

Теоретические основы химической технологии, 2019, T. 53, № 1, стр. 15-22
Исследование распределения ди-2-этилгексилфосфорной кислоты в экстракционной системе, используемой в центробежном экстракционном полупротивоточном генераторе изотопа иттрия-90 медицинского назначения
А. Ю. Цивадзе 1, А. Т. Филянин 1, *, О. А. Филянин 1, Н. А. Данилов 1
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
Москва, Россия
* E-mail: a.filyanin@yandex.ru
Поступила в редакцию 19.09.2018
После доработки 28.09.2018
Принята к публикации 27.09.2018
Аннотация
Изучен метод распределения ди-2-этилгексилфосфорной кислоты между додеканом, изооктаном, гексаном и пятимолярной соляной кислотой с целью максимального удаления из реэкстракта иттрия-90. Предложена дополнительная его промывка этими растворителями, для чего разработан специальный одноступенчатый центробежный аппарат с подвижной легкой фазой. Количественное содержание иттрия-90 в конечном продукте определялось электролитическим методом. Малые концентрации ди-2-этилгексилфосфорной кислоты в водной фазе определяли экстракционно-спектрофотографическим методом.
ВВЕДЕНИЕ
Все более широкое применение в медицине находят короткоживущие радионуклиды и меченные ими соединения [1]. Радионуклидная диагностика используется для обнаружения различных заболеваний человека на ранних стадиях; метод радионуклидной терапии, основанный на использовании открытых источников α- и β-излучения, способных создавать высокие локальные дозы облучения, используется для предупреждения болей при костных метастазах, для лечения метастаз печени, легких, рака почки, раковых поражений головного мозга, предстательной железы и других онкологических заболеваний. Радиоактивный изотоп иттрий-90 широко используется в медицине для терапевтических целей [2]. Это связано с тем, что он обладает оптимальными ядерно-физическими характеристиками и имеет небольшой период полураспада – 64.2 ч, является практически чистым β-излучателем – 99.98% и обладает большой энергией излучения – 2.27 МэВ, способной создавать высокие локальные дозы облучения. Радионуклид 90Y образуется в результате распада 90Sr по следующей схеме [3]:
Источником получения стронция-90 являются концентраты продуктов деления изотопов урана-233, 235, 238 и плутония-239. Обычно не удается за одну операцию выделить стронций-90 требуемой степени чистоты, и необходимо проведение дополнительного процесса очистки, который чаще всего осуществляется сорбционными методами [4, 5].
Главное требование к изотопу иттрия-90 – его глубокая очистка от материнского изотопа стронция-90 и примесей тяжелых металлов. Эта задача наиболее эффективно решается экстракционным путем при использовании фосфорорганических кислот, например ди-2-этилгексилфосфорной кислоты (Д2ЭГФК). Практически во всех случаях используются лабораторные процедуры с применением делительных воронок, хроматографических процессов и сублимационных методов [6]. При этом необходимо отметить, что для проведения различных технологических процессов широко используются центробежные аппараты [7, 8]. В предлагаемом варианте в основу положен разработанный ранее [9] экстракционный метод получения высокочистого иттрия-90, основанный на применении специально разработанного экстракционного центробежного полупротивоточного генератора иттрия-90, состоящего из двух блоков [10].
Первый блок – двухступенчатый экстракционный центробежный полупротивоточный аппарат с подвижной легкой фазой [11, 12], в котором первая ступень является экстракционной, вторая – промывной. Экстрактор предназначен для выделения иттрия-90 0.25 М раствором Д2ЭГФК в додекане из азотнокислого раствора (0.5 М HNO3) равновесной смеси 90Sr с 90Y и промывки экстракта 0.5 М HNO3.
Второй блок – одноступенчатый экстракционный центробежный полупротивоточный аппарат с неподвижной легкой органической фазой. Он предназначен для проведения двух последовательных операций: глубокой отмывки иттрия-90 от следов стронция-90 0.1 М соляной кислотой и последующей реэкстракции иттрия-90 5–6 М HCl.
Главное достоинство подобного сочетания двух разных экстракционных центробежных полупротивоточных аппаратов заключается в том, что блок I обеспечивает практически полное извлечение 90Υ, содержание стронция-90 в котором составляет ~0.1%. При этом объем экстракта иттрия-90, получаемый на блоке I, в 2–2.5 раза меньше объема исходного раствора, подаваемого на первую ступень, т.е. процесс выделения иттрия-90 проходит одновременно с его концентрированием. Второй блок дает возможность осуществить отмывку экстракта иттрия-90 от следов материнского изотопа стронция-90 до любой степени чистоты, что зависит только от относительного объема пропущенного промывного раствора. Затем на этом же аппарате проводится процесс реэкстракции 90Y, полнота которой зависит также только от относительного объема пропущенной 5 М HCl.
Одним из недостатков разработанного экстракционного полупротивоточного экстракционного генератора иттрия-90 медицинского назначения является возможность загрязнения конечного реэкстракта иттрия-90 экстрагентом (Д2ЭГФК), в результате чего значительно уменьшается выход конечного продукта в виде хлорида иттрия, который используется для перевода в лекарственную форму.
Поскольку литературные данные по растворимости Д2ЭГФК в подобных экстракционных системах отсутствуют, то необходимо было:
1. изучить распределение Д2ЭГФК между додеканом и 5 М HCl, найти растворимость Д2ЭГФК в 5 М HCl;
2. попытаться подобрать растворитель из представленных парафиновых углеводородов и изучить распределение микроколичеств Д2ЭГФК между этими растворителями и 5 М HCl;
3. смоделировать удаление растворенной Д2ЭГФК из реэкстракта 90Y (5 М HCl) путем полупротивоточной промывки ее выбранным растворителем.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Чтобы выполнить поставленную задачу, необходимо было смоделировать процесс получения реэкстракта 90Y на созданном ΙΙІ блоке одноступенчатого экстракционного центробежного аппарата с подвижной легкой фазой, принципиальная схема которого представлена на рис. 1.
Рис. 1.
Принципиальная схема одноступенчатого экстракционного центробежного полупротивоточного аппарата с подвижной легкой фазой (третий блок): 1 – трубка отбора; 2 – неподвижный фланец; 3 – вращающийся корпус; 4 – камера отбора; 5 – кольцевая перегородка; 6 – камера расслаивания; 7 – разделительное кольцо; 8 – центральная трубка; 9 – кольцевой зазор; 10 – мешалка; 11 – камера смешения.
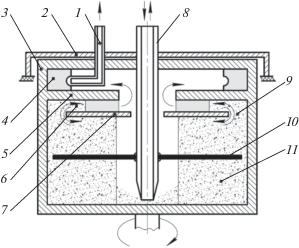
Третий блок экстракционного генератора иттрия-90 работает следующим образом. Реэкстракт иттрия-90 в 5 М HCl, полученный на втором блоке, через центральную трубку 8 подается в определенном объеме в камеру смешения 11. После заполнения аппарата запускают электродвигатель, который приводит во вращение экстрактор. Затем включают перистальтический насос и начинают подавать в качестве органического растворителя изооктан с расходом 5–6 мл/мин через центральную трубку 8 в камеру смешения 11. Равномерное перемешивание жидкостей в камере смешения 11 обеспечивается мешалкой 10. Образовавшаяся эмульсия из смесительной камеры 11 через кольцевой зазор 9 попадает в камеру расслаивания 6, где под действием центробежных сил она расслаивается на легкую и тяжелую фазы. При этом обедненная по легкой фазе эмульсия через кольцевой зазор 9 из камеры расслаивания 6 возвращается снова в смесительную камеру 11, создавая рециркуляцию в аппарате. По мере поступления легкой фазы (изооктана) она доходит до внутреннего радиуса, определяемого кольцевой перегородкой 5, и поступает в камеру отбора 4, откуда с помощью трубки отбора 1 выводится из аппарата. Затем экстрактор останавливается и очищенный реэкстракт иттрия-90 в 5 М HCl с помощью вакуума и центральной трубки 8 выводится из аппарата и передается на упаривание.
Краткая характеристика использованных в ходе работы реактивов. В работе использовались стандартные реактивы (табл. 1), выпускаемые в промышленности квалификации х. ч. и ч. д. а.; дополнительной очистки не подвергались за исключением технической Д2ЭГФК.
Таблица 1.
Характеристика реактивов, используемых в работе
| № | Наименование | ГОСТ, техническая характеристика | |
|---|---|---|---|
| 1 | Азотная кислота | х. ч. | ГОСТ 4461-67 |
| 2 | Гексан | х. ч. | МРТУ 6-09-2937-66 |
| 3 | Додекан | х. ч. | МРТУ 6-09-4615-67 |
| 4 | Натрий гидроокись | х. ч. | ГОСТ 54328-66 |
| 5 | Соляная кислота | х. ч. | ГОСТ 3118-67 |
| 6 | Ди-2-этилгексилфосфорная кислота (Д2ЭГФК) | техн. | ТУ 6-09-782-76 |
| 7 | Ацетон | ч. д. а. | ГОСТ 2603-71 |
| 8 | Родамин Ж | – | ФС42-2619-89 |
| 9 | 2.2.4-Триметилпентан | х. ч. | LAB-SKIN |
Растворы Д2ЭГФК готовили с добавлением к навеске концентрированной кислоты соответствующих количеств органических растворителей: гексана (tb = 68.7°С), изооктана (tb = 99.2°С) и додекана (tb = 216.3°С) [13].
В работе были использованы радиоактивные изотопы 90Y и 90Sr, приведенные в табл. 2.
Таблица 2.
Радиоизотопы, используемые в работе
| № | Изотоп | Тип распада | Период полураспада | E, МэВ (выход β-частицы, %) |
|---|---|---|---|---|
| 1 | 90Sr | β– | 28.5 лет | 0.546 (100) |
| 2 | 90Y | β– | 64.2 ч | 2.274 (99.98) 0.513 (9.016) |
Методика проведения опытов и основные методы анализа. Проведен радиохимический анализ и получены данные по распределению 90Y и 90Sr между водными растворами HCl различной концентрации и 0.25 М Д2ЭГФК. Предварительно подготовленную экстракционную систему, например, 0.1 М HCl и 0.25 М Д2ЭГФК, помещали в 2 делительные воронки (№ 1 и 2) – 5 мл водной и 5 мл органической фазы в каждую. В третьей делительной воронке (№ 3) находились изотопы 90Sr и 90Y (107 Бк/мл) в экстракционной системе 5 М HCl + 1 М Д2ЭГФК в додекане. Воронку № 3 встряхивали, и после расслаивания 90Y переходил в органическую фазу в соответствии с коэффициентом распределения D(Y) > 1000, а 90Sr в соответствии с коэффициентом распределения D(Sr) < 0.001 оставался в водной фазе. Капилляром отбирали порцию Д2ЭГФК в додекане, содержащую 90Y, и вносили ее в воронку № 1, в которой в дальнейшем определяли D(Y) по счету в водной и органической фазах в воронке № 1. Из водной фазы воронки № 3 капилляром отбирали порцию 5 М HCl, содержащую 90Sr, и вносили ее в воронку № 2, где определяли коэффициент распределения 90Sr по счету водной и органической фаз в воронке № 2.
Концентрации Д2ЭГФК в органической фазе (>0.1 М) и HCl в водной фазе определяли титрованием 0.21 Н раствором щелочи (NaOH) с индикатором фенолфталеином.
Малые концентрации Д2ЭГФК, растворенные в водной фазе, определяли экстракционно-спектрофотометрическим методом, а именно путем измерения оптической плотности комплекса Д2ЭГФК с родамином Ж при 520 нм.
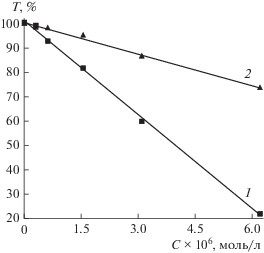
Для определения концентрации Д2ЭГФК непосредственно в додекане, изооктане и гексане были проведены дополнительные исследования. Метод основан на изменении коэффициентов пропускания исследуемого раствора при изменении концентрации вещества в растворе. Для построения калибровочного графика (рис. 2) использовали 5 стандартных растворов Д2ЭГФК в бензоле с концентрацией 0.3 × 10–6–6.2 × 10–6 моль/л.
Разработана методика проведения опытов по распределению Д2ЭГФК между водными растворами 5 М HCl и растворами Д2ЭГФК в гексане, изооктане и додекане, которую проводили в делительных воронках вместимостью 150 мл при температуре 25 ± 2°С.
Для экстракции отбирали 35 мл 5 М HCl и 35 мл соответствующих растворов Д2ЭГФК в исследуемом растворителе и перемешивали вручную в течение трех мин (время, достаточное для установления равновесия). Затем из водной фазы отбиралась проба 5 М HCl с растворенной Д2ЭГФК, которую помещали в чистую делительную воронку. Объем водной фазы доводили до 35 мл 1 М HCl. В ту же воронку добавляли 35 мл бензола. Содержимое встряхивали в течение 3 мин, и после расслаивания органическую фазу (~30 мл) центрифугировали и помещали в чистую делительную воронку. В эту же воронку добавляли 30 мл 0.02% раствора родамина Ж в ацетатном буфере (pН 6.46), встряхивали в течение трех минут. Образующийся окрашенный комплекс родамина Ж с Д2ЭГФК в бензоле центрифугировали и измеряли оптическую плотность бензольного раствора при 514 нм на спектрофотометре СФ-26.
В экспериментах были использованы следующие измерительные приборы и оборудование:
спектрофотометр СФ-26;
спектрофотометр GENESYS 2;
pН-метр – ионометр “ЭКОТЕСТ 120” с комбинированным электродом;
весы технические лабораторные;
весы аналитические электронные KERN ALS 220-4;
весы торсионные типа ВТ на 500 мг;
центрифуга ОПН-8 с ротором РУ-Л;
ΙΙІ блок центробежного одноступенчатого генератора 90Y медицинского назначения.
Изучение распределения Д2ЭГФК в экстракционной системе, используемой в генераторе 90Y медицинского назначения. Целью настоящей работы было изучение распределения Д2ЭГФК в экстракционной системе 5 М HCl–парафиновые углеводороды (додекан, изооктан, гексан) и максимальное удаление Д2ЭГФК из реэкстракта иттрия-90, поступающего на упаривание. Достижение поставленной цели позволит увеличить выход конечного продукта иттрия-90 в виде хлорида иттрия на 30–40%, что будет способствовать улучшению экономических показателей процесса.
Именно эта экстракционная система (0.25 М Д2ЭГФК–5 М HCl) используется на ΙΙІ блоке экстракционного одноступенчатого генератора изотопа 90Y медицинского назначения. Выполнение этой работы позволит усовершенствовать технологию выделения 90Y в форме YCl3, которая используется при синтезе медицинских препаратов, меченных изотопом 90Y.
Центробежный полупротивоточный экстракционный генератор позволяет в пределах 2 ч получать изотоп 90Y с содержанием 90Sr не более 10–6% с выходом не менее 95% по сравнению с исходной его активностью, поступающей вместе со 90Sr на первую ступень Ι блока генератора. Однако с целью получения изотопа иттрия и именно в форме YCl3, необходимой для синтеза медицинских препаратов, остаток от упаренного реэкстракта 5 М HCl, содержащего 90Y, смывали безводным спиртом. Из спиртового раствора иттрий осаждали на катоде электролизом. С катода иттрий смывали 0.1 М HCl. При этом на катоде осаждалось не более 50% 90Y (по активности) от его содержания в спиртовом растворе.
Было высказано предположение, что на катоде осаждается лишь та часть иттрия, которая находилась в хлоридной форме, а остается та часть иттрия, которая находилась в малодиссоциированной форме, например, в виде комплекса с Д2ЭГФК (Y[HR]3), растворенной в реэкстракте, после его контакта с 0.25 М Д2ЭГФК в додекане.
В качестве экстрагента используется раствор 0.25 М Д2ЭГФК в додекане.
Доступная для использования Д2ЭГФК содержит примеси нейтральных алкилфосфатов, примеси пиро- и моно-2-этилгексилфосфорной кислотой (М2ЭГФК), а также посторонних металлов, поэтому она предварительно подвергалась очистке с тем, чтобы концентрация Д2ЭГФК в конечном продукте была 2.7–2.8 моль/л и содержание М2ЭГФК не превышало 1.5%. Содержание М2ЭГФК определяется ее относительно высокой растворимостью в водной фазе и высокой скоростью ее радиолиза с образованием в качестве конечного продукта H3PO4, которая в свою очередь будет связывать иттрий в плохо экстрагируемую, малодиссоциированную форму фосфата иттрия YPO4.
Для очистки Д2ЭГФК может быть использована любая из известных методик, гарантируемых получение конечного продукта требуемого качества. Нами была использована методика, разработанная в Академии химзащиты, в основу которой положены щелочной гидролиз пирофосфатных примесей и различие растворимостей натриевых солей Д2ЭГФК и кислой натриевой соли М2ЭГФК в CCl4.
Анализ кривой потенциометрического титрования полученной Д2ЭГФК (рис. 3) показал, что с помощью этой методики содержание М2ЭГФК в очищенной Д2ЭГФК уменьшилось до 1.27%, выход составил 65%, концентрация полученной кислоты составляла 2.7 моль/л, плотность (измерили с помощью торсионных весов) 0.975 кг/м3. Таким образом, очищенная Д2ЭГФК соответствует требованиям регламента. Однако главным параметром, определяющим применение очищенной партии Д2ЭГФК, являются величины коэффициентов распределения (D) и разделения (β) иттрия и стронция. Поэтому путем разбавления полученной Д2ЭГФК был приготовлен ее раствор в додекане с концентрацией 0.25 моль/л и определены величины коэффициентов распределения и разделения Y и Sr при экстракции их из растворов соляной кислоты различной концентрации. Как видно из табл. 3, при концентрации HCl 0.1 моль/л (условия, соответствующие параметрам первой ступени ΙΙ блока) D(Y) > 5000, а D(Sr) ≈ 0.04.
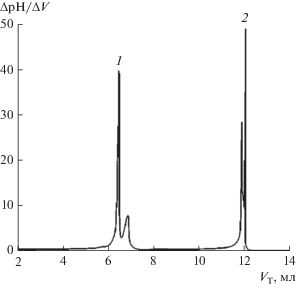
Таблица 3.
Величины коэффициентов распределения D и разделения β(Y/Sr) в зависимости от концентрации HCl в равновесной водной фазе
| Caq.ph(HCl), моль/л | D(Y) | D(Sr) | β(Y/Sr) |
|---|---|---|---|
| 0.1 | >5000 | 0.04 | >1.25 × 105 |
| 0.25 | >5000 | 0.025 | >2 × 105 |
| 0.5 | 50 | 0.01 | 5 × 103 |
| 1.0 | 4 | 0.008 | 5 × 103 |
| 2.0 | 0.8 | 0.003 | 2.67 × 102 |
| 3.0 | 0.04 | – | – |
| 5.0 | 0.009 | ~0.001 | 9 |
Такие величины коэффициентов распределения обеспечивают дополнительную отмывку 90Y от 90Sr и практически полностью позволяют удерживать иттрий в органической фазе. На втором блоке осуществляется реэкстракция иттрия 5 М HCl. При этом величина коэффициента распределения D(Y) ≈ 0.009, что позволяет на 99% извлечь иттрий из 0.25 М Д2ЭГФК.
Таким образом, полученная партия Д2ЭГФК полностью соответствует экстрагенту, используемому в центробежном экстракционном генераторе изотопа Y и именно с таким экстрагентом было изучено распределение Д2ЭГФК между водной (5 М HCl) и органической (парафиновые углеводороды) фазами.
Изучение распределения Д2ЭГФК между 5 М HCl и парафиновыми углеводородами. Как уже отмечалось, наличие в реэкстракте иттрия (5 М HCl), поступающем на упаривание, Д2ЭГФК является нежелательным явлением.
Упаривание проводили под вакуумом в стеклянных сосудах с внешним обогревом теплоносителя (горячая вода). При этом 5 М HCl полностью упаривается, а Д2ЭГФК, имеющая высокую температуру кипения, с большой долей вероятности останется в выпарном аппарате. При последующем растворении в безводном спирте оставшаяся Д2ЭГФК неизбежно провзаимодействует с иттрием с образованием прочного слабо диссоциирующего комплекса и выход иттрия-90 в виде YCl3 уменьшится.
Поэтому целесообразно удалить растворенную в 5 М HCl Д2ЭГФК до минимально возможной концентрации, до упаривания. Поскольку при получении 90Y в экстракционном центробежном генераторе в качестве растворителя используется додекан, то первоначально было изучено распределение Д2ЭГФК в экстракционной системе 5 М HCl–0.25 М раствор Д2ЭГФК в додекане.
Из табл. 4 видно, что растворимость Д2ЭГФК в 5 М HCl уменьшается по мере уменьшения ее концентрации в додекане. Величины коэффициентов распределения уменьшаются в том же направлении.
Таблица 4.
Распределение Д2ЭГФК между 5 М HCl и додеканом
| Corg.ph(Д2ЭГФК), моль/л | Caq.ph(Д2ЭГФК), моль/л ×105 | D(Д2ЭГФК) |
|---|---|---|
| 0.05 | 0.80 | 6250 |
| 0.15 | 2.39 | 6280 |
| 0.25 | 2.50 | 10 000 |
| 0.35 | 2.72 | 12 870 |
| 0.88 | 2.83 | 31 095 |
| 1.20 | 3.19 | 37 620 |
| 2.70 | 31.0 | 8710 |
Определение величины коэффициентов распределения Д2ЭГФК при очень малых ее концентрациях в органической фазе (~10–5 моль/л) и определение возможности удаления растворенной в 5 М HCl Д2ЭГФК проводили следующим образом. В делительную воронку помещали 35 мл 5 М HCl после предварительного контакта ее с 0.25 М раствором Д2ЭГФК в додекане (в результате чего в водную фазу переходило ~2.3 × 10–5 М Д2ЭГФК) и добавляли 35 мл додекана. После перемешивания фаз в течение трех мин и последующего расслаивания определяли концентрацию Д2ЭГФК в равновесной водной фазе (5 М HCl), которая составила Caq.ph = 2.1 × 10–6 моль/л. По разнице между Cin(Д2ЭГФК) и Ceq(Д2ЭГФК) определяли концентрацию ее в додекане, которая составляла 2.09 × 10–5 моль/л. Величина очистки Д2ЭГФК в таком случае составила
Эта величина D в дальнейшем использовалась при расчете выходной кривой при моделировании противоточного процесса. При повторной промывке 5 М HCl равным объемом додекана остаточная концентрация Д2ЭГФК в 5 М HCl составляла <1 × 10–7 моль/л (предел обнаружения). Однако существует возможность попадания додекана, используемого для удаления растворенной в 5 М HCl Д2ЭГФК, в аппарат для упаривания реэкстракта. В случае использования высокотемпературного метода такое упаривание не представляет опасности. При низкотемпературном же упаривании существует опасность попадания додекана, температура кипения которого составляет 216.3°С, в конечный продукт YCl3, что нежелательно. Поэтому для удаления Д2ЭГФК, растворенной в 5 М HCl, были выбраны 2 представителя парафиновых углеводородов: гексан (tb = 68.7°С) и изооктан (tb = 99.24°С). Температура кипения гексана ниже температуры кипения воды, температура кипения изооктана близка к температуре кипения воды. Таким образом, использование названных растворителей гарантирует полное удаление их в процессе обоих видов упаривания. С этой целью было исследовано распределение Д2ЭГФК между 5 М HCl и гексаном, а также между 5 М HCl и изооктаном (табл. 5).
Таблица 5.
Распределение Д2ЭГФК между 5 M HCl и гексаном, а также между 5 М HCl и изооктаном
| Гексан | Изооктан | ||||
|---|---|---|---|---|---|
| Corg.ph, моль/л | Caq.ph, моль/л ×105 | D | Corg.ph, моль/л | Caq.ph, моль/л ×105 | D |
| 1.19 × 10–5 | 0.14 | 8.5 | 1.1×10–5 | 0.11 | 9.9 |
| 0.05 | 1 | 5000 | 0.05 | 0.9 | 5560 |
| 0.27 | 2.06 | 13 110 | 0.10 | 1.42 | 7420 |
| 0.53 | 2.39 | 22 180 | 0.26 | 1 | 26 000 |
| 0.80 | 3.90 | 20 510 | 0.47 | 4 | 11 750 |
| 1.04 | 5.00 | 20 800 | 1.05 | 7 | 15 000 |
| 2.72 | 30.0 | 9070 | 2.23 | 23 | 9670 |
На рис. 4 изображен спектр, снятый с холостого раствора изооктана, предварительно проконтактировавшего с родамином Ж. Спектр снимали, чтобы удостовериться, что в случае изооктана замеры пропускания проб непосредственно в органической фазе можно проводить при 520 нм, как и в случае с бензолом.
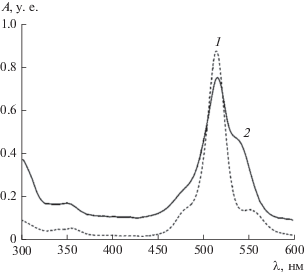
Из представленных в табл. 5 данных следует, что распределение Д2ЭГФК между 5 М HCl и гексаном, а также между 5 М HCl и изооктаном аналогично распределению ее между 5 М HCl и додеканом. Коэффициенты распределения Д2ЭГФК при ее малой концентрации в 5 М HCl (~10–5 моль/л) близки к 10. Величина коэффициента распределения D(Д2ЭГФК) = 10 была использована в дальнейшем для расчета выходной кривой в опытах с полупротивоточной центробежной ступенью генератора (рис. 5).
Рис. 5.
Расчетная кривая зависимости относительной концентрации Д2ЭГФК в 5 М HCl от объема пропущенного промывного раствора.
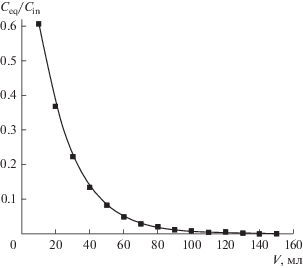
Из табл. 6 видно, что по мере увеличения объема гексана содержание Д2ЭГФК в органической фазе, а следовательно, и в находящейся в равновесии с органической, водной фазе, убывает. После пропускания ~100 мл гексана через неподвижную водную фазу (5 М HCl) экстрактор был остановлен, и 5 М HCl была из него удалена. Анализ Д2ЭГФК в удаленной кислоте показал, что ее концентрация ниже предела обнаружения 1 × 10–7 моль/л, т.е. ее концентрация гарантировано снизилась примерно в 100 раз. Для уменьшения содержания Д2ЭГФК в 5 М HCl в 1000 раз достаточно пропускания 150 мл гексана. Аналогичные результаты были получены с додеканом и изооктаном.
ЗАКЛЮЧЕНИЕ
Изучено распределение Д2ЭГФК между 5 М HCl и следующими парафиновыми углеводородами: додеканом, изооктаном и гексаном. Показано, что полупротивоточным методом можно очистить реэкстракт иттрия-90 от растворенной Д2ЭГФК не менее чем на 2 порядка (до 10–7 моль/л). В качестве органического растворителя желательно использовать изооктан. Для улучшения экономических показателей технологической схемы получения иттрия-90 медицинского назначения предполагается усовершенствовать ее путем внедрения дополнительной промывной экстракционной полупротивоточной ступени перед этапом упаривания для промывки от остаточных количеств Д2ЭГФК.
Авторы выражают благодарность инженеру Ю.В. Шумиловой принимавшей активное участие в проведении данных исследований.
ОБОЗНАЧЕНИЯ
| A | оптическая плотность, у. е. |
| C | концентрация, моль/л |
| D | коэффициент распределения |
| E | энергия, МэВ |
| T | пропускание, % |
| t | температура, °C |
| V | объем, мл |
| β | коэффициент разделения |
| λ | длина волны, нм |
ИНДЕКСЫ
Список литературы
Kowalsky R.J, Perry J.R. Radiopharmaceuticals in Nuclear Medicine Practice. V. 1. Norwalk: Appleton & Lange, 1987.
Шаповалов В.В, Мельниченко Н.А, Нерозин Н.А. и др. Экстракционно-хроматографическое выделение 90Y для медицинских целей // Радиохимия. 2012. Т. 54. № 4. С. 357.
Схемы распада радионуклидов. Энергия и интенсивность излучения. Ч. 1. М.: Энергоатомиздат, 1987.
Маслова Г.Б, Кудрявцева С.П, Гелис В.М, Козлитин Е.А, Полякова Н.И. Выделение и очистка стронция-90 сорбционным методом из растворов от переработки ядерного топлива. I. Извлечение стронция-90 // Радиохимия. 1975. Т. 37. № 5. С. 465.
Кудрявцева С.П, Маслова Г.Б, Полякова Н.И. Выделение и очистка стронция-90 сорбционным методом из растворов от переработки ядерного топлива. II. Получение чистого стронция-90 // Радиохимия. 1995. Т. 37. № 5. С. 470.
Воротынцев В.М., Малышев В.М., Петухов А.Н. Глубокая очистка веществ методом противоточной сублимации // Теорет. основы хим. технологии. 2018. Т. 52. № 5. С. 548.
Шваб А.В., Зятиков П.Н., Садретдинов Ш.Р., Чепель А.Г. Моделирование процесса фракционного разделения частиц в воздушно-центробежном классификаторе // Теорет. основы хим. технологии. 2010. Т. 44. № 6. С. 641.
Ахмадиев Ф.Г., Зиннатуллин Н.Н. Математическое моделирование процесса разделения двухфазных смесей в центробежном сгустителе // Теорет. основы хим. технологии. 2014. Т. 48. № 2. С. 214.
Кодина Г.Е, Корпусов Г.В, Филянин А.Т. Получение радионуклида 90Y высокой чистоты на специально созданных центробежных полупротивоточных экстракторах // Радиохимия. 2002. Т. 44. № 1. С. 61.
Šrank J., Melichar F., Filyanin A.T., Tomeš M., Beran M. Preparation of 90YCl3 radiopharmaceutical precursor for nuclear medicine using technology of centrifugal extractors // Applied Radiation and Isotopes. 2010. V. 68. № 12. P. 2163.
Корпусов Г.В., Филянин А.Т., Сальникова Е.В. Центробежный экстрактор. Пат. 827105 СССР. 1981.
Цивадзе А.Ю., Филянин А.Т., Корпусов Г.В., Коди-на Г.Е., Филянин О.А. Центробежный экстрактор. Пат. 47244 РФ. 2005.
Мартынов Б.В. Справочник по экстракции. Т. 3. Экстракция органическими кислотами и их солями. М: Атомиздат, 1978.
Дополнительные материалы отсутствуют.
Инструменты
Теоретические основы химической технологии