

Успехи современной биологии, 2019, T. 139, № 3, стр. 211-220
ДНК-штрихкодирование: методы и подходы
С. В. Шеховцов 1, 2, 3, *, И. Н. Шеховцова 4, С. Е. Пельтек 1
1 Институт цитологии и генетики СО РАН
Новосибирск, Россия
2 Институт биологических проблем Севера ДВО РАН
Магадан, Россия
3 Новосибирский государственный университет
Новосибирск, Россия
4 Центральный сибирский ботанический сад СО РАН
Новосибирск, Россия
* E-mail: shekhovtsov@bionet.nsc.ru
Поступила в редакцию 13.11.2018
После доработки 25.11.2018
Принята к публикации 28.11.2018
Аннотация
Метод ДНК-штрихкодирования был предложен 15 лет назад и набрал значительную популярность. В этой работе дается обзор методических подходов в данной области и их прогресса за прошедшие годы. Рассматриваются прямой и обратный подходы к ДНК-штрихкодированию и их перспективность в связи с развитием методов секвенирования.
ВВЕДЕНИЕ
ДНК-штрихкодирование отсчитывает свое начало с момента выхода статьи “Biological identifications through DNA barcodes” в 2003 г. (Hebert et al., 2003a). При этом на тот момент само по себе применение последовательностей ДНК для определения видовой принадлежности новым назвать было нельзя: считается, что принципы использования нуклеиновых кислот и белков для филогенетических исследований введены в 1965 г. (Zuckerkandl, Pauling, 1965), а использовать ДНК для идентификации видов предложено в 1982 г. (Nanney, 1982) в статье о Tetrahymena (в которой речь шла еще не о последовательностях, а о ДНК–ДНК-гибридизации и картах рестрикции). Секвенирование ДНК достигло довольно больших масштабов в 1990-е гг. и было совершенно рутинным занятием к 2003 г. Тем не менее, выдвинутое в 2003 году предложение (Hebert et al., 2003a) привлекло большое внимание, так как был введен новый красивый термин “ДНК-штрихкодирование” и была предложена амбициозная цель подвергнуть подобной процедуре все 10–15 млн потенциально существующих видов животных (Hammond, 1992; Hawksworth, Kalin-Arroyo, 1995) при обещанной 100%-ной эффективности идентификации, а также при возможности использовать метод для обнаружения новых видов.
Последний пункт вызвал особенно бурную реакцию у систематиков, так как позволял неспециалистам вторгаться в систематику групп и изменять ее (несмотря на то, что данный подход многократно использовался ранее самими систематиками). Недовольство вызвало и то, что авторы предполагали, что ДНК-штрихкодирование неизбежно превзойдет морфологический анализ. Последовали критические статьи (DeSalle et al., 2005; Will et al., 2005; Cameron et al., 2006) и ответы на них (Hebert, Gregory, 2005; Hajibabaei et al., 2007). Данные работы обсуждаться здесь не будут, так как метод ДНК-штрихкодирования давно устоялся, стали очевидными и его достоинства, и ограничения.
Хеберт с соавт. предполагали (Hebert et al., 2003a), что на получение ДНК-штрихкодов 10–15 млн видов животных уйдет 20 лет. Несколько позже (Ratnasingham, Hebert, 2007) авторы оценивали, что 100 современных центров секвенирования (а сейчас их функционирует гораздо больше) смогут решить подобную задачу за 10 лет. На уровне современных технологий предполагаемые 10 млн сиквенсов можно получить гораздо быстрее. В недавней работе (Hebert et al., 2016) было исследовано 1 085 000 образцов, для 939 000 из которых были получены последовательности ДНК. Тем не менее, очевидно, что исходно заявленные цели ДНК-штрихкодирования еще далеки от достижения. В данной статье мы рассмотрим некоторые методические аспекты ДНК-штрихкодирования и перспективы применения технологий секвенирования нового поколения.
МОЛЕКУЛЯРНЫЕ МАРКЕРЫ ДЛЯ ДНК-ШТРИХКОДИРОВАНИЯ
Для ДНК-штрихкодирования принципиально иметь подходящую маркерную последовательность ДНК, удовлетворяющую ряду условий: она должна легко и надежно амплифицироваться (иными словами, необходимо иметь универсальные праймеры, подходящие широкому кругу видов); быть достаточно изменчивой, чтобы различать близкородственные виды, но в то же время иметь не слишком большую скорость замен внутри видов (Шнеер, 2009а,б). Еще одно важное пожелание к ДНК-штрихкоду – отсутствие изменчивости по длине, которая может привести к проблемам с выравниванием и идентификацией.
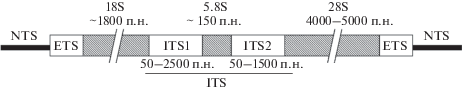
Более того, большинство эукариот – диплоиды, то есть имеют в геноме по две копии каждого локуса. Если последовательности двух аллелей отличаются, на секвенограммах в соответствующих местах будут совпадающие пики (в случае точечных замен) или сдвиги (в случае инсерций/делеций). В результате маркеры обычно выбирают среди незначительной части генома клетки, а именно в геномах органелл (митохондрий и пластид), которые являются гаплоидными, хотя в некоторых случаях различающиеся геномы могут сосуществовать в одном организме (гетероплазмия), а также в кластерах ядерной рибосомальной РНК (рРНК). Последние имеют несколько преимуществ. Как известно, рибосомы эукариот включают в себя четыре функциональных рРНК: 28S, 18S, 5.8S и 5S. Так как рРНК составляет около половины РНК клетки, каждый из генов, кодирующих данные рРНК, представлен множеством (от десятков до десятков тысяч) копий (Richard et al., 2008). При этом первые три гена объединены в одну транскрибируемую единицу (рисунок), то есть они транскрибируются в виде одной длинной молекулы, представляющей из себя гены, окруженные внешними (ETS, external transcribed spacers) и внутренними (ITS, internal transcribed spacers) спейсерами, которая затем разрезается на отдельные рРНК. Копии таких транскрибируемых единиц расположены тандемно и разделены нетранскрибируемыми спейсерами NTS (non-transcribed spacer). Спейсеры (внешние и внутренние) имеют более высокую скорость замен; более того, внутри генов рРНК выделяют несколько функциональных доменов. Таким образом, в пределах кластера чередуются более и менее консервативные участки, что дает возможность подбирать различные маркерные последовательности для разделения таксонов разного ранга, при этом консервативные участки дают возможность подбирать универсальные праймеры. Как уже было сказано, каждый локус генома эукариот представлен в двух копиях, последовательности которых могут отличаться. Но хотя копий рРНК сотни, они обычно идентичны друг другу, что является следствием так называемой согласованной эволюции; копии единицы кластера РНК гомогенизируются за счет генной конверсии или же неравного кроссинговера (Eickbush, Eickbush, 2007). Кроме того, так как трансляция – один из самых важных клеточных процессов, считается, что для генов рРНК почти не характерен горизонтальный перенос (Gogarten, Townsend, 2005). Таким образом, как будет показано ниже, почти все маркеры для ДНК-штрихкодирования относятся либо к геномам органелл, либо к кластеру ядерной рРНК.
Животные
В случае с животными выбор подходящего маркера был сделан быстро: мишенью ДНК-штрихкодирования стал фрагмент митохондриального гена цитохром оксидазы I (cox1 или COI). Выбор митохондриальной ДНК очевиден, так как у Metazoa она характеризуется очень высокой скоростью замен, в несколько раз выше по сравнению с ядерным геномом, а также отсутствием рекомбинации, затрудняющей анализ. Причины этого, а также возможные исключения подробно изложены в обзорах (Ballard, Whitlock, 2004; Bernt et al., 2013). В принципе, любой митохондриальный ген можно было бы использовать с той же целью почти с тем же успехом (ген cox1 – не самый быстро эволюционирующий ген мтДНК животных), однако решающим стало следующее обстоятельство: в данном гене существуют консервативные участки, к которым можно подобрать универсальные праймеры, подходящие к большому числу видов. Так, универсальные праймеры (Folmer et al., 1994; Lobo et al., 2013) успешно работают на самых разных типах Metazoa. Кроме того, существуют и более специфические наборы праймеров для отдельных групп (Hebert et al., 2004; Radulovici et al., 2009; Ivanova et al., 2013). Участок, ограниченный этими праймерами, составляет около 650 п.н., а это как раз длина надежного чтения стандартной реакции секвенирования по Сэнгеру, то есть можно получить максимум информации за одну реакцию секвенирования. Иными словами, стандартный фрагмент гена cox1 оказался идеальным маркером для Metazoa, за исключением базальных ветвей, в пределах которых он не всегда обладает необходимым разрешением (Huang et al., 2008; Shearer, Coffroth, 2008; Vargas et al., 2013).
Растения
Растениям с подходящими для ДНК-штрихкодирования маркерами повезло гораздо меньше. Несмотря на огромные усилия, действительно подходящего маркера подобрать не удалось. В отличие от животных, мтДНК растений имеет очень низкую скорость замен по сравнению с ядерным и пластидным геномами при довольно высокой скорости перестроек (Wolfe et al., 1987; Drouin et al., 2008), в связи с чем митохондриальные гены для ДНК-штрихкодирования растений не применяются. Ядерные гены имеют (в среднем) более высокую скорость замен по сравнению с пластидными, однако их использование затрудняется сложностью подбора универсальных праймеров, а также тем, что большинство растений – палеополиплоиды (Jiao et al., 2011; Li et al., 2015b). В особенности это касается покрытосеменных, где все семейства, за исключением нескольких базальных ветвей, прошли как минимум два события удвоения генома (Soltis et al., 2015). Пластидные геномы стали компромиссным выбором: скорость замен в них несколько ниже, чем в ядерных, однако набор генов более или менее постоянен, и ко многим из них можно подобрать универсальные праймеры. Таким образом, в настоящее время в качестве маркеров для ДНК-штрихкодирования растений используются более десятка пластидных генов и последовательности ядерного рибосомального кластера.
Тем не менее, оказалось, что при использовании отдельных маркеров количество успешно разделяемых при помощи ДНК-штрихкодирования видов в пределах отдельных родов и семейств иногда не превышает 50% (Fazekas et al., 2008; Hollingsworth et al., 2011). Настолько низкое разрешение, очевидно, не может быть приемлемо, в связи с чем обычно используют несколько маркеров одновременно. Были сделаны независимые попытки стандартизировать набор маркеров для ДНК-штрихкодирования растений (Chase et al., 2007; Kress, Erickson, 2007; Ford et al., 2009). В конце концов, рабочая группа по ДНК-штрихкодированию растений (CBOL Plant Working Group, 2009) приняла в качестве стандарта фрагменты двух пластидных генов: большой субъединицы рибулозо-1,5-бифосфаткарбоксилазы (rbcL) и матюразы K (matK). Это компромиссное решение: маркер для ДНК-штрихкодирования должен легко амплифицироваться универсальными праймерами и характеризоваться высокой скоростью замен. В предложенной комбинации rbcL легко амплифицируется, но имеет наименьшую скорость эволюции среди пластидных генов, в то время как последовательность matK быстро эволюционирует, но плохо амплифицируется (Li et al., 2015a). В связи с вышесказанным данные по этим маркерам обычно дополняют вспомогательными маркерами, из которых наиболее часто используют различные пластидные последовательности (спейсеры между генами trnH и psbA, atpF и atpH, psbK и psbI; фрагменты генов rpoC1, rpoB, accD, ndhJ, ycf1), различные фрагменты ядерного рибосомального кластера, а также таксон-специфические ядерные гены (Wang et al., 2011; Shekhovtsov et al., 2012). Предлагается также использовать в качестве супер-баркода весь пластидный геном в целом (Kane, Cronk, 2008; Li et al., 2015a). При использовании технологий секвенирования нового поколения этот подход становится вполне доступным; некоторым препятствием является необходимость выделения органелл (для этого надо иметь живые растения), однако возможно собирать пластидные геномы и по результатам секвенирования тотальной ДНК (Nock et al., 2011).
Грибы
Для грибов ДНК-штрихкодирование стало еще более актуальным методом, чем для животных и растений. Число видов грибов велико. На сегодня описано около 100 000 видов, в то время как по разным оценкам общее число видов грибов составляет от 700 000 до нескольких миллионов (Hawksworth, 1991; Schmit, Mueller, 2007; Begerow et al., 2010), причем систематике грибов уделяют заметно меньше внимания, чем животным и растениям. При этом большинство видов грибов отличается сравнительной морфологической простотой. Неудивительно, что множество исследований ДНК обнаружили значительную генетическую изменчивость у различных групп грибов (Weiss et al., 2004; Crespo, Lumbsch, 2010).
Как и у растений, у грибов последовательность митохондриального гена cox1 не стала основным маркером для ДНК-штрихкодирования. Причин этому несколько: отсутствие консервативных последовательностей, пригодных для подбора праймеров; малое количество нуклеотидных отличий между близкородственными видами; присутствие интронов – до 18 у Agaricus bisporus (Férandon et al., 2013); отсутствие митохондрий у некоторых групп (Bullerwell, Lang, 2005; Gilmore et al., 2009). Это место занял ITS, транскрибируемый спейсерный регион рРНК: спейсеры ITS1 и ITS2, разделенные геном 5.8S рРНК (рисунок). Плюс этой последовательности в том, что ее фланкируют консервативные гены 18S и 28S рРНК, в которых легко подобрать универсальные праймеры. Последовательность ITS имеет самую высокую успешность амплификации по сравнению с прочими маркерами (Schoch et al., 2014; Xu, 2016). У данного маркера, правда, есть и определенные недостатки: в некоторых случаях он не дает достаточного разрешения (Xu et al., 2000); иногда встречается гетерогенность между копиями в геноме. Тем не менее, Международный Консорциум по ДНК-штрихкодированию Грибов (International Fungal Barcoding Consortium) признал ITS главным маркером для ДНК-штрихкодирования грибов (Schoch et al., 2014).
Протисты
Сказанное выше о перспективности ДНК-штрихкодирования для грибов в равной степени относится и к протистам. Число описанных видов, по разным оценкам, варьирует от 40 тыс. (Hoef-Emden et al., 2007) до 75 тыс. (Pawlowski et al., 2012). Общее предполагаемое число видов пока с трудом поддается оценке и может составлять примерно от 150 тыс. до 1.5 млн (Adl et al., 2005).
Как и в случае грибов, наиболее подходящей мишенью для ДНК-штрихкодирования протистов является кластер рРНК. Однако протисты – группа парафилетическая и гораздо более разнообразная генетически. В связи с этим в качестве главного маркера был взят не регион ITS, а ген 18S рРНК, точнее, его домен V4 (Pawlowski et al., 2012). Кроме того, часто применяют и другие маркеры, например, ITS или вариабельные домены 5'-конца 28S рРНК; митохондриальный ген cox1 – для протистов, имеющих митохондрии; для фотосинтезирующих протистов используются пластидные гены, например, 23S рРНК и rbcL.
Прокариоты
К прокариотам (бактериям и археям) термин “ДНК-штрихкодирование” почти никогда не применяется. Тем не менее, мы не можем не упомянуть о том, что определение и описание новых видов и штаммов прокариот в настоящее время в большой степени строится на 16S рРНК, входящей в состав малой субъединицы рибосом и гомологичной 18S рРНК эукариот (Stackebrandt, Goebel, 1994). Иными словами, ДНК-штрихкодирование протистов и прокариот имеет одну основу. Правда, в связи с малым размером генома последних, подобные работы все больше заменяются метагеномными исследованиями, а в последнее время – секвенированием полных геномов из единичных особей прокариот (Rinke et al., 2013).
ДНК-ШТРИХКОДИРОВАНИЕ: ПРЯМОЙ ПОДХОД
Обычно при ДНК-штрихкодировании явно или неявно применяют так называемый прямой подход: образцы предварительно сортируют по морфологическим признакам, проводится выделение и секвенирование выбранного маркера и сравнение его последовательности с референсной библиотекой.
Успех ДНК-штрихкодирования в наибольшей степени зависит от качества обработки выборки и референсной библиотеки. В наилучшем варианте работа будет выглядеть таким образом: несколько исследовательских лабораторий в разных учреждениях работает над определенной группой. Полученные результаты сверяются, и лишь в случае совпадения результаты принимаются. Все морфологические определения производятся систематиками, специализирующимися по данной группе организмов, причем в качестве референсных используются и типовые образцы. В большинстве случаев, правда, это сложно или вовсе невозможно из-за большого их возраста, методов хранения, ведущих к деградации ДНК, или невозможности изъятия материала для ДНК-анализа без ухудшения внешнего вида образца. Тем не менее, есть множество примеров использования различных методик для анализа образцов ДНК из типовых и музейных образцов (Rohland et al., 2004; Hajibabaei et al., 2006; Gilbert et al., 2007).
Более того, даже если секвенировать типовые образцы для всех видов, достоверное определение требует еще и анализа внутривидовой изменчивости. Таким образом, действительно надежным источником данных можно считать работы профессиональных систематиков, использующих секвенирование ДНК лишь как вспомогательный метод в дополнение к детальному морфологическому анализу. Подобные работы включают в себя исследование генетической изменчивости изучаемого вида по всему ареалу, а также сестринских видов.
При работе с исследуемой выборкой ее обычно явно или неявно предварительно сортируют на морфологически различающиеся группы, так называемые морфовиды (morphospecies), или отдельные таксономические единицы (recognizable taxonomic units, RTU) (Krell, 2004). Затем проводят выделение ДНК, ПЦР и секвенирование.
Если все вышеописанные процедуры выполнены наилучшим образом, остается проблема анализа последовательностей. Например, мы получили последовательность от некоего неидентифицированного образца и сравниваем его с референсной библиотекой, которая (представим себе идеальный случай) составлена из последовательностей типовых образцов абсолютно всех видов рода. Скорее всего, сравниваемая последовательность будет отличаться от всех референсных последовательностей некоторым количеством замен. Как определить, к какому виду она относится? Допустим, наш образец гораздо ближе к одному виду (одна замена), чем к остальным (10–20 замен); мы скажем, что мы определили наш образец до вида. Если же нет (скажем, от одного вида он отличается пятью заменами, а от другого – шестью), заключим, что, может быть, обнаружили новый вид.
Получается, что эту задачу мы решаем, не имея четких критериев, в то время как неплохо было бы иметь формализуемый алгоритм. Еще с самых первых работ по ДНК-штрихкодированию использовали программы для построения филогенетических деревьев методом ближайшего соседа (neighbor-joining), такие как MEGA, PAUP и т.д. Соответственно, образец можно считать принадлежащим к некому виду в том случае, если его последовательность входит в кластер, образованный референсными последовательностями данного вида. Такой подход называют методом, основанным на расстояниях (distance-based methods).
Часто упоминают правило так называемого barcoding gap (штрихкодового промежутка): межвидовые различия должны превышать внутривидовые более чем в 10 раз (Hebert et al., 2004; Čandek, Kuntner, 2015). Правило это опять же сформулировано приблизительно. Для одних групп животных это правило работает, для других – нет (Huemer et al., 2014; Čandek, Kuntner, 2015); но даже в первом случае бросается в глаза большой охват видов и крайне малый – внутривидовой изменчивости. Скорее всего, если взять для каждого вида выборку из различных точек с границ ареала, величина промежутка сильно снизится. В то же время, как показано выше, при ДНК-штрихкодировании растений выявляемые различия настолько низки, что правило barcoding gap, очевидно, не работает.
Существует и альтернативный подход (DeSalle et al., 2005), который заключается в использовании характерных последовательностей нуклеотидов. В наиболее простом варианте применительно к двум сестринским видам одного рода он выглядит так: если, например, данная последовательность содержит А в позиции 180, Т в позиции 350 и С в позиции 600, данный образец следует определить как вид Х, а если в тех же позициях стоят соответственно G, С и Т, то это вид Y. Очевидно, что можно придумать множество нестрогих вариантов подобных критериев. Такой подход называют основанным на признаках (character-based).
Понятно, что какой бы подход мы не использовали, можно встретить образец, не укладывающийся в критерии. Соответственно, при работе с малоизученными (с точки зрения внутривидовой генетической изменчивости) видами приходится время от времени пересматривать критерии.
Таким образом, кроме возможных методических ошибок, ДНК-штрихкодирование включает в себя два этапа преобразования выборки: произвольный выбор образцов на стадии ее формирования и морфологического анализа (мы берем в анализ только те образцы, которые мы выбрали по субъективным и не всегда явно сформулированным критериям) и на стадии разделения полученных последовательностей ДНК на виды или иные категории, например, OTU (operational taxonomic units) (Blaxter et al., 2005). В зависимости от принятых на этих этапах методик результаты анализа могут заметно отличаться.
Проиллюстрируем эти проблемы на примере хорошо известных организмов – дождевых червей. Эта группа достаточно неплохо изучена и не слишком богата видами: в фауне России всего 57 видов и подвидов (Всеволодова-Перель, 1997). Однако первые же молекулярно-генетические работы на самых широко распространенных и хорошо изученных видах показали, что в каждом из них можно выделить несколько кластеров гаплотипов COI, различающихся на 10–15% (King et al., 2008; Shekhovtsov et al., 2013). Такой уровень является достаточно высоким даже для различных видов в пределах одного рода (Hebert et al., 2003b). Напрашивается вывод о том, что это виды-двойники, однако данные по ядерной ДНК или не позволяют сделать окончательного заключения, или полностью опровергают это предположение (Shekhovtsov et al., 2016; Martinsson et al., 2017). Проблема усугубляется частыми случаями неверного определения материала, связанными с тем, что значительная внутривидовая морфологическая изменчивость перекрывается межвидовыми различиями, а также случаями интрогрессии мтДНК (Dupont et al., 2016). Создание референсной библиотеки затруднено тем, что дождевых червей фиксируют в формалине, который сшивает молекулы ДНК и белков, делая их недоступными для анализа.
Таким образом, ДНК-штрихкодирование внесло заметный вклад в изучение экологии дождевых червей, показав, что реальное видовое разнообразие заметно выше, чем считалось на основании только морфологического анализа: оценочное видовое разнообразие некоторых регионов оказалось выше на 30–100% (Шеховцов и др., 2016; Porco et al., 2018; Shekhovtsov et al., 2018). В то же время такие работы не позволяют использовать получаемые ДНК-штрихкоды для однозначной видовой идентификации образца. Таким образом, ДНК-штрихкодирование выполняет свою задачу с точки зрения экологов, но не систематиков.
ДНК-ШТРИХКОДИРОВАНИЕ ПРИ ПОМОЩИ МЕТОДОВ СЕКВЕНИРОВАНИЯ ДНК НОВОГО ПОКОЛЕНИЯ И ОБРАТНЫЙ ПОДХОД
Хотя секвенирование по Сэнгеру (Sanger et al., 1977) все еще преобладает в работах по ДНК-штрихкодированию, с момента выпуска первой коммерчески успешной платформы в 2005 г. (Kulski, 2016) методы секвенирования ДНК нового поколения NGS (next-generation sequencing) не только стали общепринятыми в исследованиях геномов и транскриптомов, но и находят все большее применение в прочих областях. Приборы, основанные на различных технологиях NGS, способны выдавать суммарно от миллионов до 1012 нуклеотидов за один запуск, причем цена одного нуклеотида на много порядков ниже, чем при традиционном секвенировании по Сэнгеру.
Конфликт с целями ДНК-штрихкодирования в том, что методы NGS предназначены для получения больших объемов данных о последовательностях для одного образца, а ДНК-штрихкодирование нуждается в получении последовательности коротких фрагментов ДНК для большого количества образцов.
Существуют подходы, позволяющие решить это противоречие.
Первый – метабаркодинг – проведение ДНК-штрихкодирования не одной особи, а целого сообщества. Для этой цели выделяют суммарную ДНК, содержащую фрагменты ДНК множества видов, амплифицируют ее при помощи универсальных праймеров, секвенируют ее и по полученным данным реконструируют состав сообщества.
Примерами применения такого подхода являются, например, исследования сообществ протистов (Geisen et al., 2015; Massana et al., 2015; Pawlowski et al., 2016), однако данный метод может детектировать и крупные многоклеточные организмы. Таким образом была исследована фауна амфибий и костистых рыб в различных водоемах (Valentini et al., 2016). Для этого через фильтр пропускали до 100 л воды, проводили ПЦР с собранной ДНК с последующим секвенированием. Параллельно с этим проводили традиционную оценку видового разнообразия путем визуального наблюдения и ловли особей. По данным авторов, для большинства водоемов метабаркодинг выявлял заметно большее число видов, чем традиционные методы оценки, что говорит о его большой перспективности для экологических исследований. Другая область применения данного метода – исследование состава диеты при помощи фекалий (De Barba et al., 2014; Kaunisto et al., 2017) или содержимого пищеварительной системы (McClenaghan et al., 2015; Harms-Tuohy et al., 2016).
Нельзя не отметить, что для метабаркодинга зачастую используется деградированная ДНК, в связи с чем обычные маркеры для ДНК-штрихкодирования в данном случае не подходят. Для данных работ существуют универсальные праймеры, амплифицирующие короткие фрагменты ДНК (70–200 п.н.) ядерной, рибосомальной, митохондриальной или пластидной ДНК, которые содержат достаточное количество информации для идентификации видов (Taberlet et al., 2012, 2018).
Другой подход к применению методов NGS для ДНК-штрихкодирования в качестве первых этапов включает обычное выделение ДНК и амплификацию необходимого маркера. При этом, однако, в праймеры добавляют специфическую последовательность-метку. После проведения секвенирования данная метка позволяет соотнести каждую полученную последовательность с исходным образцом.
Стоимость секвенирования при помощи методов NGS велика, однако с каждым годом уменьшается, в связи с чем данный подход становится все более привлекательным. Так, по оценкам (Wang et al., 2018), ДНК-штрихкодирование одного образца в Канадском центре ДНК-штрихкодирования составляет около 18 $, в то время как в работе данных авторов секвенирование одного образца на платформе Illumina MiSeq оценивается в 0.5 $. В данной оценке есть некоторая субъективность, так как авторы включили в эту сумму только стоимость реактивов без учета трудозатрат и стоимости прибора. Тем не менее, высокая стоимость секвенирования при помощи методов NGS компенсируется заметно более низкими трудозатратами, так что при одновременном исследовании нескольких сотен или тысяч образцов может иметь преимущество перед секвенированием по Сэнгеру.
Одно из главных отличий ДНК-штрихкодирования на платформах NGS состоит в том, что мы получаем не одну последовательность, а десятки тысяч, которые методами биоинформатики объединяются в одну консенсусную последовательность. Эта особенность позволяет детектировать случаи гетероплазмии (то есть присутствия в геноме нескольких функциональных копий данной последовательности – молекул митохондриальной или пластидной ДНК или различных копий в кластере ядерной рРНК), а также случаи присутствия NUMTs и NUPTs (ядерных митохондриальных и пластидных псевдогенов) или иных контаминирующих последовательностей (Cruaud et al., 2017). Таким образом, можно уменьшить количество ошибок и увеличить количество получаемой функциональной информации. Более того, в работе (Cruaud et al., 2017) от 13 до 28% ДНК-штрихкодов (для различных использованных маркеров) было получено из реакций ПЦР, продукты которых не были видны на электрофореграммах, то есть в обычных исследованиях не были бы учтены при анализе.
Еще более важное преимущество данного подхода – снятие ограничений на количество образцов. Если стоимость обычной работы прямо пропорциональна количеству образцов, то при NGS мы платим за запуск прибора вне зависимости от числа взятых в анализ образцов. Этим, например, воспользовались при исследовании (Wang et al., 2018) видового разнообразия муравьев. Процедура предварительной сортировки выборки и идентификации образцов ставит работу в зависимость от взгляда конкретного систематика, что может привести к некоторым искажениям в результатах, которые уже невозможно будет скорректировать на последующих этапах. Обратный подход к ДНК-штрихкодированию дает возможность непосредственно оценивать реальное видовое и генетическое разнообразие.
ЗАКЛЮЧЕНИЕ
Несмотря на обильную критику, метод ДНК-штрихкодирования устоялся и находит все новые области применения. Хотя с момента его введения методы секвенирования ДНК заметно продвинулись, традиционный прямой подход все также ограничен малой скоростью морфологического анализа. В то же время методы секвенирования нового поколения дают возможность принципиально ускорить данный процесс за счет применения обратного подхода, то есть без предварительной сортировки и идентификации образцов. Таким образом возможно достичь заявленных глобальных целей ДНК-штрихкодирования, но без точной привязки к таксономии.
Список литературы
Всеволодова-Перель Т.С. Дождевые черви фауны России. Кадастр и определитель. М.: Наука, 1997. 102 с.
Шеховцов С.В., Базарова Н.Э., Берман Д.И. и др. ДНК-штрихкодирование: сколько видов дождевых червей живет на юге Западной Сибири? // Вавилов. журн. генет. селек. 2016. Т. 20. № 1. С. 125–130.
Шнеер В.С. ДНК-штрихкодирование видов животных и растений, способ их молекулярной идентификации и изучения биоразнообразия // Журн. общ. биол. 2009а. Т. 70. № 4. С. 296–315.
Шнеер В.С. ДНК-штрихкодирование – новое направление в сравнительной геномике растений // Генетика. 2009б. Т. 45. № 11. С. 1436–1448.
Adl S.M., Simpson A.G., Farmer M.A. et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists // J. Eukar. Microbiol. 2005. V. 52. № 5. P. 399–451.
Ballard J.W.O., Whitlock M.C. The incomplete natural history of mitochondria // Mol. Ecol. 2004. V. 13. № 4. P. 729–744.
Begerow D., Nilsson H., Unterseher M., Maier W. Current state and perspectives of fungal DNA barcoding and rapid identification procedures // App. Microbiol. Biotechnol. 2010. V. 87. № 1. P. 99–108.
Bernt M., Braband A., Schierwater B., Stadler P.F. Genetic aspects of mitochondrial genome evolution // Mol. Phylogen. Evol. 2013. V. 69. № 2. P. 328–338.
Blaxter M., Mann J., Chapman T. et al. Defining operational taxonomic units using DNA barcode data // Philos. Transact. R. Soc. B. Biol. Sci. 2005. V. 360. № 1462. P. 1935–1943.
Bullerwell C.E., Lang B.F. Fungal evolution: the case of the vanishing mitochondrion // Curr. Opin. Microbiol. 2005. V. 8. P. 362–369.
Cameron S., Rubinoff D., Will K. Who will actually use DNA barcoding and what will it cost? // Syst. Biol. 2006. V. 55. № 5. P. 844–847.
Čandek K., Kuntner M. DNA barcoding gap: reliable species identification over morphological and geographical scales // Mol. Ecol. Res. 2015. V. 15. № 2. P. 268–277.
CBOL Plant Working Group. A DNA barcode for land plants // PNAS USA. 2009. V. 106. P. 12794–12797.
Chase M.W., Cowan R.S., Hollingsworth P.M. et al. A proposal for a standardised protocol to barcode all land plants // Taxon. 2007. V. 56. P. 295–299.
Crespo A., Lumbsch H.T. Cryptic species in lichen-forming fungi // IMA Fungus. 2010. V. 1. № 2. P. 167–170.
Cruaud P., Rasplus J.Y., Rodriguez L.J., Cruaud A. High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy // Sci. Rep. 2017. V. 7.https://doi.org/10.1101/073304
De Barba M., Miquel C., Boyer F. et al. DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet // Mol. Ecol. Res. 2014. V. 14. № 2. P. 306–323.
DeSalle R., Egan M.G., Siddall M. The unholy trinity: taxonomy, species delimitation and DNA barcoding // Philos. Transact. R. Soc. B. Biol. Sci. 2005. V. 360. № 1462. P. 1905–1916.
Drouin G., Daoud H., Xia J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants // Mol. Phylogen. Evol. 2008. V. 49. № 3. P. 827–831.
Dupont L., Porco D., Symondson W.O.C., Roy V. Hybridization relics complicate barcode-based identification of species in earthworms // Mol. Ecol. Res. 2016. V. 16. № 4. P. 883–894.
Eickbush T.H., Eickbush D.G. Finely orchestrated movements: evolution of the ribosomal RNA genes // Genetics. 2007. V. 175. № 2. P. 477–485.
Fazekas A.J., Burgess K.S., Kesanakurti P.R. et al. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well // PLoS One. 2008. V. 3. № 7. P. e2802.
Férandon C., Xu J., Barroso G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences // Fung. Genet. Biol. 2013. V. 55. P. 85–91.
Folmer O., Hoeh W.R., Black M.B., Vrijenhoek R.C. Conserved primers for PCR amplification of mitochondrial DNA from different invertebrate phyla // Mol. Mar. Biol. Biotechnol. 1994. V. 3. P. 294–299.
Ford C.S., Ayres K.L., Haider N. et al. Selection of candidate DNA barcoding regions for use on land plants // Bot. J. Linn. Soc. 2009. V. 159. P. 1–11.
Geisen S., Laros I., Vizcaíno A. et al. Not all are free-living: high-throughput DNA metabarcoding reveals a diverse community of protists parasitizing soil metazoa // Mol. Ecol. 2015. V. 24. № 17. P. 4556–4569.
Gilbert M.T.P., Moore W., Melchior L., Worobey M. DNA extraction from dry museum beetles without conferring external morphological damage // PLoS One. 2007. V. 2. № 3. P. e272.
Gilmore S.R., Gräfenhan T., Louis-Seize G., Seifert K.A. Multiple copies of cytochrome oxidase 1 in species of the fungal genus Fusarium // Mol. Ecol. Res. 2009. V. 9. Suppl. 1. P. 90–98.
Gogarten J.P., Townsend J.P. Horizontal gene transfer, genome innovation and evolution // Nat. Rev. Microbiol. 2005. V. 3. № 9. P. 679–687.
Hajibabaei M., Singer G.A., Hebert P.D.N., Hickey D.A. DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics // Tr. Genet. 2007. V. 23. № 4. P. 167–172.
Hajibabaei M., Smith M., Janzen D.H. et al. A minimalist barcode can identify a specimen whose DNA is degraded // Mol. Ecol. Res. 2006. V. 6. № 4. P. 959–964.
Hammond P. Species inventory // Global biodiversity: status of the earth’s living resources / Ed. B. Groombridge. L.: Chapman & Hall, 1992. P. 17–39.
Harms-Tuohy C.A., Schizas N.V., Appeldoorn R.S. Use of DNA metabarcoding for stomach content analysis in the invasive lionfish Pterois volitans in Puerto Rico // Mar. Ecol. Prog. Ser. 2016. V. 558. P. 181–191.
Hawksworth D.L. The fungal dimension of biodiversity: magnitude, significance, and conservation // Mycol. Res. 1991. V. 95. P. 641–655.
Hawksworth D.L., Kalin-Arroyo M.T. Magnitude and distribution of biodiversity // Global biodiversity assessment / Ed. V.H. Heywood. Cambridge: Cambridge Univ. Press, 1995. P. 107–191.
Hebert P.D.N., Cywinska A., Ball S.L. Biological identifications through DNA barcodes // Proc. R. Soc. B. Biol. Sci. 2003a. V. 270. № 1512. P. 313–321.
Hebert P.D.N., Gregory T.R. The promise of DNA barcoding for taxonomy // Syst. Biol. 2005. V. 54. № 5. P. 852–859.
Hebert P.D.N., Ratnasingham S., de Waard J.R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species // Proc. R. Soc. B. Biol. Sci. 2003b. V. 270. Suppl. 1. P. S96–S99.
Hebert P.D.N., Ratnasingham S., Zakharov E.V. et al. Counting animal species with DNA barcodes: canadian insects // Philos. Transact. R. Soc. B. Biol. Sci. 2016. V. 371. № 1702. P. 20150333.
Hebert P.D.N., Stoeckle M.Y., Zemlak T.S., Francis C.M. Identification of birds through DNA barcodes // PLoS Biol. 2004. V. 2. № 10. P. e312.
Hoef-Emden K., Küpper F.C., Andersen R.A. Meeting report: sloan foundation workshop to resolve problems relating to the taxonomy of microorganisms and to culture collections arising from the barcoding initiatives. Portland ME, November 6–7, 2006 // Protist. 2007. V. 158. № 2. P. 135–137.
Hollingsworth P.M., Graham S.W., Little D.P. Choosing and using a plant DNA barcode // PLoS One. 2011. V. 6. № 5. P. e19254.
Huang D., Meier R., Todd P.A., Chou L.M. Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding // J. Mol. Evol. 2008. V. 66. № 2. P. 167–174.
Huemer P., Mutanen M., Sefc K.M., Hebert P.D.N. Testing DNA barcode performance in 1000 species of European Lepidoptera: large geographic distances have small genetic impacts // PLoS One. 2014. V. 9. № 12. P. e115774.
Ivanova N.V., Zemlak T.S., Hanner R.H., Hebert P.D.N. Universal primer cocktails for fish DNA barcoding // Mol. Ecol. Not. 2013. V. 7. P. 544–548.
Jiao Y., Wickett N.J., Ayyampalayam S. et al. Ancestral polyploidy in seed plants and angiosperms // Nature. 2011. V. 473. № 7345. P. 97–100.
Kane N.C., Cronk Q. Botany without borders: barcoding in focus // Mol. Ecol. 2008. V. 17. P. 5175–5176.
Kaunisto K.M., Roslin T., Sääksjärvi I.E. et al. Pellets of proof: first glimpse of the dietary composition of adult odonates as revealed by metabarcoding of feces // Ecol. Evol. 2017. V. 7. № 20. P. 8588–8598.
King R.A., Tibble A.L., Symondson W.O.C. Opening a can of worms: unprecedented sympatric cryptic diversity within British lumbricid earthworms // Mol. Ecol. 2008. V. 17. № 21. P. 4684–4698.
Krell F.T. Parataxonomy vs. taxonomy in biodiversity studies – pitfalls and applicability of ‘morphospecies’ sorting // Biodiv. Conserv. 2004. V. 13. № 4. P. 795–812.
Kress W.J., Erickson D.L. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH–psbA apacer region // PLoS One. 2007. V. 2. P. e508.
Kulski J.K. Next-generation sequencing – an overview of the history, tools, and “omic” applications // Next generation sequencing – advances, applications and challenges / Ed. J.K. Kulski. London: Intech, 2016. P. 3–60.
Li X., Yang Y., Henry R.J. et al. Plant DNA barcoding: from gene to genome // Biol. Rev. 2015a. V. 90. № 1. P. 157–166.
Li Z., Baniaga A.E., Sessa E.B. et al. Early genome duplications in conifers and other seed plants // Sci. Adv. 2015b. V. 1. № 10. P. e1501084.
Lobo J., Costa P.M., Teixeira M.A. et al. Enhanced primers for amplification of DNA barcodes from a broad range of marine metazoans // BMC Ecol. 2013. V. 13. № 1. P. 34.
Martinsson S., Rhodén C., Erséus C. Barcoding gap, but no support for cryptic speciation in the earthworm Aporrectodea longa (Clitellata: Lumbricidae) // Mitoch. DNA. Pt A. 2017. V. 28. № 2. P. 147–155.
Massana R., Gobet A., Audic S. et al. Marine protist diversity in European coastal waters and sediments as revealed by high–throughput sequencing // Env. Microbiol. 2015. V. 17. № 10. P. 4035–4049.
McClenaghan B., Gibson J.F., Shokralla S., Hajibabaei M. Discrimination of grasshopper (Orthoptera: Acrididae) diet and niche overlap using next-generation sequencing of gut contents // Ecol. Evol. 2015. V. 5. № 15. P. 3046–3055.
Nanney D.L. Genes and phenes in Tetrahymena // Bioscience. 1982. V. 32. № 10. P. 783–788.
Nock C.J., Waters D.L., Edwards M.A. et al. Chloroplast genome sequences from total DNA for plant identification // Plant Biotec. J. 2011. V. 9. P. 328–333.
Pawlowski J., Audic S., Adl S. et al. CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms // PLoS Biol. 2012. V. 10. № 11. P. e1001419.
Pawlowski J., Lejzerowicz F., Apotheloz-Perret-Gentil L. et al. Protist metabarcoding and environmental biomonitoring: time for change // Eur. J. Protistol. 2016. V. 55. P. 12–25.
Porco D., Chang C.H., Dupont L. et al. A reference library of DNA barcodes for the earthworms from Upper Normandy: biodiversity assessment, new records, potential cases of cryptic diversity and ongoing speciation // Appl. Soil Ecol. 2018. V. 124. P. 362–371.
Radulovici A.E., Sainte-Marie B., Dufresne F. DNA barcoding of marine crustaceans from the estuary and gulf of St Lawrence: a regional-scale approach // Mol. Ecol. Res. 2009. V. 9. Suppl. 1. P. 181–187.
Ratnasingham S., Hebert P.D.N. BOLD: the barcode of life data system (www. barcodinglife. org) // Mol. Ecol. Not. 2007. V. 7. P. 355–364.
Richard G.F., Kerrest A., Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes // Microbiol. Mol. Biol. Rev. 2008. V. 72. № 4. P. 686–727.
Rinke C., Schwientek P., Sczyrba A. et al. Insights into the phylogeny and coding potential of microbial dark matter // Nature. 2013. V. 499. № 7459. P. 431–437.
Rohland N., Siedel H., Hofreiter M. Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens // Biotechniques. 2004. V. 36. № 5. P. 814–820.
Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors // PNAS USA. 1977. V. 74. № 12. P. 5463–5467.
Schmit J., Mueller G. An estimate of the lower limit of global fungal diversity // Biodiv. Conserv. 2007. V. 16. P. 99–111.
Schoch C.L., Robbertse B., Robert V. et al. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi // Database: J. Biol. Databases Cur. 2014.https://doi.org/10.1093/database/bau061
Shearer T.L., Coffroth M.A. Barcoding corals: limited by interspecific divergence, not intraspecific variation // Mol. Ecol. Res. 2008. V. 8. № 2. P 247–255.
Shekhovtsov S.V., Berman D.I., Bazarova N.E. et al. Cryptic genetic lineages in Eisenia nordenskioldi pallida (Oligochaeta, Lumbricidae) // Eur. J. Soil Biol. 2016. V. 75. P. 151–156.
Shekhovtsov S.V., Golovanova E.V., Peltek S.E. Cryptic diversity within the Nordenskiold’s earthworm, Eisenia nordenskioldi subsp. nordenskioldi (Lumbricidae, Annelida) // Eur. J. Soil Biol. 2013. V. 58. P. 13–18.
Shekhovtsov S.V., Shekhovtsova I.N., Peltek S.E. Phylogeny of Siberian species of Carex sect. Vesicariae based on nuclear and plastid markers // Nord. J. Bot. 2012. V. 30. № 3. P. 343–351.
Shekhovtsov S.V., Sundukov Y.N., Blakemore R.J. et al. Identifying earthworms (Oligochaeta, Megadrili) of the Southern Kuril islands using DNA barcodes // Anim. Biodiv. Conserv. 2018. V. 41. № 1. P. 9–17.
Soltis P.S., Marchant D.B., van de Peer Y., Soltis D.E. Polyploidy and genome evolution in plants // Curr. Opin. Genet. Dev. 2015. V. 35. P. 119–125.
Stackebrandt E., Goebel B.M. Taxonomic note: a place for DNA–DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology // Int. J. Syst. Evol. Microbiol. 1994. V. 44. № 4. P. 846–849.
Taberlet P., Bonin A., Zinger L., Coissac E. Environmental DNA: for biodiversity research and monitoring. Oxford: Oxford Univ. Press, 2018. 272 p.
Taberlet P., Coissac E., Hajibabaei M., Rieseberg L.H. Environmental DNA // Mol. Ecol. 2012. V. 21. № 8. P. 1789–1793.
Valentini A., Taberlet P., Miaud C. et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding // Mol. Ecol. 2016. V. 25. № 4. P. 929–942.
Vargas S., Schuster A., Sacher K. et al. Barcoding sponges: an overview based on comprehensive sampling // PLoS One. 2013. V. 7. № 7. P. e39345.
Wang Q., Yu Q.S., Liu J.Q. Are nuclear loci ideal for barcoding plants? A case study of genetic delimitation of two sister species using multiple loci and multiple intraspecific individuals // J. Syst. Evol. 2011. V. 49. № 3. P. 182–188.
Wang W.Y., Srivathsan A., Foo M. et al. Sorting specimen-rich invertebrate samples with cost-effective NGS barcodes: validating a reverse workflow for specimen processing // Mol. Ecol. Res. 2018. V. 3. P. 490–501.
Weiss M., Selosse M.A., Rexer K.H. et al. Sebacinales: a hitherto overlooked cosm of heterobasidiomycetes with a broad mycorrhizal potential // Mycol. Res. 2004. V. 108. № 9. P. 1003–1010.
Will K.W., Mishler B.D., Wheeler Q.D. The perils of DNA barcoding and the need for integrative taxonomy // Syst. Biol. 2005. V. 54. № 5. P. 844–851.
Wolfe K.H., Li W.H., Sharp P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs // PNAS USA. 1987. V. 84. № 24. P. 9054–9058.
Xu J. Fungal DNA barcoding // Genome. 2016. V. 59. № 11. P. 913–932.
Xu J., Vilgalys R., Mitchell T.G. Multiple gene genealogies reveal recent dispersion and hybridization in the human pathogenic fungus Cryptococcus neoformans // Mol. Ecol. 2000. V. 9. № 10. P. 1471–1481.
Zuckerkandl E., Pauling L. Molecules as documents of evolutionary history // J. Theor. Biol. 1965. V. 8. № 2. P. 357–366.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии