалам, сохраняющим норму, можно использовать метод проектора присоединенных плоских волн. В дополнение к полной энергии рассчитываются также силы и напряжения, чтобы можно было проводить оптимизацию геометрии и молекулярную динамику ab initio. С помощью ABINIT можно рассчитывать молекулы, наноструктуры и твердые вещества с любым химическим составом, в нем есть полная база атомных потенциалов.
В дополнение к нахождению основного состояния вещества, в ABINIT реализована теория возмущений в рамках функционала плотности для вычисления функций отклика, включая расчет фононных состояний, диэлектрического отклика, реакции на деформации, различные нелинейные отклики, в том числе пьезоэлектрический отклик, рамановские сечения и электрооптический отклик. В ABINIT также можно вычислять свойства возбужденного состояния с помощью зависящей от времени теории функционала плотности и теории возмущений многих тел, использующих приближение GW и уравнение Бете–Солпитера.
2.4. WIEN2k
Программный пакет WIEN2k позволяет выполнять расчеты электронной структуры твердых тел с использованием теории функционала плотности [19]. Он основан на методе линеаризованной присоединенной плоской волны (LAPW), который является одним из наиболее точных методов для расчета электронной структуры кристаллов, так как не использует различные приближения для описания потенциала.
Для учета обмена и корреляции в пакете используется как приближение локальной спиновой плотности (ПЛСП), так и приближение обобщенного градиента (ОГП). Для расчета валентных состояний релятивистские эффекты учитываются либо с применением скалярного релятивистского подхода, либо с помощью вариационного метода, включающего спин-орбитальные взаимодействия. Основные состояния рассматриваются полностью релятивистским подходом. Чтобы улучшить процедуру линеаризации (то есть увеличить гибкость базиса) и сделать возможным последовательную обработку полуостовных и валентных состояний в одном энергетическом окне (для обеспечения ортогональности), могут быть добавлены дополнительные базисные функции (“локальные орбитали”).
С использованием WIEN2k можно рассчитать полную энергию, силы на атомах, энергию Ферми и вес каждого зонного состояния, спин-орбитальные взаимодействия, плотность состояний, рентгеновские спектры поглощения и излучения, рентгеноструктурные факторы и оптические свойства.
2.5. EMTO
EMTO-CPA – программный пакет для расчета электронной структуры твердых тел, основанный на формализме точных маффин–тин-орбиталей [20, 21]. Учет полной зарядовой плотности обеспечивает точность расчетов, сопоставимую с методами с полным потенциалом. Основным преимуществом EMTO является возможность использования приближения когерентного потенциала для эффективного расчета свойств неупорядоченных структур, например высокоэнтропийных сплавов. Реализованный в этом пакете метод неупорядоченных локальных моментов (DLM) можно использовать для моделирования магнитного разупорядочения, такого как пагамагнетизм. Отметим, что оригинальные результаты расчетов для бинарных, тройных и четырехкомпонентных ОЦК-сплавов на основе системы Fe–Cr, приведенные в данном обзоре, выполнены именно этим методом.
Точность кодов ТФП лежит в основе научной достоверности и воспроизводимости значительной части текущей работы в области естественных и технических наук, и, следовательно, это имеет последствия, которые выходят далеко за рамки традиционного сообщества исследователей электронной структуры. В работе [22] было проведено сравнение результатов расчетов электронной структуры для чистых элементов с использованием различных программных пакетов для первопринципного моделирования. Поскольку вид обменно-корреляционного функционала точно неизвестен, были использованы различные приближения для его описания. После того как конкретный обменно-корреляционный функционал выбран, математическая задача полностью определяется как система уравнений Кона–Шэма, решение которых дает волновые функции и энергии, из которых можно оценить полную электронную энергию системы. Разнообразие схем численного решения было реализовано в различных программных пакетах. Можно ожидать, что, поскольку они решают одни и те же уравнения, они все должны давать одинаковые ответы для данной кристаллической структуры, но анализ литературных источников показывает, что это предположение далеко не всегда верно. Однако, когда сравнение различных программных пакетов проводится с использованием одного и того же обменно-корреляционного функционала, они должны давать одинаковую точность. К сожалению, конкретные результаты могут варьироваться от одного кода к другому из-за приближений, которые не связаны с функционалом обмен–корреляция. Эти приближения уменьшают вычислительную нагрузку, но ограничивают точность.
В работе [22] для 69 различных программных пакетов была определена погрешность D в расчете зависимости энергии от объема для чистых элементов, при этом выполнен один и тот же протокол эталонных тестов с различными кодами ТФП. Параметры этих уравнений состояния (EOS), такие как параметр решетки или объемный модуль, обычно используются для оценки точности. Таким образом, создан комплексный тест на точность вычислений в различных пакетах первопринципного моделирования.
Вначале была определена погрешность по отношению к полноэлектронным методам. Обычно эти методы требуют существенно больших вычислительных затрат, но подходы, основанные на использовании электронов, часто считаются стандартом для расчетов ТФП, поскольку они реализуют потенциал без изменения. Сравнивая методы псевдопотенциала или PAW с полноэлектронными кодами, можно получить представление о панели ошибок, связанной с каждой схемой оптимизации. Значения погрешности D между различными полноэлектронными методами отражают остающиеся расхождения в кодах, такие как разная трактовка скалярно-релятивистских членов или небольшие различия в численных методах.
По сравнению с программными пакетами, реализующими полноэлектронные потенциалы, подходы, использующие различные “псевдооптимизации”, как правило, быстрее, потому что в них рассматривается меньшее количество состояний и не производится явное построение и диагонализация гамильтоновой матрицы. Среди них для методов PAW и ультрамягких псевдопотенциалов требуется меньше базисных функций, чем для полноэлектронных методов, сохраняющих норму, но такие расширенные функции, как теория линейного отклика или гибридные функционалы, иногда могут быть недоступны из-за повышенной сложности реализации. Тем не менее все оптимизированные коды дают значения, хорошо согласующиеся с результатами полноэлектронных расчетов.
На основе анализа литературных данных можно сделать вывод о высокой надежности и эффективности квантово-механического компьютерного моделирования, проводимого с использованием пакетов компьютерных программ, реализующих методы ТФП. Результаты моделирования станут важным компонентом базы данных “Материалы для атомной энергетики”.
Ниже будет дана более развернутая информация о базах данных и о том, как методы машинного обучения и высокопроизводительных расчетов могут быть применены для предсказания компонентного состава материалов, их структуры, а также требуемых свойств.
Проанализируем основные базы данных результатов первопринципных расчетов.
3. БАЗЫ ДАННЫХ РЕЗУЛЬТАТОВ ПЕРВОПРИНЦИПНЫХ РАСЧЕТОВ
3.1. Materials Project
База данных Materials Project11 [23] – основная программа Инициативы генома материалов, которая использует высокопроизводительные вычисления для раскрытия свойств всех известных неорганических материалов. Materials Project (MP) объединяет в себе инструменты для высокопроизводительных вычислений, распространение данных через Интернет и инструменты для анализа с открытым исходным кодом, что позволяет ученым-исследователям по-новому взглянуть на проблему обнаружения материалов. Главная задача проекта – это ускорение дизайна материалов путем создания открытых систем для совместной работы, ориентированных на каждый этап процесса проектирования вычислительных материалов – создание данных, их проверка, распространение и дизайн. Данный проект призван создать основу, которая может быть расширена до новых областей. По мере разработки новых теоретических методов их можно подключить к процессу дизайна материалов и применить ко всем материалам в базе данных. Предпринимаются усилия, направленные на прогнозирование поверхностных энергий, упругих постоянных, точечных дефектов и свойств конечной температуры с использованием больших наборов данных и новых алгоритмов. Веб-распространение и интерфейсы отображения расширяются до новых наборов данных, таких как экспериментальные данные для сравнения с вычисленными результатами. Наконец, функция “вычисления по требованию” позволит ученым напрямую вносить новые идеи в базу данных. MP не только является конечным продуктом, но и представляет собой развивающийся ресурс, который, как ожидается, станет более полезным благодаря постоянно растущим наборам данных и эффекту от взаимодействия пользователей в Сети. Инициатива распространения большого количества точной информации в сообществе разработчиков материалов должна значительно ускорить и дать возможность открывать лучшие материалы для будущих систем экологически чистой энергии, компонентов экологически чистого строительства, передовой электроники, а также для улучшения здоровья и благополучия общества.
3.2. Лаборатория открытия новых материалов (лаборатория NOMAD)
Еще один проект платформы для обмена и использования вычислительных данных о материалах – это Лаборатория открытия новых материалов (NOMAD Laboratory22) [24]. Лаборатория NOMAD разделяет методологию FAIR [25] (findable, accessible, interoperable, reusable) распространения данных, что значит легкость в нахождении, доступность, совместимость и многоразовость. Эти характеристики крайне важны при обмене данными. Очевидно, если все входные и выходные файлы вычислений являются общими, вычисления не нужно повторять снова и снова, от чего выигрывает все сообщество. Кроме того, если большая группа исследователей имеют доступ к большим данным, то эти данные могут использоваться совершенно новыми исследовательскими методами, то есть методами искусственного интеллекта, для обнаружения ранее не обнаруженных, инновационных материалов. Лаборатория NOMAD обрабатывает, курирует и размещает вычислительные данные о материалах, рассчитанные с помощью всех важных материаловедческих программных пакетов, доступных сегодня, и делает эти данные доступными, предоставляя несколько взаимосвязанных инструментов для работы с данных. Совместно с разработчиками кода или опытными пользователями продолжает добавляться поддержка новых программных пакетов. Общая цель заключается в том, чтобы продвигать материаловедение, позволяя исследователям в области фундаментальной науки и техники понимать и использовать данные материалов для выявления лучших, неисследованных материалов.
У лаборатории NOMAD существует множество сервисов, один из которых (репозиторий NOMAD) содержит необработанные данные в том виде, в котором они созданы используемым компьютерным кодом, и эти данные составляют основу всех сервисов NOMAD. Архив NOMAD содержит нормализованные данные, то есть данные открытого доступа из Репозитория NOMAD, преобразованные в общий, независимый от кода формат. Другими словами, многочисленные парсеры считывают и переводят всю информацию, содержащуюся во входных и выходных файлах. Это обеспечивает одно из положений методологии FAIR, а именно то, что данные из разных источников могут сравниваться и, следовательно, совместно обрабатываться различными инструментами NOMAD (и другими). Инструменты Визуализации NOMAD обеспечивают удаленную визуализацию многомерных данных NOMAD через специальную инфраструктуру, разработанную в лаборатории NOMAD. Централизованный сервис позволяет пользователям в интерактивном режиме выполнять комплексные задачи по визуализации данных на своих компьютерах без необходимости установки специального оборудования или программного обеспечения. Пользователи имеют доступ к данным и инструментам, используя стандартные устройства (ноутбуки, смартфоны), независимо от их местоположения, кроме того, имеется возможность исследования данных в виртуальной реальности (VR). Энциклопедия NOMAD – это общедоступная веб-платформа, которая, взаимодействуя с Архивом NOMAD, помогает искать свойства разных материалов. Например, такие как структурные особенности, механические и термические свойства, электронные и магнитные свойства, реакцию на свет и многое другое. Также NOMAD предоставляет инструменты для работы с большими данными. Это Аналитический инструментарий NOMAD, который предоставляет набор примеров и инструментов, чтобы продемонстрировать, как данные материалов могут быть превращены в знания.
Долгосрочная перспектива Лаборатории NOMAD состоит в том, чтобы предоставить карты материалов, которые будут показывать, какие материалы и с каким составом требуются для определенного применения, будь то термоэлектрические материалы, материалы для солнечных батарей, сверхпроводники, топологические изоляторы или другие системы, представляющие интерес.
3.3. AFLOW (Automatic flow)
Достижения в области вычислительного материаловедения создают новые возможности для открытия и оптимизации структур, включая обнаружение неожиданных соединений и метастабильных структур, электронной структуры, свойств поверхности и наночастиц. Практическая реализация этих возможностей требует систематической генерации и классификации соответствующих вычислительных данных высокопроизводительными методами. Авторы [26] представили AFLOW (Automatic flow) – программную среду для высокопроизводительного расчета свойств кристаллической структуры сплавов, интерметаллидов и неорганических соединений. Программное обеспечение AFLOW доступно для научного сообщества на веб-сайте консорциума по исследованию материалов aflowlib.org. Его инструменты для анализа и манипулирования геометрической и электронной структурой также доступны для онлайн на веб-сайте.
Высокопроизводительный фреймворк AFLOW – это набор инструментов, написанный на С++, под работу с системами UNIX. Он подготовлен для работы поверх программных пакетов для первопринципных расчетов, и оптимизирован под работу с VASP и Quantum Espresso. Главная его особенность заключается в том, что он может работать параллельно и в нескольких потоках. Код предлагает несколько опций для запуска пакета ТФП, либо на одной структуре, либо на наборах структур. AFLOW позволяет автоматически вычислять набор физических наблюдаемых по заданному классу или большую базу данных структур с небольшим вмешательством человека для настройки входных файлов, выполнения вычислений, сопоставления и построения результатов. Также AFLOW может сам генерировать, запускать, корректировать и доводить до сходимости множество расчетов без вмешательства человека. Кроме того, база данных AFLOW включает в себя несколько миллионов суперструктур вида ОЦК и ГЦК (содержащих до 20 атомов на ячейку) и такое же количество суперструктур вида ГПУ (все из которых содержат до 24 атомов на ячейку). AFLOW предлагает инструменты для анализа и взаимодействия со структурами, полезные для пользователей, которым не нужно выполнять высокопроизводительные вычисления или создавать базы данных для анализа данных.
3.4. База данных квантовых материалов (OQMD)
Еще один проект, открытая база данных квантовых материалов (OQMD) [27], содержит более 200 000 рассчитанных ТФП кристаллических структур и находится в свободном доступе для публичного использования. OQMD представляет собой обширную базу данных высокопроизводительных ТФП-расчетов, состоящую из предсказанных ТФП кристаллографических параметров и энергий формирования для более 200 000 экспериментально наблюдаемых структур из Международной базы данных кристаллической структуры (ICSD) [28] и теоретических структур-прототипов. Расчеты ТФП были выполнены для каждой уникальной записи в ICSD, по состоянию на август 2013 года база данных насчитывала 32 489 рассчитанных структур. OQMD выполняет две основные функции: в качестве большого набора данных известных структур, из которого оптимальные материалы могут быть найдены, и в качестве точного описания химических потенциалов и выпуклых оболочек convex hull простых и сложных систем. В качестве большой базы данных ТФП-расчетов OQMD можно использовать для сравнения прогнозируемых свойств ТФП с экспериментальными измерениями для оценки точности подхода ТФП.
3.5. Использование результатов первопринципных расчетов в создании новых материалов
Данные первопринципных расчетов и созданные базы данных уже используются для предсказаний компонентного состава и структуры новых материалов. Например, высокопроизводительный анализ фосфатных материалов для использования в литий-ионных аккумуляторах в качестве катодов был проведен в работе [29]. В ней были исследованы емкость, напряжение, удельная энергия, плотность энергии и термическая стабильность для тысячи соединений. Были проанализированы их структурные факторы, влияющие на напряжение, а также исследована безопасность их использования в аккумуляторах. Кроме того, в результате работы было обнаружено, что для фосфатов выделение кислорода происходит при более низкой температуре, чем для оксидов. Вычислительная база данных тысяч известных и виртуальных фосфатных материалов, созданная в результате этой работы, должна помочь экспериментальному процессу разработки новых катодных материалов сфокусироваться на наиболее перспективных направлениях.
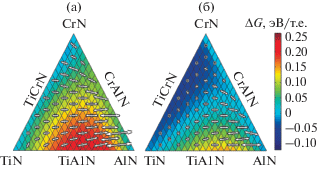
Нитриды металлов представляют собой класс материалов, в которых огромное разнообразие физических явлений позволяет синтезировать сплавы, очень привлекательные для приложений, начиная от электроники и спинтроники до конструкционных материалов. В области многофункциональных нанокомпозитных покрытий квазибинарные нитриды, такие как (Ti–Al)N или (Cr–Al)N, демонстрируют уникальные сочетания высокой твердости, высокой температуры плавления и отличной электропроводности и значительно превосходят по характеристикам исходные материалы TiN и CrN. Начав с квантово-механических расчетов из первых принципов [30], авторы предложили путь проектирования для следующего поколения нитридных сплавов с использованием концепции многокомпонентного легирования, основанного на самоорганизации на наномасштабе через образование метастабильных промежуточных продуктов во время спинодального распада. На рис. 3 продемонстрировано теоретически, что квазитройные сплавы TizCryAlxN спинодально распадаются на богатые (TiCr)N- и (CrAl)N-области. Эксперимент подтвердил, что спинодальное разложение приводит к упрочнению при старении, в то время как присутствие Cr в фазе AlN задерживает образование вредной фазы вюрцита, что приводит к значительному улучшению термической стабильности по сравнению с обоими соответствующими исходными нитридами, а также квазибинарными сплавами.
Рис. 3.
Теоретическое предсказание локальных тенденций спинодального распада в cистеме TizCryAlxN при 0 К (a) и при 1300 К (б); стрелки представляют наиболее благоприятное направление и их величина пропорциональна движущей силе спинодального распада для данного состава; ΔG – свободная энергия смешения [30].
В работе [31] высокопроизводительный ТФП- расчет был направлен на определения всех возможных термодинамически стабильных реакций литирования всех силицидов, фосфидов и станнидов переходных металлов из базы данных энергий образования ТФП-расчетов. В результате были предсказаны ключевые показатели производительности: потенциал ячейки, изменение объема при добавлении лития, а также гравиметрическая и объемная емкость. Для определения наиболее перспективных реакций были применены фильтры для отбора только тех реакций, гравиметрическая емкость которых была больше 372 и меньше 1200 мАч/г, а средний потенциал ячейки составлял от 0.25 до 0.75 В по сравнению с Li. После чего остались 16 возможных реакций из анодных материалов, в том числе: Co2P, CoP, Cr3P, CrP, FeLiP, MnP, Ni3P, PTi, Mn2P, CoSi2, NiSi, NiSi2, Si2Ti и CoSn. Из них PTi, LiSiNi2 и CoSi2 являются наиболее привлекательными кандидатами для замены графитового углерода среди силицидов, станнидов и фосфидов переходных металлов.
Новые металлооксидные катодные покрытия, способные поглощать фтористо-водородную кислоту (HF) и улучшать электрохимические характеристики литий-ионных аккумуляторов, были получены в работе [32] с использованием разработанного фреймворка. Ab initio термодинамический фреймворк для обнаружения новых катодных покрытий основывался на четырех параметрах: энтальпии поглощения HF, объемной и гравиметрической способностях оксидов поглощать HF и циклических потерях Li в компонентах покрытия. Было продемонстрировано, что часть циклического Li может быть потеряна во фториды металлов из-за того, что их литирующие напряжения примерно на 1.0–1.5 В выше, чем у соответствующих оксидов. Обнаружено, что улучшение в сохранении емкости, наблюдаемое в экспериментах, когда частицы катода покрыты оксидами металлов, такими как Al2O3, ZrO2, TiO2 или MgO, согласуется с термодинамическими расчетами. Спрогнозировано, что трехвалентные оксиды d-металлов Sc2O3, Ti2O3, V2O3, Cr2O3, Mn2O3 и Y2O3 могут составить конкуренцию используемым в настоящее время покрытиям Al2O3 и MgO.
В [33] предложен генетический алгоритм для предсказания кристаллической структуры в комбинации с первопринципной структурной оптимизацией, который был использован для исследования твердого азота под высокими давлениями. Исследовав структуру твердого азота под давлением 80 ГПа, были выявлены четыре уже обнаруженные теоретически и экспериментально низкоэнергетические немолекулярные структуры (CG, C2/c, черный фосфор и Cmcm-цепь), но также была обнаружена метастабильная односвязная трехмерная структура. Эта структура стабильна и имеет энтальпию только на 0.17 эВ/атом выше, чем у фазы CG при 80 ГПа, кроме того, она не наблюдалась в ранее предложенных алгоритмах поиска [34].
Метод для прогнозирования наиболее твердых кристаллических структур в заданной химической системе был разработан в [35]. Базисом данного метода является эволюционный алгоритм USPEX [36], который в сочетании с моделью твердости, дополненной моделью связи-валентности и теорией графов, позволяет правильно описать твердость слоистых, молекулярных и низкосимметричных кристаллических структур. С применением данного метода к C и TiO2 был получен ряд низкоэнергетических углеродных структур с твердостью, немного меньшей, чем у алмаза, и доказано, что TiO2 в любом из его возможных полиморфов не может быть самым твердым оксидом, так как его твердость оказалась довольно низкой $( < {\kern 1pt} 17$ ГПа). Также было найдено несколько sp3-аллотропов углерода с очень высокой твердостью, простой структурой и достаточно низкой энергией, которые могут быть синтезированы. Предполагается, что концепция гибридной глобальной оптимизации, которая используется здесь для твердости, может быть использована для оптимизации других свойств материалов.
Новые сверхпроводящие и полупроводниковые соединения Fe–B были предсказаны с помощью эволюционного поиска и высокопроизводительных ab initio расчетов в работе [37]. Эволюционный поиск новых соединений выявил набор основных состояний, таких как oP10-FeB4 и oP12-FeB2, а первопринципные расчеты показали, что они термодинамически стабильнее, чем уже известные a-B и oP8-FeB, более чем на 25 мэВ/атом. Представленный анализ структурных и электронных свойств показал, как фазы стабилизируются и какие физические свойства они должны продемонстрировать после их синтеза. oP10-FeB4 может стать еще одним исключением из правил Матиаса, которые рекомендуют избегать магнитных элементов при разработке новых сверхпроводников. Это соединение имеет высокий уровень плотности электронных состояний на уровне Ферми, приводящий к общему значению ${{\lambda }_{{{\text{tot}}}}} = 0.80$ и неожиданно высокой Tc, равной 15–20 К. oP12-FeB2, по прогнозам, будет первым диборидным полупроводником с шириной запрещенной зоны приблизительно 0.5 эВ. Предлагаемые материалы могут также продемонстрировать привлекательные механические свойства, поскольку для MxB1 –x с $x < 0.5$ твердость имеет тенденцию быть выше.
В [38] методом ab initio эволюционного поиска была исследована эволюция системы CaB6 под давлением. Было показано, что единственная известная кристаллическая структура в большом семействе гексаборидов металлов (простая кубическая тип cP7 структура, открытая более 80 лет назад) превращается в тетрагональную конфигурацию tI56, состоящую из 24 атомов бора. С помощью данного метода были интерпретированы экспериментальные рентгенограммы, что позволило определить структуру tI56 (28 атомов на элементарную ячейку) без какого-либо ввода параметров. Открытие первого соединения MB${{{\text{\;}}}_{6}}$ не-cP7-типа, закаленного до давления окружающей среды, может помочь в разработке интерметаллидов на основе бора с новыми электронными, магнитными и сверхпроводящими свойствами благодаря тщательному выбору металла и условий синтеза. Идентификация структуры tI56-CaB6 в эволюционном поиске показала возможность работы со все более крупными системами и незнакомыми структурными типами без какого-либо ввода структурных параметров из эксперимента.
4. МЕТОДЫ МАШИННОГО ОБУЧЕНИЯ В СОЗДАНИИ НОВЫХ МАТЕРИАЛОВ
Благодаря успешной разработке и активному использованию вычислительных методов моделирования материалов на основе ТФП, которые с каждым годом становятся все более точными и эффективными, позволяя рассчитывать соединения, металлы, сплавы и молекулы широкому кругу пользователей, резко увеличилось количество и качество данных о материалах, полученных из первых принципов. Более того, рост мощности компьютеров и их доступность приумножили количество данных во много раз. Благодаря этому также стали возможны высокопроизводительные расчеты больших групп материалов для отбора наиболее походящих кандидатов для эксперимента [29–31]. Поэтому сейчас имеются огромные объемы данных о материалах, как вычислительных, так и экспериментальных, которые доступны свободно в онлайн-базах данных [39, 24, 26, 27 ]. Как оказалось, применяя методы машинного обучения к накопленным данным, можно извлечь много нового: неизвестные и инновационные материалы, структуры и соединения.
Сейчас на методы машинного обучения возлагают надежды по смене парадигмы обнаружения новых материалов [40, 41], поскольку методы машинного обучения путем анализа больших объемов данных дают возможность отойти от проведения экспериментов и расчетов “наугад” и предсказывать требуемые материалы с заданными свойствами напрямую. Несмотря на то что методы машинного обучения начали применяться в науке о материалах относительно недавно, уже опубликованы обзоры на эту тему [40–42].
Количество и качество данных – это то, от чего зависит успех работы методов машинного обучения. Для обмена и хранения данных существует много баз данных, например, таких как Materials Project [23, 39], NOMAD [24], AFLOW [26], OQMD [27] и других [28, 43–45]. Необходимостью для баз данных является принятие методологии FAIR [25].
4.1. Категории методов машинного обучения
На данный момент существует большое разнообразие методов машинного обучения, которые уже были применены для анализа данных о материалах [41]. Методы машинного обучения можно разделить на три категории: обучение с учителем, обучение без учителя и обучение с подкреплением. Обучение с учителем является одним из самых распространенных методов машинного обучения в сфере науки о материалах. В таком виде обучения выбранный алгоритм машинного обучения тренируется на наборе данных с известным свойством, после чего предсказывает это свойство другому набору данных. Перед обучением необходимо настроить модель, чтобы она воспринимала набор данных как набор признаков, используемых алгоритмом в процессе обучения. Для проверки того, как настроена модель, проводится ее валидация – в идеале на наборе данных для кросс-валидации, выборке, отдельной от тестовой и обучающей.
4.1.1. Метод регуляризации Тихонова. Существуют разные семейства алгоритмов, одна из таких групп – это линейные алгоритмы. Один из них – метод регуляризации Тихонова, или метод регрессии гребня (ridge regression), – заключается в аппроксимации многомерной линейной зависимости методом линейных квадратов с использованием параметра регуляризации. Улучшенная версия этого алгоритма называется методом ядерной регрессии гребня (KRR), получается вследствие добавления специальной функции – так называемой функции ядра. Метод опорных векторов (SVM) также принадлежит семейству линейных методов, как и методы регрессии гребня и ядерной регрессии гребня. Этот метод находит гиперплоскости, которые разделяют набор данных на классы. Методы SVM и KRR обладают одинаковой эффективностью, кроме того, что SVM обучается и предсказывает быстрее KRR на средних объемах данных. В случае больших наборов данных оба метода будут неэффективны. Еще один метод регрессии, так называемый метод “лассо” (LASSO, Least Absolute Shrinkage and Selection Operator), пытается улучшить эффективность путем введения дополнительной регуляризации, накладывающей ограничения на выбор параметров. Несмотря на некоторые улучшения, данный метод тоже становится неэффективным в случае больших объемов данных.
4.1.2. Деревья принятия решений. Класс алгоритмов, известных как деревья принятия решений, – это алгоритмы, которые в общих чертах выглядят как графы в виде деревьев. Каждый узел в которых представляет из себя какое-либо логическое решение, разделяющее данные на два класса. Метод случайных лесов (RF) или экстремальных деревьев, вместо того чтобы строить одно древо решений, строит несколько независимых деревьев на случайном наборе данных. Эти методы довольно быстрые и эффективные и позволяют обрабатывать большие наборы данных. Кроме того, более слабые методы деревьев решений используются в комбинации с методами бустинга, такими как градиентный бустинг и адаптивный бустинг, чтобы в конечном итоге получить сильную модель.
4.1.3. Нейронные сети. Нейронные сети также применяются для анализа данных о материалах. Нейронная сеть – это модель, созданная по типу человеческих нейронов, состоит зачастую из нескольких слоев нейронов: входного, определенного количества спрятанных слоев и выходного слоя. Успешно применяются сверточные нейронные сети, которые отличаются от обычных введением дополнительных сверточных слоев, а также глубокие нейронные сети, т.е. сети, у которых больше 5 спрятанных слоев. Кроме того, могут быть использованы полностью сгенерированные сети, так называемые генеративно-состязательные сети.
4.2. Примеры применения машинных методов в материаловедении
Методы машинного обучения уже не один раз доказали свою эффективность, с их помощью предсказывались кристаллические структуры [33, 35], сверхпроводящие соединения [37, 38], новые перовскиты [46]. Также методы машинного обучения были использованы для предсказания упругих и термодинамических свойств [47, 48], запрещенных зон [49–51], температуры сверхпроводимости [52] и многих других свойств материалов. Обширный тест методов машинного обучения на более чем 25 000 кубических системах перовскита был выполнен в работе [46]. Для этого были сгенерированы все возможные соединения со стехиометрией ABX3 в кубической структуре перовскита, при этом в рассмотрение были взяты 64 элемента Периодической системы, кроме инертных газов и лантаноидов. Равновесная геометрия и энергия этих твердых фаз затем оценивались с использованием теории функционала плотности. После чего разные методы МО были протестированы, из которых самым точным оказался метод чрезвычайно случайных деревьев. Расчеты показали, что наиболее важными характеристиками являются два числа, а именно, период и группа элементов в периодической таблице (приводя к ошибке ~$140$ мэВ/атом для обучающего набора из 20 000 образцов). Достичь ошибки 121 мэВ/атом можно было, добавив девять дополнительных свойств. Также было показано, что ошибка уменьшается с размером обучающего набора. Для достижения средней точности 130–100 мэВ/атом потребуется около 20 000–30 000 обучающих образцов. Однако ошибка уменьшается медленно для больших наборов. Кроме того, эта ошибка значительно больше, если в состав входят элементы из первого ряда или несколько переходных металлов (которые часто дают сложные магнитные свойства). Было предположено, как методы машинного обучения можно применить для ускорения высокопроизводительных исследований на основе ТФП. Используя как пример случай перовскитов, авторы выполнили расчет ТФП около 20 000 кубических перовскитов со случайными составами, обучили регрессор с использованием чрезвычайно случайных деревьев, и использовали этот регрессор для прогнозирования энергетического расстояния до оболочки convex hull для оставшихся ~230 000 возможных соединений, не выполняя для них явных расчетов ТФП и удалив все системы из оболочки, которые лежат выше определенной энергии отсечки (например, установив отсечку при 700 мэВ/атом можно восстановить 99.4% всех систем, которые стабильны в пределах ТФП), и рассчитали в ТФП эти соединения. В статье это составило дополнительные 41 000 вычислений ТФП, что привело к экономии времени вычислений на 73%. Фактически, чтобы восстановить 99% всех стабильных систем, можно сэкономить 78% вычислительных ресурсов, в то время как для восстановления 95% – можно сэкономить 86%.
Предсказание тройных соединений с составом AB2C2 методами машинного обучения и высокопроизводительными первопринципными расчетами было проведено в [53]. Было выбрано два наиболее распространенных интерметаллических прототипа для этой композиции, а именно: tI10-CeAl2Ga2 и tP10-FeMo2B2. Результат показал, что существует 2108 термодинамически стабильных систем, из которых только 215 можно найти в онлайн-базах данных. Как и ожидалось для этих двух прототипов, подавляющее большинство стабильных систем являются металлическими и немагнитными. Тесты показали, что общая доля ложноотрицательных значений была приблизительно 9% для tI10 и около 0% для tP10. Из анализа результатов авторами было предположено, что доля ложноотрицательных значений в 9% может быть уменьшена, улучшив прогнозирование модели. Это может быть достигнуто путем обработки систем, содержащих элементы второго ряда и магнитные элементы, для которых ошибка предсказания значительно выше, отдельно от других соединений. Это может быть достигнуто, например, путем смещения начального тренировочного набора для включения большей части этих элементов.
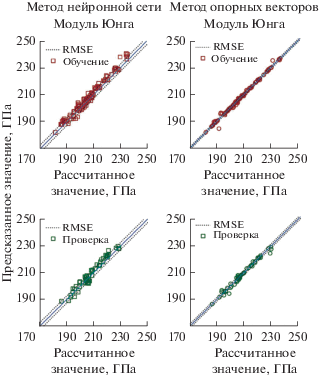
Несмотря на то что высокопроизводительные расчеты из теории функциональной плотности стали распространенным инструментом для открытия материалов, они ограничены относительно большими вычислительными затратами. В статье [54] исследуют использование данных ТФП из высокопроизводительных вычислений для создания более быстрых, суррогатных моделей с машинным обучением, которые можно использовать для направления новых поисков. Разработанный метод работает с использованием моделей дерева решений для отображения рассчитанных по ТФП энтальпий образования на набор параметров, состоящих из двух различных типов: зависящие от композиции параметры свойств элементов в сочетании с параметрами, полученными из диаграмм Вороного кристаллической структуры соединения. В результате была получена модель, которая имеют половину ошибки перекрестной проверки и аналогичную скорость обучения моделей, созданных с помощью методов кулоновской матрицы и функции частичного радиального распределения. Для набора данных из 435 000 энергий образования, взятых из Открытой базы данных квантовых материалов (OQMD) [24], модель достигает средней абсолютной ошибки 80 мэВ/атом при перекрестной проверке, которая ниже, чем приблизительная ошибка между вычисленным ТФП и экспериментально измеренной энтальпией образования и ниже на 15% среднего абсолютного отклонения тренировочного набора. Также было продемонстрировано, что метод может точно оценить энергию образования материалов вне тренировочного набора и использоваться для идентификации материалов с особенно большими энтальпиями образования. Нами также были получены обнадеживающие результату по предсказанию модуля упругости многокомпонентных ОЦК-сплавов на основе системы Fe–Cr, представленные на рис. 4.
Рис. 4.
Сравнение предсказанного значения модуля Юнга многокомпонентных ОЦК-сплавов на основе системы Fe–Cr с точными значениями на этапе обучения и проверки; сравнения представлены для модели нейронной сети и метода опорных векторов; пунктирные линии – соответствующие стандартные отклонения.
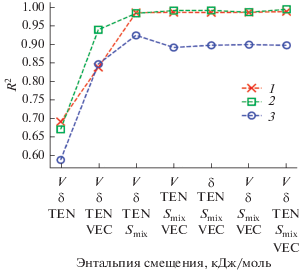
Предполагается, что при условии высокой точности прогнозирования с использованием метода машинного обучения, как, например, видно на рис. 5 для модуля Юнга E, энергии смешения ΔHmix и отношения модуля сдвига к модулю всестороннего сжатия G/B, часто используемого в качестве феноменологического критерия для описания хрупкости или пластичности материалов, и возможности использования больших обучающих наборов данных этот метод можно будет использовать для идентификации новых перспективных материалов с комбинацией привлекательных свойств при низких вычислительных затратах.
Рис. 5.
Зависимость точности прогнозирования модуля Юнга E (1), энергии смешения ΔHmix (2) и отношения модула сдвига к модулю всестороннего сжатия G/B (3) от различной комбинации признаков, использованных в обучении модели нейронной сети для многокомпонентных ОЦК-сплавов на основе системы Fe–Cr; результаты коэффициента детерминации R 2 представлены для модели нейронной сети; зависимость дает информацию о важности выбора признаков (показаных по оси абсцисс) при обучении модели.
Материалы на основе перовскита стали повсеместно распространены во многих технологически важных областях: от катализаторов в твердооксидных топливных элементах до светопоглощающих слоев в солнечной фотоэлектрической промышленности. Термодинамическая фазовая стабильность является ключевым параметром, который определяет, будет ли материал синтезирован и может ли он разрушиться при определенных условиях. Фазовая стабильность может быть рассчитана с использованием теории функционала плотности, но значительные вычислительные затраты делают такой расчет потенциально невозможным при скрининге большого количества возможных соединений. Поэтому в [55] была разработана модель машинного обучения для прогнозирования термодинамической фазовой стабильности оксидов перовскита с использованием набора данных, состоящего из энергий более 1900 оксидов перовскита, рассчитанных из первых принципов. Фазовая стабильность была определена с использованием анализа convex hull, с энергией выше convex hull ${{E}_{{{\text{hull}}}}}$. Авторы сгенерировали набор из 791 признаков, основанных на данных о свойствах элементов, которые соотносятся со значением ${{E}_{{{\text{hull}}}}}$ каждого соединения перовскита, и обнаружили, что при выборе признаков 70 лучших признаков были достаточными для получения наиболее точных моделей без существенного переобучения. Был выбран классификатор экстремальных деревьев как лучшая модель для классификации, а ядерная регрессия гребня – как лучшая модель для регрессии из пяти моделей кандидатов при сравнении эффективности перекрестной проверки. Наилучшая оценка F1, достигнутая для классификации, составила 0.881 (+/–0.032), а наилучшая среднеквадратическая ошибка для регрессии ${{E}_{{{\text{hull}}}}}$ составила 28.5 (+/–7.5) мэВ/атом. Модель была проверена с помощью отдельных тестов экстраполяции на пяти подмножествах, целевой схемы перекрестной проверки, в результате чего модель показала хорошую эффективность классификации и регрессии в тестовых случаях, даже когда информация о составе в наборе обучающих данных была ограничена. Кроме того, разработанная модель была применена к сгенерированным вручную соединениям перовскита и сравнена с расчетами ТФП. Модель смогла дать точные прогнозы для соединений, содержащих элементы, которые часто отбираются в наборе обучающих данных, что делает эту модель полезной для прогнозирования стабильности оксидов перовскита в пространстве композиций, актуальных для высокоактивных катодов твердооксных топливных элементов. Учитывая быструю скорость прогнозирования, модель продемонстрировала потенциал для быстрого скрининга новых материалов-кандидатов в большом пространстве композиции с помощью подходов машинного обучения.
В [56] показано, что глубокие нейронные сети, использующие только два дескриптора – электроотрицательность и ионные радиусы, могут предсказать ТФП энергии образования гранатов C3A2D3O12 и перовскитов ABO3 с низкими средними абсолютными ошибками 7–10 мэВ/атом и 20–34 мэВ/атом соответственно, что находится в пределах точности ТФП. Ключевое преимущество разработанных моделей нейронных сетей заключается в том, что они опираются только на чрезвычайно небольшое число дескрипторов. В качестве дескрипторов не рассматриваются никакие структурные степени свободы за пределами ионных радиусов конкретного вида и упорядочения катионов в смешанных оксидах. Это резко контрастирует с большинством моделей машинного обучения в литературе, использующих большое количество коррелированных дескрипторов, которые делают такие модели очень восприимчивыми к переобучению, или силовым полям машинного обучения, которые могут включать в себя структурные и атомные степени свободы, что означает потерю переносимости на другие составы. Однако разработанные модели нейронных сетей по-прежнему ограничиваются композициями гранатов и перовскитов (со смешиванием катионов или без них) без вакансий, хотя дополнительные расширения на другие прототипы общей кристаллической структуры и для учета вакансий возможны. Кроме того, показано, как модели, прогнозирующие энергии образования, могут быть объединены с существующими большими общедоступными базами данных вычисленных энергий ТФП для прогнозирования ${{E}_{{{\text{hull}}}}}$ и, следовательно, фазовой стабильности. Эти возможности могут быть использованы для эффективной обработки больших пространств химических составов несмешанных и смешанных кристаллов, чтобы идентифицировать стабильные составы и их порядок, что значительно ускоряет возможность открытия новых материалов.
4.3. Адаптивный дизайн материалов
Как ускорить процесс поиска требуемых материалов, используя подход адаптивного дизайна материалов, сочетающий в себе эксперименты, ТФП-расчеты и методы машинного обучения, было показано в работе [57]. Стратегия базировалась на подходе активного обучения, на каждом цикле модель обучалась на наборе ТФП и экспериментальных данных, после чего предсказывала следующие эксперименты или расчеты, чтобы эффективно перемещаться по сложному пространству поиска. Кроме того, в модели были использованы интерфейсы и глобальная оптимизация, что помогало соблюдать баланс между целью поиска материалов, которые обладали лучшими свойствами, и необходимостью исследовать части пространства поиска с меньшим количеством точек выборки и большей неопределенностью. Авторы демонстрируют результаты модели, находя сплавы с памятью формы на основе NiTi с очень низким тепловым гистерезисом ${\Delta }T$, причем обнаруженный сплав Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 обладал наименьшим ${\Delta }T$ (1.84 К). Были синтезированы и охарактеризованы 36 предсказанных композиций (на основе 9 циклов обратной связи) из потенциального пространства 8 000 000 композиций. Из них у 14 сплавов был меньший ${\Delta }T$, чем у любого из 22 в исходном наборе данных.
Фреймворк, основанный на методах машинного обучения, который решает проблемы в области материаловедения, где наборы данных разнообразны, но имеют скромные размеры, был разработан в [47]. Авторы демонстрируют, как их фреймворк МО прогнозирует объемные модули и модули сдвига (K и G соответственно) для поликристаллических неорганических соединений, используя данные о 1940 соединениях из базы данных рассчитанных модулей упругости для металлов, полупроводников и изоляторов. Их фреймворк МО объединяет GBM-Locfit (многомерную локальную регрессию в рамках механизма повышения градиента), 10-кратную перекрестную проверку с консервативным критерием риска и разнообразный набор составных и структурных дескрипторов, которые обобщаются на k-е соединения. Таким образом, после одного прогона ТФП для определения lg(V) и энергии когезии ${{E}_{{\text{c}}}}$ прогнозы для K и G могут быть сделаны для любого неорганического поликристаллического k-го соединения. Кроме того, дескрипторы K и G, описанные здесь, уже доступны на веб-сайте Materials Project для всех материалов, для которых полные упругие тензоры еще не были рассчитаны методами ТФП.
В [48] данные ab initio-расчетов из хранилища AFLOW объединяются с моделями Количественных отношений структур материалов к свойствам QMSPR, чтобы предсказать важные свойства: классификацию металл/изолятор, энергию запрещенной зоны, объемный модуль/модуль сдвига, температуру Дебая и теплоемкость. Точность прогноза хорошо согласуется с качеством обучающих данных практически для любого стехиометрического неорганического кристаллического материала, полученные данные также сопоставимы с доступными термомеханическими экспериментальными данными. Хотя сами модели демонстрируют отличную прогностическую способность с небольшими отклонениями, анализ выбросов показывает, что структуры предсказываются хуже всего. Это неудивительно, поскольку истинные условия стабильности (например, высокое давление/высокая температура) еще предстоит определить. По оценкам Базы данных неорганических кристаллических структур ICSD, структуры более чем 7000 материалов (примерно 4%) основаны на расчетах, а не на реальных экспериментах. Такие открытия показывают еще одно приложение для моделирования МО, быстрой и надежной обработки больших наборов данных. Применимость разработанных методов QMSPR распространяется на все 230 пространственных групп и подавляющее большинство элементов в периодической таблице.
То, как инструмент машинного обучения может предсказывать упругие свойства цеолитов, показано в [58]. Авторы разработали подход машинного обучения, опираясь только на геометрические особенности, которые связаны с локальной геометрией, структурой и пористостью цеолита, чтобы предсказать объемные и сдвиговые модули цеолитов с точностью, превышающей точность подходов силового поля. Методология подхода заключается в комбинировании регрессора повышения градиента, с использованием деревьев регрессии и набора локальных, структурных и пористых свойств для генерации цеолитных материалов. Разработка этой модели выявила несколько корреляций, которые связывают характерные особенности цеолита с упругими свойствами. Модуль обучения на наборе ТФП-данных позволяет точно предсказывать модули K и G для любого цеолитного соединения. Для демонстрации мощности разработанного метода были рассчитаны модули для 590 448 гипотетических цеолитов. Это крупномасштабное исследование показывает общие черты полиморфов SiO2 и демонстрирует явное снижение максимального сдвигового сопротивления при увеличении объема пор. Кроме того, авторы предполагают, что разработанный подход может быть легко модифицирован для описания новых и появляющихся баз данных пористых материалов (металлоорганические системы и молекулярные кристаллы) с обнаружением новых материалов, которые также являются механически стабильными.
Использование методов машинного обучения для ускорения дизайна кристаллических материалов обычно требует построенных вручную векторов, признаков или сложного преобразования координат атома для ввода кристаллической структуры, что либо ограничивает модель определенными типами кристаллов, либо затрудняет предоставление химических свойств. В работе [59] был разработан фреймворк сверточных нейронных сетей графа кристалла, чтобы напрямую изучить свойства материала по связи атомов в кристалле, обеспечивая универсальное и интерпретируемое представление кристаллических материалов. Разработанный метод обеспечивает высокоточное предсказание свойств, рассчитанных из теории функционала плотности для восьми различных свойств кристаллов с различными типами структур и составов после обучения на наборе из 104 точек данных. Кроме того, разработанный фреймворк интерпретируемый, потому что можно определить вклад из локальной химической среды в глобальные свойства. В качестве примера подход был применен к разработке новых перовскитов и показал, что информация, извлеченная из модели, согласуется с общими представлениями о химических свойствах материалов и значительно уменьшает пространство поиска для высокопроизводительного скрининга.
Запрещенные зоны для более чем 200 новых соединений халькопирита с ранее не испытанными химическими составами предсказаны в работе [50]. Метод ансамблевого анализа данных, включающий методы регрессии Обыкновенных наименьших квадратов (OLS), Разреженных частичных наименьших квадратов (SPLS) и Эластичной сети/оператора наименьшей абсолютной усадки и оператора выбора (LASSO) в сочетании с методами грубого набора (RS) и анализа главных компонентов (PCA) был использован для разработки надежных количественных моделей типа структура–свойство (QSAR) для прогнозирования ширины запрещенной зоны. Результатом регрессионного анализа является прогнозируемая ширина запрещенной зоны для новых соединений на основе модели, использующей дескрипторы, наиболее связанные с запрещенной зоной. После чего использовались алгоритмы ранжирования признаков: для оценки связи между шириной запрещенной зоны и химическими дескрипторами, используемыми в прогностических моделях, и для понимания причины выбросов в прогнозах. Было обнаружено, что для прогнозирования ширины запрещенной зоны соединений халькопирита ABC2 наиболее важными дескрипторами являются атомный номер элементов B и C, температура плавления элемента C и радиус псевдопотенциала элемента C. На основе методологии ансамблевого прогнозирования были идентифицированы два соединения-выброса, на чью ширину запрещенной зоны наиболее сильно повлияла температура плавления элемента C.
4.4. Дескрипторы квантовых структур металлов
Возможность делать быстрые и точные прогнозы для запрещенных зон двойных перовскитов AA'BB'O6 представляет большой практический интерес для ряда применений. В [49] представлена надежная модель машинного обучения вместе с набором простых дескрипторов для эффективного и точного предсказания электронной структуры запрещенных зон двойных перовскитов. Предложенный оптимальный набор дескрипторов был идентифицирован в результате поисков по большой части пространства признаков, в котором было более ~1.2 миллиона дескрипторов, сформированных путем объединения простых элементных признаков, таких как электроотрицательность, потенциалы ионизации, энергетические уровни электронной структуры и валентные орбитальные радиусы составляющих атомов. Разработанная здесь модель статистического обучения на основе ядерной регрессии гребня была обучена и протестирована на базе данных, состоящей из информации о запрещенных зонах ~1300 двойных перовскитов, рассчитанных с помощью функционала GLLB-SC в рамках теории функционала плотности. Одним из наиболее важных выводов о химических свойствах, обнаружившихся в результате работы с полученной моделью, является то, что структура запрещенной зоны в основном контролируется (и, следовательно, может быть эффективно изучена) самыми низкими занятыми энергетическими уровнями элементов A и электроотрицательностью элементов B. Средняя среднеквадратичная ошибка тестового набора для перекрестно проверенной модели только с четырьмя основными признаками оказалась равной 0.5 эВ и дополнительно снижается до ~0.37 эВ (0.36 эВ), если использовать модель с 16 признаками. Качество прогнозирования обученной модели вне выборки дополнительно демонстрируется ее способностью прогнозировать запрещенные зоны нескольких одиночных перовскитов. Также было показано, что эффективность прогнозирования модели можно визуально структурировать, построив несколько двумерных контурных карт признаков-свойств. Авторы считают, что подход МО, представленный в статье, является общим и может применяться к любому классу материалов в ограниченном химическом пространстве с заданной кристаллической структурой, чтобы делать эффективные предсказания запрещенных зон. Такая стратегия прогнозирования может быть практически полезна при первоначальном отборе для высокопроизводительного выявления перспективных кандидатов.
Еще одна модель для предсказания ширины запрещенной зоны неорганических твердых веществ на основе только их состава была разработана в [51]. Эта модель использует классификацию опорных векторов, чтобы сначала отделить металлы от неметаллов, после чего следует количественное прогнозирование ширины запрещенной зоны неметаллов с использованием регрессии опорных векторов (SVR). Превосходная точность регрессионной модели (способна отличать металлы от неметаллов с точностью ~92%) достигается при использовании обучающего набора, состоящего полностью из экспериментальных данных о измеренных 3896 запрещенных зонах, собранных из литературы, с использованием только композиционных дескрипторов. Анализ запрещенных зон с использованием новой модели SVR (Eg,SVR) показывает отличное согласие с Eg,exp. Среднее абсолютное отклонение составляет 0.75 эВ, а среднеквадратичная ошибка модели – 1.46 эВ, что указывает на то, что погрешность этой модели находится примерно посередине между PBE-функционалом и гибридным функционалом. Среди выбранных соединений метод работает превосходно для таких соединений, как CuSbS2 (ошибка 1%), PbTe (ошибка 5%) и ZnO (ошибка 1%). Единственное место, для которого метод машинного обучения, по-видимому, работает плохо, – это предсказание сверхширокой запрещенной зоны у LiF. Тем не менее ошибка в этом случае все еще составляет только 30% с прогнозируемой величиной 9.87 эВ, в то время как Eg,exp = 14.2 эВ. Таким образом, авторы разработали модель на основе SVR, которая может надежно предсказать ширину запрещенной зоны любого материала при значительном снижении вычислительных затрат по сравнению с более сложными теоретическими методами. Также авторы использовали свою модель для оценки ширины запрещенной зоны 94 095 соединений, собранных в Pearson’s Crystal Database (PCD).
Кестиритовые I2-II-IV-V4 полупроводники являются перспективными солнечными поглотителями для фотоэлектрических применений. Ширина запрещенной зоны полупроводников, ее характер, прямой или косвенный, являются фундаментальными свойствами, определяющими эффективность фотоэлектрического устройства. В [60] авторы, используя комбинацию точных вычислений из первых принципов и машинного обучения, предсказали свойства запрещенной зоны для 1586 полупроводников кестерита I2-II-IV-V4. Выполнив гибридно-функциональные первопринципные расчеты для подмножества из 200 соединений, они обучили модели МО прогнозировать величину и характер запрещенной зоны. Модель, основанная на регрессии опорных векторов, предсказывает величину щели запрещенной зоны со среднеквадратичной ошибкой 283 мэВ. Классификатор зоны на прямую/непрямую был аппроксимирован с использованием логистической регрессии и имеет точность 89%. Предсказания модели идентифицируют 242 материала с шириной запрещенной зоны в оптимальном диапазоне 1.2–1.8 эВ. Проверив их стабильность, авторы предполагают, что 34 из этих материалов являются синтезируемыми; 25 из этих материалов фактически имели ширину запрещенной зоны в диапазоне 1.2–1.8 эВ, что было подтверждено с использованием первопринципных расчетов.
Авторы [61] предложили методологию, которая может ускорить процесс поиска материалов, обладающих требуемыми свойствами, путем обучения моделей по экспериментальным данным по мере их сбора, чтобы предсказывать, какой эксперимент должен быть выполнен следующим. В результате работы методология последовательного обучения, основанная на методе случайных лесов с оценками неопределенности, была предложена. Неопределенность рассчитывалась с использованием скорректированной на смещение инфинитезимальной оценки метода “складного ножа” и оценки метода “складного ножа” после начальной загрузки, обе модели показали хорошую калибровку. Этот результат важен сам по себе, поскольку хорошо откалиброванные оценки неопределенности имеют решающее значение для моделей, управляемых данными, в материаловедении и других инженерных приложениях. Эти результаты представляют собой некоторые из первых оценок границ неопределенности случайных лесов для научных приложений. Фреймворк Лесов с оценками неопределенности для последовательного обучения FUELS может применяться в широком спектре приложений с большим количеством свободных параметров. В этой статье авторы исследовали его эффективность на четырех контрольных примерах из материаловедения: максимизация магнитной деформации магнитокалорических материалов, максимизация критической температуры сверхпроводников, максимизация добротности ZT термоэлектрика и максимизация усталостной прочности стали. Во всех этих тестовых случаях экспериментальный дизайн был параметризован с использованием от двадцати до шестидесяти различных признаков, что было возможно благодаря хорошему масштабированию FUELS в многомерное пространство. Во всех четырех случаях FUELS значительно превзошел случайные предположения, для поиска оптимального кандидата потребовалось в три раза меньше необходимых измерений, чем в среднем случайном предположении. Кроме того, были сравнены три разные стратегии выбора следующего кандидата фреймворка FUELS: максимальное ожидаемое улучшение (MEI), максимальная неопределенность (MU) и максимальная вероятность улучшения (MLI). В указанных тестовых случаях MLI неизменно демонстрировал наивысшую производительность, MEI показывал неоднозначные результаты, когда требовалось большое исследование параметрического пространства, и MU работал плохо, когда модель случайного леса могла делать точные прогнозы после того, как была аппроксимирована только на нескольких обучающих точках.
4.5. Предсказание критических температур машинными методами
Критическая температура сверхпроводников была изучена несколькими разработанными методами машинного обучения в работе [52]. На основе информации из базы данных SuperCon [62] исходные упрощенные химические свойства генерировались с помощью программного пакета Magpie. В качестве первого применения методов МО материалы были разделены на два класса в зависимости от того, выше или ниже 10 К критическая температура. Для прогнозирования класса сверхпроводников была создана непараметрическая модель классификации случайных лесов. Классификатор показал отличную производительность с точностью вне выборки и показателем F1 около 92%. Затем несколько успешных моделей регрессии случайных лесов были созданы для прогнозирования значения Tс, включая отдельные модели для трех подгрупп, т.е. соединений купратов, материалов на основе железа и соединений с низким Tс. Изучая предикторы каждого семейства сверхпроводников, было получено представление о физических механизмах, отвечающих за сверхпроводимость среди различных групп. Благодаря использованию кристаллографических/электронных признаков из онлайн-хранилищ AFLOW, модели МО были улучшены. После объединения всех моделей в одну систему был произведен поиск новых неорганических сверхпроводников по всей базе данных неорганических кристаллических структур ICSD. В результате модель определила 35 оксидов в качестве материалов-кандидатов. Некоторые из них химически и структурно аналогичны купратам (хотя во время обучения модели не была предоставлена четкая структурная информация). Еще одна особенность, которая объединяет почти все эти материалы, – наличие плоских или почти плоских энергетических зон чуть ниже энергии самого высокого занятого электронного состояния. Данная работа демонстрирует важную роль, которую модели ML могут играть в исследованиях сверхпроводимости. Данные, собранные в течение нескольких десятилетий в SuperCon и других соответствующих базах данных, используются моделями ML и позволяют понять связь между химическим составом/структурой материалов и сверхпроводимостью материалов.
Температура плавления оказывает большое влияние на высокотемпературные свойства и пределы рабочей температуры керамики со сверхвысокими температурами (UHTC). Чтобы обойти проблему измерения сверхвысокой температуры плавления в работе Nan Qu и др. В [63] был предложен новый метод прогнозирования температуры плавления UHTC с использованием машинного обучения. Набор данных, основанный на экспериментальной информации, включал в себя более десяти тысяч данных о температуре плавления и был взят из базы данных тугоплавких сплавов FactSage. В основе разработанной модели лежит искусственная нейронная сеть с обратным распространением (BPANN). Разработанная модель была применена к системе UHTC Hf-C-N для прогнозирования температуры плавления. После чего была проведена взаимная проверка прогноза температуры плавления UHTC с расчетами из теории функционала плотности, что подтвердило прогнозирование модели. Точность предсказания полученного фреймворка составила около 90%.
Таким образом, на основе анализа литературных данных можно сделать вывод о перспективности и высоком потенциале разработки баз данных результатов первопринципного моделирования и использования методов машинного обучения для научно обоснованной разработки новых материалов атомной энергетики.
Фундаментальной основой разработки материалов с заданным и управляемым комплексом свойств являются диаграммы состояния многокомпонентных систем и сопутствующая информация о физико-химическом взаимодействии, которая в диаграммах состояния не отражена, однако определяет их строение (кристаллическая структура твердых фаз и термодинамические свойства твердых и жидкой фаз). Знание химического и фазового составов и структуры сплавов, концентрационно-температурных интервалов стабильности фаз и процессов их образования позволяют сознательно выбирать условия термообработки материалов. Очевидно, что свойства материалов существенно зависят от фазового состава, характера морфологии структуры, термообработки, растворимости компонентов в фазах и т. д., т.е. от тех вопросов, на какие дает ответ диаграмма состояния. Поэтому крайне важно получить систематическую информацию о характере и температуре фазовых равновесий, растворимости компонентов в фазах, кристаллической структуре соединений. Проанализированные данные о строении диаграмм состояния многокомпонентной системы Fe–Ni–Cr–Al–Mo–C–N–W–Nb–V, а также ограничивающих в широком интервале температурах, включая кристаллизацию, о структуре и свойствах формируют теоретический фундамент, необходимый для разработки материалов с заданным комплексом свойств. Они в основном полезны для понимания процессов, происходящих в сплавах, позволяют сознательно выбрать оптимальное содержание основных и легирующих компонентов и условия термообработки материалов.
5. ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА БИНАРНЫХ СПЛАВОВ
5.1. Диаграммы состояния бинарных систем Fe–Cr
Квантово-механические расчеты на основе ТФП активно использовались для моделирования сплавов на основе системы Fe–Cr. Бинарные сплавы Fe–Cr являются основой для многих важных промышленных сталей. Сплавы, содержащие более 10 ат. % Cr, представляют большой практический интерес в качестве конструкционных сталей благодаря их прочностным характеристикам и коррозионной стойкости. При больших концентрациях хрома, близких к 20 ат. % Сr, данные стали претерпевают спинодальный распад при температурах, близких к 800 К, что приводит к ухудшению механических и коррозионных свойств. Исследование фазовой стабильности и возможных механизмов охрупчивания является весьма актуальной задачей. Поскольку эти сплавы широко применяются в качестве труб охлаждения в корпусных ядерных реакторах, где присутствует высокое давление, важно также исследовать влияние внешнего давления на фазовую стабильность и спинодальный распад сплавов Fe–Cr.
Большинство первопринципных исследований, посвященных фазовому равновесию в сплавах на основе Fe–Cr, указывают, что химические взаимодействия, обусловленные во многих случаях магнетизмом, играют важнейшую роль в термодинамической стабильности этих сплавов. Одной из интересных особенностей системы Fe–Cr, связанной со спинодальным распадом, является наличие сплавов с аномальной стабильностью, связанной с возникновением эффективного взаимодействия в первой координационной сфере, которое приводит к упорядоченному состоянию в сплавах с высоким содержанием железа. Данный эффект был предсказан теоретически с использованием обобщенного метода возмущений в приближении сильной связи (GPM) в работе [64].
Первопринципные расчеты сплавов Fe–Cr в рамках модели GPM предсказывают переход от положительного к отрицательному значению эффективного парного потенциала взаимодействия в первой координационной сфере для концентраций около 25 ат. % Cr. Отрицательный знак указывает, что кластеризация может легко произойти в этом диапазоне концентраций: кластеризация фактически наблюдалась при дифракции нейтронов даже в диапазоне концентраций Cr 15 ат. % [65]. С другой стороны, при малых концентрациях Cr большой и положительный парный потенциал взаимодействия в первой координационной сфере указывает на возможное существование химического ближнего порядка. Таким образом, в рамках модели GPM сплавы Fe–Cr представляют собой систему, в которой парный потенциал взаимодействия может изменять свой знак при изменении концентрации. Модель GPM позволяет описывать энергию упорядочения в разупорядоченных системах только в рамках парных взаимодействий. В этой модели энергия упорядочения может быть описана через кластерное разложение при ограничении только парными вкладами, если в качестве основного состояния выбран полностью разупорядоченный сплав, описанный, например, в приближении когерентного потенциала (ПКП). Следовательно, для каждой концентрации “эффективные” потенциалы парного взаимодействия можно рассчитать как функцию характеристик зонной структуры неупорядоченного сплава. Несмотря на приближения в использованные в модели GPM, полученные в работе теоретические [64] результаты кажутся весьма удовлетворительными и позднее были подтверждены в измерениях атомного ближнего порядка методом диффузионного нейтронного рассеяния [66].
На основе первопринципных расчетов энтальпии образования неупорядоченных ОЦК-сплавов Fe–Cr в ферромагнитном состоянии в работе [67] была обнаружена аномальная стабильность сплавов при низких концентрациях Cr. Похожие результаты были получены в последующих теоретических расчетах [68–71]. Вычисления показывают, что энтальпия смешения отрицательна при концентрациях Cr ниже 6%. В то же время она становится положительной при более высоких концентрациях Cr и достигает максимального значения 10 кДж/моль при эквимолярном составе. Помимо того, обнаружено, что объемные модули, как теоретические, так и экспериментальные, обладают особенностью при 6 ат. % Cr, то есть в том же интервале концентраций, где энергия смешения является отрицательной [71].
5.1.1. Фазовые равновесия. Диаграмма состояния системы Fe–Cr является основой многих материалов, имеющих практическое значение. Многочисленные исследования посвящены изучению свойств сплавов этой системы. Известной особенностью системы Fe–Cr является расслоение в ОЦК-фазе при низких температурах. Внутри купола расслоения сплавы имеют тенденцию разделяться на ОЦК-фазу, обогащенную Cr($\alpha {\kern 1pt} '$), и ОЦК-фазу, обогащенную Fe(α) [72]. Было обнаружено, что благодаря этому разделению фаз сплавы Fe–Cr проявляют повышенную твердость и пониженную пластичность после старения, которая известна как “охрупчивание 475°C” [73].
Подробный анализ имеющейся в литературе информации о фазовых превращениях и термодинамических свойствах фаз системы Fe–Cr проведен в работах [74–76]. Термодинамическое описание системы приведено в работах [74–80]. Автор [79] пересмотрел термодинамические параметры для жидкой фазы, полученные в работе [74], для лучшего описания в системах высокого порядка. Однако оценка [79] не воспроизводит равновесия жидкость–твердое тело в системе Fe–Cr, а в последней оценке [80] термодинамическое описание жидкой фазы проведено с использованием модифицированной квазихимической модели (MQM). Поэтому, несмотря на отмеченные выше недостатки термодинамической модели [74], диаграмма состояния системы Fe–Cr принята в данной работе согласно [74] и представлена на рис. 6. Это также связано с тем, что термодинамическая оценка системы Fe–Cr [74] является общепринятой и включена в базу данных TCFE10 [81] (разработчик Thermo-Calc Software AB [82]).
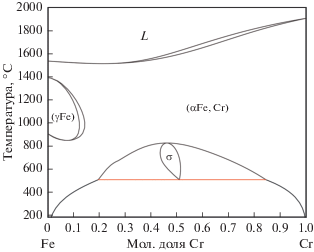
В системе Fe–Cr образуются следующие фазы: α(Fe,Cr)-фаза (ОЦК-раствор на основе чистых элементов), которая в результате распада образует α(Cr)-фазу (ОЦК-раствор на основе хрома) и α(Fe)-фазу (ОЦК-раствор на основе железа); γ(Fe)-фаза (ГЦК-раствор на основе железа); σ-фаза (собственный структурный тип CrFe). На кривых ликвидус и солидус наблюдается минимум (рис. 6). Для кристаллических фаз характерно образование областей гомогенности: узких для γ(Fe)- и σ‑фаз и протяженной для α(Fe,Cr)-фазы. На кривой растворимости хрома в γ(Fe)-фазе присутствует минимум. В низкотемпературной области диаграммы состояния находится область расслоения α(Fe,Cr)-фазы.
Фаза FeCr (σ-фаза) имеет сложную тетрагональную решетку (собственный структурный тип CrFe, tP30–$P$42/mnm) и некоторую область гомогенности. Образование σ-фазы в системе Fe–Cr впервые предположено еще в 1907 г. [83]. Однако настоящее открытие σ-фазы произошло только через 20 лет, когда авторы [84, 85] обнаружили в сплаве Fe–Cr–Ni твердую, очень хрупкую и немагнитную фазу, названную позже “сигма” [86]. При температуре 510°C σ-фаза распадается на фазы ${{\alpha }_{1}}$ и ${{\alpha }_{2}}$ по эвтектоидной реакции $\sigma \rightleftarrows {{\alpha }_{1}}$ + ${{\alpha }_{2}}$. Кристаллическая структура всех промежуточных фаз cистемы Fe–Cr представлена в табл. 1. Все нонвариантные равновесия, которые имеют место в системе Fe–Cr, приведены в табл. 2.
Таблица 1.
Кристаллическая структура и параметры решетки фаз системы Fe–Cr
| Фаза | Кристаллическая структура | Параметры решетки, Å | Примечания |
|---|---|---|---|
| $\delta $Fe | W, cI2-Im-3$m$ | a = 2.9315 | >1394°C [87] |
| $\gamma $Fe | Cu, cF4-Fm-3$m$ | a = 3.6467 | >912°C [87] |
| $\alpha $Fe | W, cI2-Im-3$m$ | a = 2.8665 | 25°C [87] |
| $\varepsilon $Fe | Mg, hP2–P 63/mmc | a = 2.468, c = 3.96 | 25°C, >13 ГПа [87] |
| Cr | W, cI2-Im-3$m$ | a = 2.8848 | 25°С [87] |
| $\sigma $, CrxFe1 – x | CrFe, tP30–P 42/mnm | a = 8.7966, c = 4.5582 | [88] |
Таблица 2.
Инвариантные равновесия в системе Fe–Cr [74]
| Инвариантное равновесие | Температура, °С | Состав, ат. % Cr | ||
|---|---|---|---|---|
| L $ \rightleftarrows (\alpha $Fe,Cr) | 1516 | 21 | 21 | – |
| ($\gamma $Fe) $ \rightleftarrows (\alpha $Fe,Cr) | 850 | 7 | 7 | – |
| ($\alpha $Fe,Cr) $ \rightleftarrows \sigma $ | 835 | 48 | 48 | – |
| $\sigma \rightleftarrows {{\alpha }_{1}} + {{\alpha }_{2}}$ | 510 | 49 | 49 | 49 |
5.1.2. Термодинамические свойства. Функции $\Delta Н$ и ${{a}_{{{\text{Me}}}}}$, рассчитанные согласно [74], приведены на рис. 7. Для энтальпий смешения и активностей компонентов α(Fe,Cr)- и γ(Fe)-фаз характерны слабые положительные отклонения от идеальности (рис. 7в–7д), что находится в полном согласии с экспериментальной информацией, представленной в работе [75]. Энтальпия смешивания жидких сплавов Fe–Cr измерялась в работах [89–92], однако результаты измерений плохо согласуются. Для энтальпий смешения расплавов в работах [91, 93, 94] были получены положительные значения, в [89] – значения, близкие к нулю, а в [90] – небольшие отрицательные значения. Результаты последней работы были использованы в [74] при проведении термодинамического описания системы (рис. 7а). Согласно экспериментальным данным, представленным в [75], поведение ${{a}_{{{\text{Me}}}}}$ в жидких сплавах близко к идеальному (рис. 7б).
Рис. 7.
Термодинамические свойства фаз системы Fe–Cr [74]: (а) – энтальпия смешения жидких сплавов; (б) – термодинамические активности компонентов жидких сплавов; (в) – энтальпия смешения α(Fe,Cr)-фазы; (г) – энтальпия смешения γ(Fe)-фазы; (д) – термодинамические активности компонентов α(Fe,Cr)- и γ(Fe)-фаз.
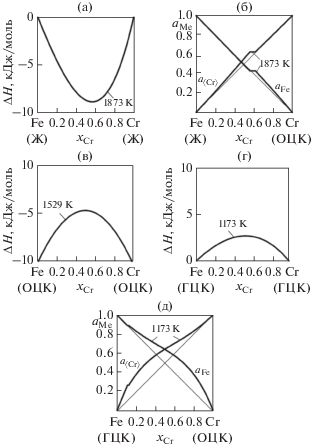
Для сплавов с высоким содержанием железа как экспериментальные, так и рассчитанные параметры решетки демонстрируют положительные отклонения от закона Вегарда. При высоких концентрациях хрома имеют место отрицательные отклонения. Кроме того, наблюдается заметная аномалия в зависимости параметра решетки от концентрации, которая присутствует как в экспериментальных, так и в теоретических результатах (рис. 8). Начальный крутой наклон концентрационной зависимости (что указывает на относительно большой эффективный размер примеси Cr в чистом Fe) резко изменяется до более низкого значения, соответствующего меньшему эффективному размеру атомов Cr при концентрациях, превышающих 7% Cr. В парамагнитном состоянии концентрационная зависимость параметра решетки сплавов Fe–Cr характеризуется отрицательными отклонениями от закона Вегарда во всем диапазоне концентраций [68].
Рис. 8.
Теоретические отклонения параметра решетки в ОЦК-сплавах Fe–Cr и Fe–Cr–Ni от правила Вегарда; результаты расчета хорошо согласуются с экспериментальными значениями [68]: 1 – Fe1 –xCrxNi5.0, 2 – Fe1 –xCrxNi10.0, 3 – Fe1 –xCrxNi15.0, 4 – Fe1 –xCrxNi20.0, 5 – Fe1 –xCrx.
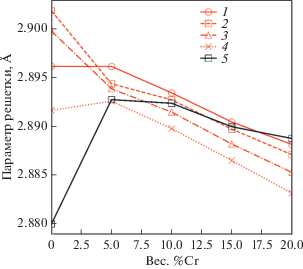
Подробное систематическое исследование термодинамических свойств Fe–Cr сплавов было проведено в работе [70] с помощью метода Монте Карло с кластерным разложением. Был исследован эффект равновесной кластеризации в зависимости от температуры и концентрации. Кластеризация начинается при 10% Cr $T = 800$ К. При более высоких температурах конфигурационная энтропия начинает сдерживать кластеризацию. Например, при $T = 1400$ К она наблюдается при 20% Cr. При еще более высокой температуре $T = 1600$ К сплавы находятся выше области несмешиваемости $\alpha {\kern 1pt} - {\kern 1pt} \alpha {\kern 1pt} '$. Рассчитанные нами энтальпии смешения, представленые на рис. 9, соответствуют этим результатам. Более того, видно, что метод первопринципного моделирования позволяет изучать эффекты легирования железа несколькими элементами одновременно в рамках единых приближений без необходимости построения новых моделей или подбора дополнительных (часто полуэмпирических) параметров.
Рис. 9.
Теоретическая энтальпия смешения ΔH твердого раствора в ОЦК-сплавах Fe–Cr и Fe–Cr–Ni как функция концентрации Cr: 1–5 – см. рис. 8.
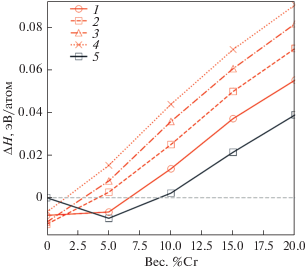
В настоящий момент большинство работ согласуются с тем, что источником аномальной стабильности в разбавленных сплавах, четко видной на рис. 9, является магнитное поведение атомов Cr, которые приобретают магнитный момент, направленный антипараллельно магнитному моменту Fe, благодаря антиферромагнитным взаимодействиям. В то же время магнитное обменное взаимодействие пары атомов Cr также является антиферромагнитным на первой координационной сфере (рис. 10). Таким образом, появление пары атомов Cr в качестве ближайших соседей приводит к нарушению локальной магнитной конфигурации, что в свою очередь приводит к постепенной потере магнитного момента атомами Cr с увеличением концентрации Cr (рис. 11) и соответствующему изменению типа эффективных химических взаимодействий [69, 71].
Рис. 10.
Теоретические значения магнитного обменного взаимодействия J гамильтониана Изинга на первой координационной сфере в ОЦК-сплавах как функция от концентрации Cr; положительные значения соответствуют ферромагнитным взаимодействиям, отрицательные – антиферромагнитным; 1 – FeCr, 2 – FeCrNi5, 3 – FeCrNi20, 4 – FeCrAl2.5, 5 – FeCrAl5.0, 6 – FeCrNi5Al2.5, 7 – Fe–Ni в FeCrNi05, 8 – Cr–Ni в FeCrNi05, 9 – Fe–Ni в FeCrNi20, 10 – Cr–Ni в FeCrNi20, 11 – Fe–Ni в FeCrNi5Al2.5, 12 – Cr–Ni в FeCrNi5Al2.5.
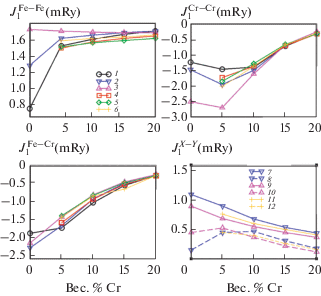
Рис. 11.
Магнитный момент на атомах Cr в неупорядоченных ОЦК-сплавах в ферромагнитном состоянии как функция концентрации Cr: 1–5 – см. рис. 8.
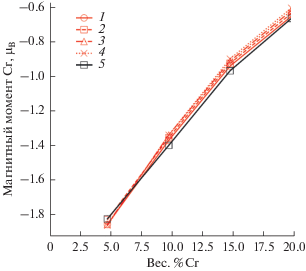
Как утверждается в вышеперечисленных работах, при более высоких концентрациях Cr взаимодействия приобретают фазово-разделительный характер, что приводит к соответствующему изменению концентрационной зависимости энтальпии образования. В частности, это приводит к изменению кривизны энтальпии образования, что может быть связано со спинодальным распадом.
Систематическое исследование влияния внешнего давления на фазовую стабильность ОЦК твердых растворов Fe–Cr представлено в работе [95]. Здесь были рассмотрены сплавы в ферромагнитном и парамагнитном состояниях при давлениях 0 и 10 ГПа. Моделирование парамагнитного состояния было выполнено с помощью приближения разупорядоченных локальных моментов (DLM). Было обнаружено, что в парамагнитном состоянии DLM давление уменьшает тенденцию к разделению фаз, в то время как оно способствует разделению фаз в сплавах с высоким содержанием железа в ферромагнитном состоянии FM. Данные энтальпии смешения из источника [95] представлены на рис. 12. Для анализа влияния давления на фазовую стабильность в данной работе была использована упрощенная модель, в которой эффективные парные взаимодействия делятся на химические и магнитные. Эта модель показывает, что влияние давления на фазовую стабильность в этой системе происходит в основном за счет уменьшения магнитного момента на Cr и, следовательно, обменного взаимодействия Fe–Cr, которое диктует аномальную стабильность сплава при нормальных условиях. Авторы утверждают, что область спинодального распада должна расширяться с ростом давления. Кроме того, расчеты показывают, что под давлением парамагнитная ГЦК-фаза становится более стабильной по сравнению с ОЦК-сплавами с ПМ в богатой железом части фазовой диаграммы.
Рис. 12.
Рассчитанная энтальпия смешения ферромагнитных (а) и парамагнитных (б) сплавов Fe–Cr как функция концентрации Cr при давлениях: 1 – 0, 2 – 5, 3 – 10, 4 – 20, 5 – 30 ГПа; на вставке – увеличенная концентрационная зависимость энтальпии смешения в ОЦК-сплавах Fe–Cr в ферромагнитном состоянии [95].
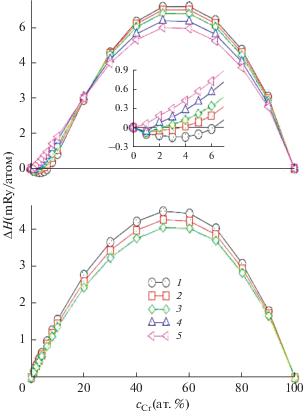
В работе [96] приводилось детальное исследование фазовой стабильности ГЦК и ОЦК магнитных бинарных сплавов Fe–Cr, Fe–Ni и Cr–Ni и тройных сплавов Fe–Cr–Ni в рамках комбинации методов ТФП, кластерного разложения и магнитного кластерного разложения. Используя базу данных, полученную в рамках ТФП, со 248 конфигурациями ГЦК и 246 конфигурациями ОЦК, авторы работы проводят оценку стабильности ГЦК- и ОЦК-фаз тройных сплавов Fe–Cr–Ni. Показано, что наблюдаемые отклонения параметров решеток от закона Вегарда в ГЦК-сплавах Fe–Cr–Ni обусловлены нелинейным изменением атомных магнитных моментов в зависимости от состава сплава. Предсказанные средние полные и локальные магнитные моменты, рассматриваемые как функции концентрации Ni в ОЦК-структуре основного состояния и ГЦК-структурах сплавов Fe–Ni, находятся в хорошем согласии с экспериментальными данными. В работе [96] показано, что тройное соединение Fe2CrNi со структурой типа ГЦК является наиболее стабильной тройной интерметаллической фазой в основном состоянии с отрицательной энтальпией образования минус 0.164 эВ/атом и обладает самой низкой температурой перехода порядок–беспорядок 650 К. Моделирование ТФП и MКР показывают, что фазовая стабильность структуры FeCrNi в первую очередь определяется сильными антиферромагнитными взаимодействиями между атомами Fe и Ni с атомами Cr.
Анализ относительной фазовой стабильности тройных ГЦК- и ОЦК-фаз при различных температурах с использованием данных энтальпий образования и свободных энергий показывает, что в высокотемпературных областях конфигурационная энтропия играет важную роль в стабилизации ГЦК-сплавов по сравнению с ОЦК-сплавами [96]. Авторы утверждают, что превосходное согласие между расчетами и экспериментальными данными по энтальпиям образования при 1600 K говорит о важности учета магнитного вклада в сплавах Fe–Cr–Ni для исправления недостатков традиционной модели кластерного разложения. В работе также приводятся расчеты параметров ближнего порядка при разных температурах, которые хорошо согласуются с экспериментальными данными по бинарным и тройным сплавам. Особое внимание было уделено сплавам Fe56Cr21Ni23, Fe38Cr14Ni48 и Fe34Cr20Ni46, близким к центру концентрационного треугольника. Значительное уменьшение параметра ближнего порядка для пар Fe–Ni в зависимости от концентрации Cr, согласуется с экспериментальными наблюдениями. Авторы работы утверждают, что этот важная особенность термодинамики сплава связана с тем, что взаимодействие между Cr, Fe и Ni является сильно антиферромагнитным, что объясняет большие отрицательные значения ближнего порядка, предсказанные для атомных пар Fe–Cr и Ni–Cr.
Анализ многокомпонентного легирования Ni, Mn и Mo на фазовую стабильность ОЦК твердых растворов Fe–Cr проведен в работе [97]. Легирование Ni повышает ударную вязкость, пластичность и устойчивость к коррозии. Мо повышает коррозионную стойкость в агрессивных средах, например в соляных и кислотных условиях. Кроме того, добавление Мо улучшает ударную вязкость нержавеющих сталей при высоких температурах. Марганец может улучшить твердость сталей.
Для сплавов Fe100-cCrc, Fe100-c-05CrcNi05 и Fe100-c-07CrcNi05Mn01Mo01 была представлена оценка энергии смешения и локальной тенденции к спинодальному распаду [97]. Показано, что для тройных и пятикомпонентных сплавов граница спинодального распада смещается с 17 ат. % Cr на 13 и 12 ат. % Cr соответственно. Энтальпии смешения ОЦК-сплавов Fe–Cr–Ni приведены на рис. 9. Энтальпии смешения Fe–Cr–Mo и Fe–Cr–Ni–Mo сплавов показаны на рис. 13. Для расчета энтальпии смешения в качестве стандартных состояний чистых компонентов были рассмотрены ферромагнитное ОЦК-железо, немагнитный ОЦК-хром, ферромагнитный ГЦК-никель, ОЦК-молибден и $\alpha $-Mn. Рассчитанные энтальпии смешения бинарной системы Fe–Cr находятся в хорошем согласии с более ранними расчетами, которые показывают аномальную фазовую стабильность ферромагнитных неупорядоченных сплавов Fe–Cr с низкими концентрациями Cr [67, 68, 71].
Рис. 13.
Энтальпии смешения Fe–Cr–Mo и Fe–Cr–Ni–Mo сплавов: 1 – Fe1–xCrxMo1.0, 2 – Fe1– xCrxNi5.0Mo1.0, 3 – Fe1 –xCrxNi10.0Mo1.0, 4 – Fe1 –xCrxNi15.0Mo1.0, 5 – Fe1 –xCrxNi20.0Mo1.0, 6 – Fe1 –xCrxMo2.0, 7 – Fe1 –xCrxNi5.0Mo2.0, 8 – Fe1 –xCrxNi10.0Mo2.0, 9 – Fe1 –xCrxNi15.0Mo2.0, 10 – Fe1–xCrxNi20.0Mo2.0, 11 – Fe1 –xCrx; значения для Fe–Cr-сплавов показаны для сравнения.
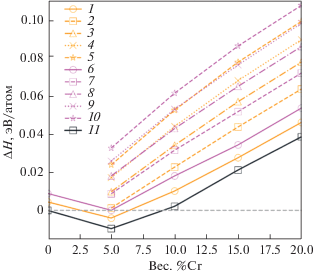
Рассчитанная энтальпия смешения ОЦК- сплава Fe95Ni05 при $T$ = 0 К равна минус 0.57 mRy [97]. Этот результат согласуется с исследованиями 57Fe мессбауэровских спектров сплавов Fe–Ni с концентрациями Ni от 1 до 5 ат. % при комнатной температуре [98]. Для сильно разбавленных тройных сплавов Fe95– сCrсNi05 энтальпия смешения немного ниже бинарной кривой Fe–Cr, при этом область с отрицательной энергией смешения сужается, а минимум кривой соответствует концентрации Cr $ \sim 3$ ат. %. Для пятикомпонентных сплавов энтальпия смешения при $T$ = 0 К положительна для всех концентраций Cr.
В работе [99] представлено исследование упругих свойств чистого железа и сплава замещения Fe–Cr с 10 ат. % Cr в зависимости от температуры с использованием первопринципных расчетов электронной структуры методом точных маффин–тин-орбиталей. В работе получены данные температурной зависимости объемного модуля упругости B и коэффициентов упругости C' и C44, и коэффициента анизотропии. Результаты, полученные для чистого ОЦК железа, находятся в хорошем согласии с экспериментальными данными. Влияние температуры на упругие свойства учитывались с помощью электронного и магнитного вкладов, а также с учетом расширения решетки. Авторы работы показывают, что степень магнитного порядка как в чистом железе, так и в сплаве Fe90Cr10 в основном определяет резкое изменение упругой анизотропии этих материалов при повышенных температурах. Эффект расширения решетки оказывается вторичным, но также очень важным для количественного моделирования.
В [100] также представлено первопринципное исследование упругих параметров ферромагнитных и парамагнитных ОЦК-сплавов Fe1 –cCrc (0 $ \leqslant $ c $ \leqslant $ 1). Получены данные концентрационных зависимостей упругих констант C11 и C44 и модулей упругости $B$ и $E$ [100]. Сравнение с экспериментальными данными показывает, что использованный теоретический подход точно описывает наблюдаемую зависимость модулей упругости от состава. В ферромагнитном твердом растворе Fe1 –cCrc оба компонента сплава имеют разные магнитные состояния по сравнению с их структурами в основном состоянии, в частности, Cr имеет большой локальный магнитный момент в сплавах с высоким содержанием железа, который исчезает выше при концентрации Сr $ \sim 50$ ат. % [100]. Авторы утверждают, что предсказанные аномальные тенденции константы упругости чувствительны к сложным взаимодействиям между магнитными и химическими эффектами. Немонотонная зависимость упругих параметров от состава оказывает существенное влияние на микромеханические свойства ферритных нержавеющих сталей.
Наравне с упругими свойствами в системе Fe–Cr, параметр решетки также имеют аномальную зависимость состава около 5 ат. % Cr [101]. В работе методом точных маффин–тин-орбиталей получена концентрационная зависимость параметра решетки ОЦК-сплавов Fe–Cr. Авторы предположили, что наблюдаемые аномалии связаны с электронным топологическим переходом в этих сплавах. В работе также представлены концентрационные зависимости коэффициентов упругости C11, C12, C' и C44, модулей упругости $B$, $G$, $E$, коэффициента Пуассона и температуры Дебая. Некоторые из этих характеристик проявляют аномальное поведение при концентрации Cr 5% (рис. 14 и 15), что согласуется с имеющимся экспериментом [102]. Представленные в работе [40] концентрационные зависимости коэффициента Пуассона, модулей упругости $B$ и $G$ хорошо согласуются с экспериментальными данными [101], хотя теоретические расчеты переоценивают модуль $E$.
Рис. 14.
Теоретическая зависимость значений упругой константы С11 от концентрации хрома в ферромагнитных ОЦК Fe–Cr и Fe–Cr–Ni сплавах: 1–5 – см. рис. 8.
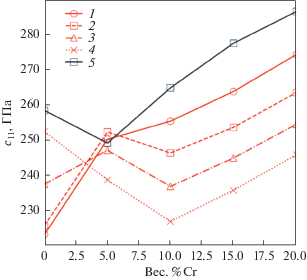
Рис. 15.
Теоретическая зависимость значений объемного модуля B от концентрации хрома в ферромагнитных ОЦК Fe–Cr и Fe–Cr–Ni сплавах: 1–5 – см. рис. 8.
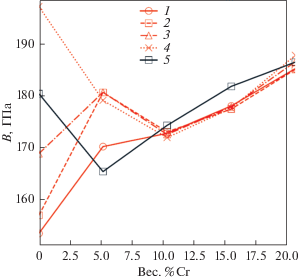
Как было упомянуто выше, сплавы на основе объемно-центрированного кубического железа составляют основу ферритных конструкционных сталей и особенно востребованы в ядерной энергетике, где радиационное излучение может сильно испортить механические свойства материала. Механизм охрупчивания, вызванного радиационным излучением, заключается в возникновении точечных дефектов, которые способствуют диффузии атомов легирующих компонент и образованию кластеров легирующих элементов в матрице сплава. Наличие легирующих элементов также может влиять на формирование кластеров точечных дефектов. Как образование кластеров точечных дефектов, так и образование кластеров с высоким содержанием легирующих элементов влияет на механические свойства сталей.
На механические свойства также может влиять превращение феррит–мартенсит. Чтобы уменьшить склонность к этому превращению в сталях заменяются легирующие элементы, способствующие активации превращения (например, Ni) другими переходными металлами (например W, Ta и Mo), которые придают сталям аналогичные макроскопические свойства (например, механические и коррозионные). Эти легирующие компоненты в твердом растворе присутствуют в небольших количествах и могут также взаимодействовать с точечными дефектами, созданными излучением. Взаимодействия дефектов и легирующих компонентов играют важную роль в эволюции микроструктуры, например радиационной деградации материалов, радиационного объемного разбухания, вызванных быстрыми нейтронами. Основной причиной разбухания, вызванного нейтронами, является переход от образования кластера легирующих элементов к образованию кластеров вакансий, то есть пор. Феноменологическая модель межузельной диффузии кластеров в Fe–Cr успешно объяснила некоторую концентрационную зависимость набухания при нейтронном облучении [103–107]. В этой модели использовались данные первопринципных расчетов, которые показали, что один собственный междоузельный атом имеет сильное дальнодействующее притягивающее взаимодействие с растворенными атомами Cr вдоль линии сжатия (и скольжения). Эффект притягивающего взаимодействия между атомами Cr в решетке и междоузельным атомом в сплаве Fe–Cr состоит в том, что петли собственного междоузельного атома воспринимают растворенный Cr как ловушку. С увеличением плотности “ловушек” скорость диффузии будет уменьшаться. Это приведет диффузию междоузельных дефектов за тот же временной масштаб, что и диффузию вакансий, и, таким образом, значительно увеличит рекомбинацию микроструктуры сплава. Когда плотность растворенного вещества становится достаточно высокой, эффективный диапазон “ловушек” начнет перекрываться и сила захвата снова уменьшится. С помощью метода молекулярной динамики в работе [68] было показано, что когезионная модель захвата в сплаве Fe–Cr воспроизводит измеренные минимумы степени разбухания, соответствующие $ \sim $9 ат. % Cr, и предполагалось, что изменение скорости диффузии петли междоузельного атома является основной причиной концентрационной зависимости разбухания в Fe–Cr.
В работе [108] приводится первопринципное исследование транспортных и диффузионных свойств шести различных легирующих элементов (Cr, Cu, Mn, Ni, P и Si) в разбавленных ОЦК-сплавах на основе железа с целью исследования потоковой связи между растворенными веществами и моновакансиями, в частности между вакансиями и индуцированными радиацией сегрегациями. Авторы разработали новый подход, в котором рассчитанные в рамках теории функционала плотности (ТФП) частоты скачков комбинируются с методом самосогласованного среднего поля, что позволяет правильно учитывать более дальнодействующие взаимодействия растворенных элементов и вакансий. В работе показано, что сопротивление вакансиям является широко распространенным явлением, которое систематически встречается в реальных бинарных сплавах, характеризующихся ничтожно малыми взаимодействиями растворенного вещества с вакансией, то есть во всех сплавах Fe(X), кроме Fe(Cr). Этот результат (особенно в случае Fe(Mn) и Fe(Ni)) контрастирует с предыдущими вычислениями. На силу сопротивления влияет сложный комбинированный эффект термодинамического и кинетического взаимодействий. Как общая тенденция, сопротивление вакансий сильнее в низкотемпературном режиме $(T < 1000$ K), включая рабочую температуру ядерных реакторов (~$573$ К). В работе показано, что профили индуцированных радиацией сегрегаций для всех модельных сплавов, за исключением Fe(Cr), демонстрируют одинаковую тенденцию: обогащение растворенного вещества при низкой температуре и истощение при высокой температуре. Эта тенденция хорошо согласуется с экспериментами, выполненными на реальных многокомпонентных ферритно-мартенситных сплавах [109]. В работе описывается модель, которая представляет собой мощный инструмент для прогнозирования диффузионных свойств в сплавах. Модель сочетает в себе точные вычисления из первых принципов в рамках самосогласованного среднего поля для получения точных транспортных коэффициентов, особенно в низкотемпературном режиме, который обычно недоступен для экспериментов.
В работе [110] было проведено исследование энергетических и магнитных свойств растворов переходных металлов в ОЦК-Fe. Представлено взаимодействие между атомами переходного металла и точечными дефектами, что в ОЦК-железе в основном определяется деформацией, обусловленной размерным фактором в 4d и 5d легирующих элементов. Показано, что вакансии связываются, а междоузельные дефекты отталкиваются. Связывающие 3d элементы обычно отталкивающе взаимодействуют с междоузельными атомами и связываются с вакансиями [110]. Легирование Cr связывает только междоузельные дефекты и поэтому должно значительно замедлить диффузию междоузельного кластера.
В работе [111] в рамках метода проектированных присоединенных плоских волн (PAW) и метода ультрамягких псевдопотенциалов (USPP) было проведено исследование энергетического и магнитного характера собственных точечных дефектов в ОЦК-Fe и ОЦК-Cr, было охарактеризовано взаимодействие атомов Cr с точечными дефектами в ОЦК-железе. Показано, что отрицательная энергия замещения Cr в ОЦК-Fe является очень важным фактором в определении энергий связи для комплексов точечных дефектов с атомами Cr. Результаты энергии смешения в разбавленных твердых растворах на основе железа, полученные двумя методами PAW и USPP, значительно отличаются друг от друга. Например, для сплава с 6.25 ат. % Cr энергия смешения в рамках методов PAW и USPP равна –1.95 и ‒14.5 мэВ [111] соответственно, в то время как метод точных маффин-тин орбиталей (EMTO-CPA) дает значение –7.59 мэВ. Стоит отметить, что при концентрации сплава 12.5 ат. % Cr в расчетах PAW и USPP энергия смешения имеет положительный знак [111].
В работе [111] для суперячеек, состоящих из 54 и 128 атомов, приводятся результаты энергии связи и образования для следующих конфигураций дефектов: пар вакансий Cr, кластеров Cr, собственных междоузельных атомов 〈100〉-Cr, 〈110〉-Cr, and 〈111〉-Cr; также представлены данные энергии миграции для атомов Fe и Cr в этих суперячейках. Авторы утверждают, что магнитные взаимодействия тесно связаны со связывающим или отталкивающим характером конфигурации дефекта. Если магнитные моменты значительно подавлены, то конфигурация становится более отталкивающей, чем если бы магнитные моменты оставались близкими к объемным значениям. Конфигурации, содержащие атомы Cr в первой координационной сфере, являются связующими, если атомы Cr имеют большие антиферромагнитные моменты. Показано, что во время релаксации каскадов смещения в Fe–Cr созданные дефекты внедрения постепенно притягиваются к растворенным атомам Cr. Обогащенные хромом кластеры (фаза $\alpha {\kern 1pt} '$) в сплавах Fe–Cr отталкивают межузельные петли и действуют как временные источники дефектов внедрения и вакансий. Увеличение энергии образования дефекта внедрения в чистом Cr по сравнению с энергией в Fe подразумевает, что диффузия внедрения будет происходить преимущественно в $\alpha $-фазе, обогащенной железом. Большое связывающее взаимодействие, обнаруженное для междоузельных комплексов вдоль направлений 〈110〉 и 〈111〉, в которых два атома Cr разделены атомом Fe. Это связано с различным типом междоузельных петель, наблюдаемых экспериментально в зависимости от содержания Cr. Показано, что связывающее взаимодействие между 〈111〉 междоузлями и атомами Cr может способствовать замедлению диффузии скользящих междоузельных петель [111]. Это должно привести к увеличению рекомбинации с вакансиями.
Из двух рассмотренных в работе [111] формализмов следует выбирать PAW, поскольку PAW более физически корректен и полученные в рамках метода PAW результаты лучше согласуются с экспериментальными данными. Это особенно верно для переходных металлов и их сплавов, где тонкие магнитные взаимодействия играют важную роль. Основное различие результатов, полученных с двумя формализмами, видно из результатов локальных магнитных моментов [111]. В случае чистого железа методы дают схожие результаты, хотя метод USPP переоценивает объемный момент. Самое большое различие в чистом Fe заключается в том, что PAW предсказывает антиферромагнитный момент на собственных междоузельных атомах 〈111〉, а USPP – нет. Для чистого хрома разница невелика, так как магнетизм не влияет в такой же степени. Однако для атомов Cr в ОЦК-Fe разница значительна. Для конфигураций с одним атомом Cr PAW предсказывает большую энергию связи на 0.1 эВ, чем USPP. Для конфигураций с двумя атомами Cr PAW предсказывает энергии связи, которые примерно на 0.3 эВ больше, чем USPP. В результате в рамках USPP большинство конфигураций с двумя Cr обладают отталкивающим взаимодействием, даже если в рамках метода PAW они характеризуются существенно связывающим взаимодействием. Экспериментальные данные по междоузельной миграции показывают, что смешанные внедрения должны мигрировать быстрее, чем внедрения чистого Fe для сплавов с низким содержанием Cr.
5.2. Диаграммы состояния бинарных систем Fe–Ni
5.2.1. Фазовые равновесия. Система Fe–Ni благодаря своему важному промышленному и технологическому значению принадлежит к одной из наиболее изученных систем. Диаграмма состояния этой системы изучалась более века, первое исследование проведено Осмондом в 1899 году [112]. Термодинамическое описание системы приведено в работах [113–117]. Диаграмма состояния системы Fe–Ni принята в данной работе согласно термодинамической оценке [116] и представлена на рис. 16 . Система характеризуется наличием непрерывного ряда твердого раствора (γFe,Ni) (ГЦК-раствор на основе чистых компонентов), твердых растворов на основе фаз (δFe) (высокотемпературный ОЦК-раствор на основе железа), (αFe) (низкотемпературный раствор ОЦК на основе железа), упорядоченных кубических фаз FeNi3 (структурный тип Cu3Au) и FeNi (структурный тип CuAu). Авторы [115], однако, пришли к выводу, что фаза FeNi является метастабильной. Кристаллическая структура и параметры решетки всех фаз системы Fe–Ni приведены в табл. 3. Основываясь на собственном исследовании и оценке опубликованных данных, авторы [115] отвергают существование третьей возможной упорядоченной фазы Fe3Ni (структура L12), поэтому она не включена в табл. 3. Все инвариантные равновесия в системе приведены в табл. 4.
Таблица 3.
Кристаллическая структура и параметры решетки фаз системы Fe–Ni
| Фаза | Кристаллическая структура | Параметры решетки, Å | Примечания |
|---|---|---|---|
| ($\delta $Fe) | W, cI2-Im-3$m$ | a = 2.9315 | >1394°C [87] |
| ($\gamma $Fe) | Cu, cF4-Fm-3$m$ | a = 3.6467 | >912°C [87] |
| ($\alpha $Fe) | W, cI2-Im-3$m$ | a = 2.8665 | 25°C [87] |
| ($\varepsilon $Fe) | Mg, hP2–P 63/mmc | a = 2.468, c = 3.96 | 25°C, >13 ГПа [87] |
| (Ni) | Cu, cF4-Fm-3$m$ | a = 3.5240 | 25°С [87] |
| FeNi3 | AuCu3, cP4-Pm-3$m$ | a = 3.5523 | [118] |
| FeNi | AuCu, tP4–P 4/mmm | a = 2.531, c = 3.579 | [119] |
Таблица 4.
Инвариантные равновесия в системе Fe–Ni [116]
| Инвариантное равновесие | Температура, °С | Состав, ат. % Ni | ||
|---|---|---|---|---|
| L + ($\delta $Fe) $ \rightleftarrows (\gamma $Fe,Ni) | 1514 | 6.4 | 5.2 | 5.7 |
| L $ \rightleftarrows (\gamma $Fe,Ni) | 1432 | 66.3 | 66.3 | – |
| ($\gamma $Fe,Ni) $ \rightleftarrows $ FeNi3 | 514 | 72.3 | 72.3 | – |
| ($\gamma $Fe,Ni) $ \rightleftarrows (\gamma $Fe,Ni)PM + ($\gamma $Fe,Ni)FM | 472 | 48.4 | 48.4 | 48.4 |
| ($\gamma $Fe,Ni)${{{\text{\;}}}_{{PM}}} \rightleftarrows (\alpha $Fe) + ($\gamma $Fe,Ni)FM | 428 | 44.4 | 6.4 | 47.5 |
| ($\gamma $Fe,Ni)${{{\text{\;}}}_{{FM}}} \rightleftarrows (\alpha $Fe) + FeNi3 | 353 | 52.5 | 5.6 | 55.5 |
| ($\alpha $Fe) + FeNi3 $ \rightleftarrows $FeNi | –38 | 0.0 | 64.0 | 62.5 |
На кривых ликвидус и солидус (γFe,Ni)-фазы присутствует минимум температуры (рис. 16 ). Для фаз (δFe), (αFe) и FeNi3 характерно образование областей гомогенности.
Низкотемпературная часть системы подробно обсуждена в работах [114, 115, 120]. Наличие разрыва смешиваемости в ГЦК-фазе (γFe,Ni) показано термодинамическими расчетами CALPHAD [113, 121] и подтверждено экспериментальными исследованиями [115]. Монотектоидное равновесие (γFe,Ni)PM = (γFe,Ni)FM + (αFe) протекает при температуре около 428°C, а при 353°С (γFe,Ni)FM. фаза распадается на (αFe) + FeNi3 по эвтектоидной реакции (γFe,Ni)FM $ \rightleftarrows (\alpha $Fe) + FeNi3. При $ \sim {\kern 1pt} 72.3$ ат. % Ni и 517°C (γFe,Ni)-фаза превращается в упорядоченную кубическую фазу FeNi3, а при температуре –38°С образуется фаза FeNi по перитектоидной реакции ($\alpha $Fe) + FeNi3 $ \rightleftarrows $ FeNi.
5.2.2. Термодинамические свойства. На рис. 17 представлены изотермы энтальпии смешения и термодинамических активностей компонентов, рассчитанные согласно [122]. Термодинамические свойства α(Fe)-фазы изучены не были.
Рис. 17.
Термодинамические свойства фаз системы Fe–Ni [122]: (а) – энтальпия смешения жидких сплавов; (б) – термодинамические активности компонентов жидких сплавов; (в) – энтальпия смешения γ(Fe,Ni)-фазы; (г) – термодинамические активности компонентов γ(Fe,Ni)-фазы; (д) – энтальпия смешения α(Fe)-фазы; (е) – термодинамические активности компонентов α(Fe)-фазы.
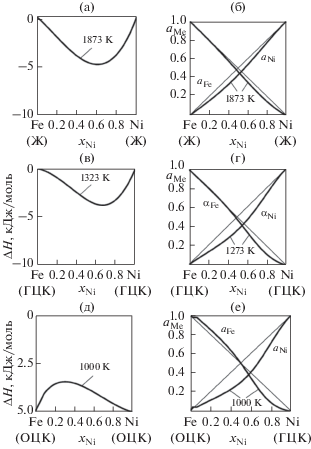
Как следует из рис. 17 а–17г, для энтальпий смешения и активности никеля расплавов и $\gamma $(Fe,Ni)-фазы наблюдаются небольшие отрицательные отклонения, активность железа в этих же фазах носит знакопеременный характер. Для ${\Delta }Н$ и ${{a}_{{{\text{Me}}}}}~\alpha $(Fe)-фазы характерны небольшие положительные отклонения от идеальности (рис. 17 д, 17е). Подобный характер взаимодействия компонентов в жидком и твердом состояниях находится в хорошем согласии с характером фазовых равновесий в системе.
5.3. Диаграммы состояния бинарных систем Сr–Ni
5.3.1. Фазовые равновесия. Диаграмма состояния системы Cr–Ni принята в данной работе согласно термодинамическому моделированию [123] и представлена на рис. 18. Система Cr–Ni при высокой температуре относится к простому эвтектическому типу, и фазовые равновесия в этой области хорошо установлены. Несмотря на близость атомных радиусов хрома и никеля, из-за неизоморфности их решеток (Cr – ОЦК, Ni – ГЦК) они образуют ограниченные твердые растворы: γ – твердый раствор на основе никеля и α – твердый раствор на основе хрома. При температурах ниже примерно 900 К образуется промежуточная фаза CrNi2, имеющая ромбоэдрическую структуру, по перитектоидной реакции, а в богатой Ni области магнитное упорядочение приводит к возникновению трикритической точки и расслоению в ГЦК-фазе (γ-фазе). Богатая никелем ГЦК-фаза (Ni) имеет очень высокую (до 50 ат. % при температуре эвтектики 1617 К) растворимость Cr, которая остается сравнительно высокой при 856 К (примерно 31 ат. %). Растворимость Ni в фазе ОЦК-Cr при температуре эвтектики составляет около 38.5 ат. % , но резко уменьшается до менее 5 ат. % при температуре около 1250 К и ниже 1 ат. % при 1000 К. Кристаллическая структура всех фаз cистемы Ni–Cr представлена в табл. 5. Все инвариантные равновесия, которые имеют место в системе Ni–Cr, приведены в табл. 6.
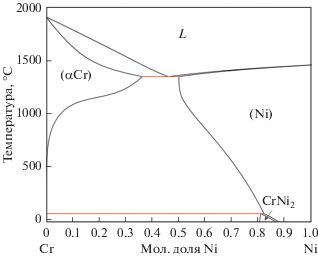
Таблица 5.
Кристаллическая структура и параметры решетки фаз системы Cr–Ni
| Фаза | Кристаллическая структура | Параметры решетки, Å | Примечания |
|---|---|---|---|
| ($\alpha $Cr) | W, cI2-Im-3$m$ | a = 2.8848 | 25°С [87] |
| ($\alpha $'Cr) | $\alpha $'Cr, tI2–I4/mmm | a = 2.882, c = 2.887 | 25°С, HP [87] |
| (Ni) | Cu, cF 4-Fm-3$m$ | a = 3.5240 | 25°С [87] |
| CrNi2 | MoPt2, oP6-Immm | a = 2.524, b = 7.571, c = 3.568 | от 60 до 76.5 ат. % Ni |
Таблица 6.
Инвариантные равновесия в системе Cr–Ni [123]
| Инвариантное равновесие | Температура, K | Состав, ат. % Cr | ||
|---|---|---|---|---|
| L $ \rightleftarrows $ (Ni) + (Cr) | 1617 | 53.5 | 50.0 | 38.5 |
| (Ni) + (Cr) $ \rightleftarrows $ NiCr2 | 856 | 31.0 | 99.5 | 33.3 |
| (Ni) парамагн. $ \rightleftarrows $ (Ni) ферромагн. + NiCr2 | 458 | 5.0 | 0.7 | 33.3 |
Единственное значение используется для температуры перитектоидной реакции образования фазы CrNi2 (Ni) + (Cr) $ \rightleftarrows $ CrNi2, а именно 863 К согласно [124] (856 К согласно принятому в данной работе термодинамическому моделированию [123]).
5.3.2. Термодинамические свойства. Данные по энтальпии в жидкой фазе для богатой никелем стороны доступны в работе [125]. Работа [126] является единственным экспериментальным источником для жидкой фазы в сплавах, богатых хромом при 2200 K, но показывает значительный разрыв с данными [125]. Однако результаты Судавцовой [126] указывают на то, что энтальпия смешения в жидкой фазе почти симметрична. На рис. 19 приведена рассчитанная в [127 ] энтальпия смешения в жидкой фазе, которая довольно хорошо соответствует относительно новым экспериментальным данным. Активность хрома в твердом состоянии при 1300 К согласно термодинамическому моделированию [127 ] приведена на рис. 19.
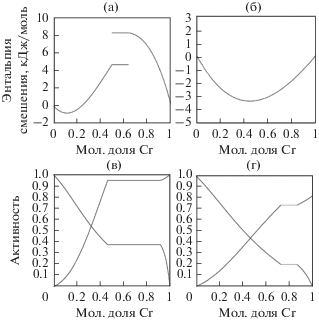
ЗАКЛЮЧЕНИЕ
В работе представлены результаты квантовых первопринципных расчетов свойств материалов, рассмотрены машинные методы искусственного интеллекта и результаты расчетов термодинамических свойств бинарных сплавов железа, хрома и никеля. В следующих работах планируется представить данные о свойствах жидкометаллических рабочих тел в различных состояниях, а также свойствах плазмы в атомных реакторах, образующейся при торможении продуктов делящегося вещества, при котором возникают заряженные частицы и неравновесная плазма.
Авторы выражают благодарность В.Е. Фортову и А.В. Дубу за многочисленные плодотворные обсуждения затронутых в обзоре тем.
Работа выполнена при финансовой поддержке Министерства образования и науки РФ (уникальный идентификатор ПНИЭР RFMEFI60719X0323).
Список литературы
Hohenberg P., Kohn W. Inhomogeneous Electron Gas // Phys. Rev. 1964. V. 136. B864.
Kohn W., Sham L.J. Self-consistent Equations Including Exchange and Correlation Effects // Phys. Rev. 1965. V. 140. A1133.
Martin R.M. Electronic Structure: Basic Theory and Practical Methods. Cambridge: University Press., 2004.
Perdew J.P., Chevary J.A., Vosko S.H. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation // Phys. Rev. B. 1992. V. 46. № 11. P. 6671.
Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Andersen K. Linear Methods in Band Theory I // Phys. Rev. B. 1975. V. 12. P. 3060.
Drittler B., Weinert M., Zeller R. et al. Vacancy Formation Energies of FCC Transition Metals Calculated by a Full Potential Green’s Function Method // Solid State Communications. 1991. V. 79. P. 31.
Huhne T., Zecha C., Ebert H. et al. Full-potential Spin-polarized Relativistic Korringa–Kohn–Rostoker Method Implemented and Applied to BCC Fe, FCC Co, and FCC Ni // Phys. Rev. B. 1998. V. 58. P. 10 236.
Payne M.C., Teter M.P., Allan D.C. et al. Iterative Minimization Techniques for ab initio Total-energy Calculations: Molecular Dynamics and Conjugate Gradients // Rev. Modern Phys. 1992. V. 64. P. 1045.
Korringa J. On the Calculation of the Energy of a Bloch Wave in a Metal // Physica. Amsterdam. 1947. V. 13. P. 392.
Kohn W., Rostoker N. Solution of the Schrödinger Equation in Periodic Lattices with an Application to Metallic Lithium // Phys. Rev. 1954. V. 94. P. 1111.
Rayleigh L. On the Instability of a Cylindrical Thread of a Viscous Liquid Surrounded by Another Viscous Fluid // Phil. Mag. 1892. V. 23. P. 481.
Lippmann B.A., Schwinger J. Variational Principles for Scattering Processes // Phys. Rev. 1950. V. 79. P. 469.
Ekholm M. Theoretical Descriptions of Complex Magnetism in Transition Metals and Their Alloys. Doct. diss. Linköping: Linköping University Electronic Press, 2012.
Kresse G., Furthmüller J. Efficient Iterative Schemes for ab initio Total-energy Calculations Using a Plane-wave Basis Set // Phys. Rev. B. 1996. V. 54. P. 11 169.
Kresse G., Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-wave Method // Phys. Rev. B. 1999. V. 59. P. 1758.
Giannozzi P., Baroni S., Bonini N. l. QUANTUM ESPRESSO: A Modular and Open-source Software Project for Quantum Simulations of Materials // J. Phys.: Cond. Mat. 2009. V. 2. № 39. 395 502.
Gonze X., Jollet F., Abreu A.F. Recent Developments in the ABINIT Software Package // Comput. Phys. Commun. 2016. V. 205. P. 106.
Blaha P. WIEN2K: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. Vienna: Technical University, 2001.
Vitos L. Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Applications. London: Springer, 2007.
Vitos L., Abrikosov I.A., Johansson B. Anisotropic Lattice Distortions in Random Alloys from First-principles Theory // Phys. Rev.Lett. 2001. V. 87. P. 156 401.
Lejaeghere K., Bihlmayer G., Björkman T. Reproducibility in Density Functional Theory Calculations of Solids // Science. 2016. V. 351. P. 6280.
Jain A., Persson K.A., Ceder G. Research Update: The Materials Genome Initiative: Data Sharing and the Impact of Collabporative ab initio Databases // Appl. Phys. Lett. Mater. 2016. V. 4. № 5. P. 053102.
Draxl C., Scheffler M. The NOMAD Laboratory: From Data Sharing to Artificial Intelligence // J. Phys.: Mater. 2019. V. 2. Iss. 3. P. 036001.
Wilkinson M., Dumontier M., Aalbersberg I.J. et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship // Sci. Data. 2016. V. 3. № 160 018. https://doi.org/10.1038/sdata.2016.18
Curtarolo S., Setyawan W., Hart G.L.W. et al. AFLOW: An Automatic Framework for High-throughput Materials Discovery // Comput. Mater. Sci. 2012. V. 58. P. 218.
Saal J.E., Kirklin S., Aykol M. et al. Materials Design and Discovery with High-throughput Density Functional Theory: The Open Quantum Materials Database (OQMD) // Jom. 2013. V. 65. Iss. 11. P. 1501.
Allen G.G.F.H., Sievers R. Crystallographic Databases. Chester: International Union of Crystallography, 1987.
Hautier G., Jain A., Ong S.P. et al. Phosphates as Lithium-ion Battery Cathodes: An Evaluation Based on High-throughput ab initio Calculations // Chemistry of Materials. 2011. V. 23. Iss. 15. P. 3495.
Lind H., Forsén R., Alling B. et al. Improving Thermal Stability of Hard Coating Films via a Concept of Multicomponent Alloying // Appl. Phys. Lett. 2011. V. 99. Iss. 9. P. 091903-1.
Kirklin S., Meredig B., Wolverton C. High-throughput Computational Screening of New Li-Ion Battery Anode Materials // Advanced Energy Materials. 2013. V. 3. Iss. 2. P. 252.
Aykol M., Kirklin S. et al. Thermodynamic Aspects of Cathode Coatings for Lithium-ion Batteries // Adv. Energy Mater. 2014. V. 4. Iss. 17. P. 1.
Yao Y., Tse J.S., Tanaka K. Metastable High-pressure Single-bonded Phases of Nitrogen Predicted via Genetic Algorithm // Phys. Rev. B: Cond. Mat. Mater. Phys. 2008. V. 77. Iss. 5. P. 1.
Zahariev F., Dudiy S.V., Hooper J. et al. Systematic Method to New Phases of Polymeric Nitrogen under High Pressure // Phys. Rev. Lett. 2006. V. 97. Iss. 15. P. 11.
Lyakhov A.O., Oganov A.R. Evolutionary Search for Superhard Materials: Methodology and Applications to Forms of Carbon and TiO2 // Phys. Rev. B: Condensed Matter and Materials Physics. 2011. V. 84. P. 2.
Glass C.W., Oganov A.R., Hansen N. USPEX-Evolutionary Crystal Structure Prediction // Computer Physics Communications. 2006. V. 175. Iss. 11–12. P. 713.
Kolmogorov A.N., Shah S., Margine E.R. et al. New Superconducting and Semiconducting Fe–B Compounds Predicted with an ab initio Evolutionary Search // Phys. Rev. Lett. 2010. V. 105. № 21. P. 1.
Kolmogorov A.N., Shah S., Margine E.R. et al. Pressure-driven Evolution of the Covalent Network in CaB6 // Phys. Rev. Lett. 2012. V. 109. Iss. 7. P. 1.
Jain A., Ong S.P., Hautier G. et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation // APL Materials. 2013. V. 1. № 1. 011002. https://doi.org/10.1063/1.4812323
Butler K.T., Davies D.W., Cartwright H. et al. Machine Learning for Molecular and Materials Science // Nature. 2018. V. 559. Iss. 7715. P. 547.
Schmidt J., Marques M.R.G., Botti S. et al. Recent Advances and Applications of Machine Learning in Solid-state Materials Science // Npj Computational Materials. 2019. V. 5. Iss. 1.
Sanchez-Lengeling B., Aspuru-Guzik A. Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering // Science. 2018. V. 361. Iss. 6400. P. 360.
Gražulis S., Daškevic A., Merkys A. et al. Crystallography Open Database (COD): An Open-access Collection of Crystal Structures and Platform for World-wide Collaboration // Nucleic Acids Res. 2011. V. 40. Iss. D. P. 420.
Groom C.R., Bruno I.J., Lightfoot M.P. et al. The Cambridge Structural Database // Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016. V. 72. P. 171.
Hachmann J., Olivares-Amaya R., Atahan-Evrenk S. et al. The Harvard Clean Energy Project: Large-scale Computational Screening and Design of Organic Photovoltaics on the World Community Grid // J. Phys. Chem. Lett. 2011. V. 2,1. № 17. P. 2241.
Schmidt J., Shi J., Borlido P. et al. Predicting the Thermodynamic Stability of Solids Combining Density Functional Theory and Machine Learning // Chemistry of Materials. 2017. V. 29. Iss. 12. P. 5090.
de Jong M., Chen W., Notestine R. et al. A Statistical Learning Framework for Materials Science: Application to Elastic Moduli of k-nary Inorganic Polycrystalline Compounds // Sci. Rep. 2016. V. 6. June. P. 1.
Isayev O., Oses C., Toher C. et al. Universal Fragment Descriptors for Predicting Properties of Inorganic Crystals // Nature Commun. 2017. V. 8. P. 1.
Pilania G., Mannodi-Kanakkithodi A., Uberuaga B.P. et al. Machine Learning Bandgaps of Double Perovskites // Sci. Rep. 2015. V. 6. P. 1.
Dey P., Bible J., Datta S. et al. Informatics-aided Bandgap Engineering for Solar Materials // Comput. Mater. Sci. 2014. V. 83. P. 185.
Zhuo Y., Mansouri Tehrani A., Brgoch J. et al. Predicting the Band Gaps of Inorganic Solids by Machine Learning // J. Phys. Chem. Lett. 2018. V. 9. Iss. 7. P. 1668.
Stanev V., Oses C., Kusne A.G. et al. Machine Learning Modeling of Superconducting Critical Temperature // Npj Computational Materials. 2018. V. 4. № 29.
Schmidt J., Chen L., Botti S. et al. Predicting the Stability of Ternary Intermetallics with Density Functional Theory and Machine Learning // J. Chem. Phys. 2018. V. 148. Iss. 24.
Ward L., Liu R., Krishna A. et al. Including Crystal Structure Attributes in Machine Learning Models of Formation Energies via Voronoi Tessellations // Phys. Rev. B. 2017. V. 96. Iss. 2. P. 1.
Li W., Jacobs R., Morgan D. Predicting the Thermodynamic Stability of Perovskite Oxides Using Machine Learning Models // Comput. Mater. Sci. 2018. V. 150. P. 454.
Ye W., Chen C., Wang Z. et al. Deep Neural Networks for Accurate Predictions of Crystal Stability // Nature Commun. 2018. V. 9. Iss. 1. P. 1.
Xue D., Balachandran P.V., Hogden J. et al. Accelerated Search for Materials with Targeted Properties by Adaptive Design // Nature Commun. 2016. V. 7. P. 1.
Evans J.D., Coudert F.X. Predicting the Mechanical Properties of Zeolite Frameworks by Machine Learning // Chemistry of Materials. 2017. V. 29(18). P. 7833.
Xie T., Grossman J.C. Crystal. Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties // Phys. Rev. Lett. 2018. V. 120. Iss. 14. P. 145.
Weston L., Stampfl C. Machine Learning the Band Gap Properties of Kesterite I2-II-IV-V4 Quaternary Compounds for Photovoltaics Applications. // Phys. Rev. Mater. 2018. V. 2. Iss. 8. P. 1.
Ling J., Hutchinson M., Antono E. et al. High-Dimensional Materials and Process Optimization Using Data-Driven Experimental Design with Well-Calibrated Uncertainty Estimates // Integrating Materials and Manufacturing Innovation. 2017. V. 6. № 3. P. 207.
NIMS. Supercon. National Institute of Materials Science, 2019.
Qu N., Liu Y., Liao M. et al. Ultra-high Temperature Ceramics Melting Temperature Prediction via Machine Learning // Ceramics International. 2019. V. 45. Iss. 15. P. 18551.
Hennion M. Chemical SRO Effects in Ferromagnetic Fe Alloys in Relation to Electronic Band Structure // J. Phys. F. 1983. V. 13. P. 2351.
Aldred A.T., Rainford B.D. Ferromagnetism in Iron-chromium Alloys. II. Neutron Scattering Studies // Phys. Rev. B. 1976. V. 14. P. 228.
Mirebeau I., Cadeville M.C., Parette G. et al. Short-range Order in FeV Alloys as Investigated by Neutron Scattering and NMR at 51V Nuclei // J. Phys. F: Met. Phys. 1982. V. 12. P. 25.
Olsson P., Abrikosov I.A., Vitos L. et al. Ab initio Formation Energies of Fe-Cr Alloys // J. Nucl. Mater. 2003. P. 84.
Olsson P., Abrikosov I. A., Wallenius J. Electronic Origin of the Anomalous Stability of Fe-rich BCC Fe–Cr Alloys // Phys. Rev. B. 2006. V. 73. P. 104416.
Klaver T.P.C., Drautz R., Finnis M.W. Magnetism and Thermodynamics of Defect-free Fe-Cr Alloys // Phys. Rev. B. 2006. V. 74. P. 094435.
Lavrentiev M.Yu., Drautz R., Nguyen-Manh D. Monte Carlo Study of Thermodynamic Properties and Clustering in the BCC Fe-Cr System // Phys. Rev. B. 2007. V. 75. P. 014208.
Korzhavyi P.A., Ruban A.V., Odqvist J. et al. Electronic Structure and Effective Chemical and Magnetic Exchange Interactions in BCC Fe-Cr alloys // Phys. Rev. B. 2009. V. 79. P. 054202.
Fisher R.M., Dulis E.J., Carroll K.G. Identification of the Precipitate Accompanying 885F Embrittlement in Chromium Steels // Trans. Am. Inst. Min. Metall. Pet. Eng. 1953. V. 197. P. 690.
Becket F.M. On Allotropy of Stainless Steels // Trans. Am. Inst. Min. Metall. Pet. Eng. 1938. V. 131. P. 15.
Andersen O., Jepsen O., Sob M. In: Lecture Notes in Physics: Electronic Band Structure and Its Applications. Berlin: Springer, 1987.
Xiong W., Selleby M., Chen Q. Phase Equilibria and Thermodynamic Properties in the Fe–Cr System // Solid State Mater. Sci. 2010. V. 35. P. 125.
Xiong W., Hedstrom P., Selleby M. et al. An Improved Thermodynamic Modeling of the Fe-Cr System down to Zero Kelvin Coupled with Key Experiments // Calphad. 2011. V. 35. № 3. P. 355.
Chart T., Putland F., Dinsdale A. Calculated Phase Equilibriums for the Chromium-iron-nickel-silicon System. I. Ternary Equilibriums // Calphad. 1980. V. 4. P. 27.
Hertzman S., Sundman B. A Thermodynamic Analysis of the Iron-Chromium System // Calphad. 1982. V. 6. P. 67.
Lee B.J. Revision of Thermodynamic Descriptions of the Iron-Chromium and Iron-Nickel Liquid Phases // Calphad. 1993. V. 17. P. 251.
Cui S., Jung I.H. Thermodynamic Modeling of the Cu–Fe–Cr and Cu–Fe–Mn Systems // Calphad. 2017. V. 56. P. 241.
Thermo-Calc Software TCFE Steels/Fe-alloys Database. Ver. 10.
Andersson J.O., Helander T., Höglund L., Shi P.F., Sundman B. Thermo-Calc and DICTRA, Computational Tools for Materials Science // Calphad. 2002. V. 26. P. 273.
Treitschke W., Tammann G. Enthalpy of Formation for σ-phase Solid Solutions at 1060 K // Anorganische Chemie. 1907. V. 55. P. 707.
Bain E.C., Griffiths W.E. An Introduction to the Iron-chromium-nickel Alloys // Trans. AIME. 1927. V. 75. № 166. P. 166.
Chevenard P. Experimental Investigations of Iron, Nickel, and Chromium Alloys // Traveaux et memoires du Bureau International des Poids et Mesures. 1927. V. 17. P. 90.
Jett E.R., Foote F. The Fe-Cr Alloy System // Metals and Alloys. 1936. V. 7. № 8. P. 207.
Massalski T.B. Binary Alloy Phase Diagrams. 2nd ed. Metals Park, Ohio: ASM International, 1990.
Yakel H.L. Jr. Atom Distributions in Sigma Phases: I. Fe and Cr Atom Distributions in a Binary Sigma Phase Equilibrated at 1063, 1013 and 923 K // Acta Crystallographica, Section B: Structural Science 1983. V. 39. P. 20.
Thiedemann U., Roesner-Kuhn M., Matson D.M. et al. Mixing Enthalpy Measurements in the Liquid Ternary System Iron-Nickel-Chromium and its Binaries // Steel Res. 1998. V. 69. P. 3.
Iguchi Y., Nobori S., Saito K. et al. A Calorimetric Study of Heats of Mixing of Liquid Iron Alloys Fe–Cr, Fe–Mo, Fe–W, Fe–V, Fe–Nb, Fe–Ta // J. Iron Steel Inst. Jpn. 1982. V. 68. P. 633.
Shumikhin V.S., Biletsky A.K., Batalin G.I. et al. Study on Thermodynamic and Kinetic Parameters of Dissolution of Solid Materials in Ferrocarbonic Melts // Arch. Eisenhuttenwes. 1981. V. 52. P. 143.
Паварс И.А., Баум Б.А., Гельд П.В. Теплофизичесик и термодинамические характеристики жидких сплавов железа с хромом // ТВТ. 1970. Т. 8. № 1. С. 67.
Баталин Г.И., Курач В.И., Судавцова В.С. Энтольпин смешивания жидких слаев системы Fe–Cr, Fe–Ti // ЖФХ. 1984. Т. 84. С. 481.
Nobori S., Saito K., Iguchi Y. et al. Measurement of Heats of Mixing for High-melting Point Metals and Molten Iron // Tetsu to Hagane. 1976. V. 62. P. 558.
Ponomareva A.V., Ruban A.V., Vekilova O.Yu. et al. Effect of Pressure on Phase Stability in Fe-Cr Alloys // Phys. Rev. B. 2011. V. 84. P. 094422.
Wróbel J.S., Nguyen-Manh D., Lavrentiev M.Y. et al. Phase Stability of Ternary FCC and BCC Fe–Cr–Ni Alloys // Phys. Rev. B. 2015. V. 91. № 2. P. 024108.
Ponomareva A.V., Ruban A.V., Mukhamedov B.O. et al. Effect of Multicomponent Alloying with Ni, Mn, and Mo on Phase Stability of BCC Fe-Cr Alloys // Acta Materialia. 2018. V. 150. P. 117.
Idczak R., Konieczny R., Konieczna Z. et al. An Enthalpy of Solution of Cobalt and Nickel in Iron Studied with 57Fe Mossbauer Spectroscopy // Acta Phys. Pol. V. 1192011 P. 37.
Razumovskiy V.I., Ruban A.V., Korzhavyi P.A. Effect of Temperature on the Elastic Anisotropy of Pure Fe and Fe0.9Cr0.1 Random Alloy // Phys. Rev. Lett. V. 107. P. 205 504.
Zhang H., Wang G., Punkkinen M.P.J. et al. Elastic Anomalies in Fe–Cr Alloys // J. Phys.: Condens. Matter. 2013. V. 25.
Zhang H., Johansson B., Vitos L. Ab initio Calculations of Elastic Properties of BCC Fe-Mg and Fe-Cr Random Alloys // Phys. Rev. B. 2009. V. 79. P. 224201.
Speich G., Schwoeble A., Leslie W. Elastic Constants of Binary Iron-base Alloys // Metall. Trans. 1972. V. 3. P. 2031.
Terentyev D., Malerba L., Barashev A.V. On the Correlation Between Self-interstitial Cluster Diffusivity and Irradiation-induced Swelling in Fe–Cr Alloys // Philos. Mag. Lett. 2005. V. 85. P. 587.
Terentyev D., Olsson P., Malerba L. et al. Characterization of Dislocation Loops and Chromium-rich Precipitates in Ferritic Iron–Chromium Alloys as Means of Void Swelling Suppression // J. Nucl. Mater. 2007. V. 362. P. 167.
Terentyev D., Malerba L., Barashev A.V. Modelling the Diffusion of Self-interstitial Atom Clusters in Fe–Cr Alloys // Philos. Mag. 2008. V. 88. P. 21.
Terentyev D., Olsson P., Klaver T.P.C. et al. On the Migration and Trapping of Single Self-interstitial Atoms in Dilute and Concentrated Fe–Cr Alloys: Atomistic Study and Comparison with Resistivity Recovery Experiments // Comput. Mater. Sci. 2008. V. 43. P. 1183.
Terentyev D., Olsson P., Malerba L. Diffusion of 3D-migrating Self-interstitial Clusters in Diluted and Concentrated Fe–Cr Alloys // J. Nucl. Mater. 2009. V. 386–388. P. 140.
Messina L., Nastar M., Garnier T. Exact ab initio Transport Coefficients in BCC Fe-X (X = Cr, Cu, Mn, Ni, P, Si) Dilute Alloys // Phys. Rev. B. 2014. V. 90. P. 104 203.
Wharry J.P., Was G.S. A Systematic Study of Radiation-induced Segregation in Ferritic-martensitic Alloys // J. Nucl. Mater. 2013. V. 442. P. 7.
Olsson P., Klaver T.P.C., Domain C. Ab initio Study of Solute Transition-metal Interactions with Point Defects in BCC Fe // Phys. Rev. B. 2010. V. 81. P. 054102.
Olsson P., Domain C., Wallenius J. Ab initio Study of Cr Interactions with Point Defects in BCC Fe // Phys. Rev. B. 2007. V. 75. P. 014110.
Osmond F. Alloys of Iron and Nickel // Minutes of the Proceedings of the Institution of Civil Engineers. 1899. V. 138. P. 312.
Xing Z., Gohil D., Dinsdale T.A. et al. NPL Report DMA: A. 1985. V. 103.
Lee B., Lee D. Formulation of the A1/L12 Atomic Ordering Energy and a Thermodynamic Analysis of the Fe–Ni System // Calphad. 1988. V. 12. P. 393.
Yang C.W., Williams D.B., Goldstein J.I. A revision of the Fe–Ni Phase Diagram at Low Temperatures (<400°C) // Journal of Phase Equilibria. 1996. V. 17. № 6. P. 522.
Cacciamani G., Dinsdale A., Palumbo M. et al. The Fe-Ni System: Thermodynamic Modelling Assisted by Atomistic Calculations // Intermetallics. 2010. V. 18. № 6. P. 1148.
Dreval L., Turchanin M., Agraval P. Thermodynamic Assessment of the Cu-Fe-Ni System // J. Alloys Compd. 2014. V. 587. P. 533.
Swartzendruber L., Itkin V., Alcock C. The Fe–Ni (Iron–Nickel) System // J. Phase Equilibria. 1991. V. 12. P. 288.
Danon J., Scorzelli R.B., Azevedo I.S., Imakuma K. Mössbauer Spectrum and Debye-Scherrer Pattern of the Ordered Phase Fe–Ni (Superstructure Llo) // Physica Scripta. 1980. V. 21. P. 223.
Rossiter P., Jago R. Towards a True Fe–Ni Phase Diagram // Mater. Res. Soc. Symp. Proc. 1984. V. 21. P. 407.
Chuang Y.Y., Chang Y.A., Schmid R. et al. Magnetic Contributions to the Thermodynamic Functions of Alloys and the Phase Equilibria of Fe-Ni System below 1200 K // Metal. Trans. A. 1986. V. 17. P. 1361.
Servant C., Sundman B., Lyon O. Thermodynamic Assessment of the Cu–Fe–Ni System // Calphad. 2001. V. 25. № 1. P. 79.
Wang Y., Cacciamani G. Thermodynamic Modeling of the Al–Cr–Ni System over the Entire Composition and Temperature Range // J. Alloys and Comp. 2016. V. 688. P. 422.
Nishiyama H. An Experimental Study on the Ordered Alloy Ni2Cr // Trans. Jpn. Inst. Met. 1969. V. 10. P. 365.
Saltykov P., Witusiewicz I., Arpshofen V.T., Seifert H.J. Enthalpy of Mixing of Liquid Al–Cr and Cr–Ni Alloys // J. Mater. Sci. Technol. 2002. V. 18 (2). P. 167.
Sudavtsova V.S., Kurach V.P., Batalin G.I. Thermochemical Properties of Molten Binary Fe–(Y, Zr, Nb, Mo) Alloys // Russ. Metall. 1987. V. 3. P. 59.
Tang F., Hallstedt B. Using the PARROT Module of Thermo-Calc with the Cr–Ni System as Example // Calphad. 2016. V. 55. P. 260.
Дополнительные материалы отсутствуют.
Инструменты
Теплофизика высоких температур