

Успехи современной биологии, 2019, T. 139, № 3, стр. 221-234
Эволюция экзонов и экзон-интронной структуры генов длинных межгенных некодирующих РНК у плацентарных млекопитающих
И. А. Сидоренко 1, И. Б. Рогозин 1, 2, В. Н. Бабенко 1, 3, *
1 Институт цитологии и генетики СО РАН
Новосибирск, Россия
2 Национальные институты здоровья
Бесезда, Роквилл Пайк, США
3 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: vl_babenko@yahoo.com
Поступила в редакцию 21.11.2018
После доработки 25.11.2018
Принята к публикации 28.11.2018
Аннотация
Гены длинных межгенных некодирующих РНК (lincRNA) в большом количестве представлены у млекопитающих, но их функции остаются в значительной степени неизвестными. Одним из возможных способов их изучения является использование крупномасштабных сравнений различных характеристик lincRNA с характеристиками кодирующих белок генов, для которых имеется множество функциональной информации. Характерной особенностью белок-кодирующих генов у млекопитающих является высокая эволюционная консервативность первичных последовательностей экзонов и экзон-интронной структуры. Хотя консервативность первичных последовательностей экзонов lincRNA не столь выражена, как у белок-кодирующих генов, но, тем не менее, она существенно выше, чем у интронов генов lincRNA. Сравнительный анализ предполагаемых позиций интронов в генах lincRNA в разных геномах млекопитающих свидетельствует о том, что некоторые интроны lincRNA сохранялись более 100 млн лет. Поэтому возможно, что первичная и/или вторичная структура этих молекул является функционально важной.
ВВЕДЕНИЕ
В последние годы растет интерес к длинным некодирующим РНК (lncRNA) – относительно новым объектам исследования в области геномики. Однако, несмотря на множество усилий, lncRNA по-прежнему имеют статус геномной “темной материи” (Ponting, Belgard, 2011; Kapusta, Feschotte, 2014). Действительно, в то время как роль других некодирующих молекул РНК (рибосомных, транспортных, малых ядерных, антисмысловых, малых ядрышковых, микро- и piwi-взаимодействующих) уже четко определена, функции lncRNA остаются в значительной степени неизвестными (Goodrich, Kugel, 2006; Mercer et al., 2009; Ng et al., 2013; Kapusta, Feschotte, 2014). Даже их определение несколько расплывчато: lncRNA – некодирующие транскрипты длиной более 200 нуклеотидов (Ponting, Belgard, 2011). Существует популярное мнение, что подавляющее большинство lncRNA является побочным продуктом фоновой транскрипции (van Bakel, Hughes, 2009; Robinson, 2010). Эта точка зрения основана на их типично низком уровне экспрессии и слабой эволюционной консервативности по сравнению с белок-кодирующими последовательностями и малыми РНК, такими как miRNA и snoRNA (Marques, Ponting, 2009). Тем не менее, некоторые из lncRNA содержат эволюционно консервативные области (Siepel et al., 2005), и большая часть lncRNA демонстрирует более низкую по сравнению со среднегеномной скорость замен и инсерций/делеций, что указывает на существование селективного отбора (Ponjavic et al., 2007; Guttman et al., 2009; Managadze et al., 2011; Guttman, Rinn, 2012; Kannan et al., 2015). Хотя величина экспрессии lncRNA часто невысока (Bertone et al., 2004; Amaral et al., 2013), комбинация различных экспериментальных подходов, примененных к транскриптомам нескольких видов, привела к массовому открытию новых транскриптов. Например, только проект FANTOM каталогизировал более 30 000 предполагаемых длинных некодирующих транскриптов в тканях мыши путем клонирования полноразмерной кДНК (комплементарная ДНК, англ. сDNA) (Liu et al., 2006). Хотя последовательности большинства lncRNA намного менее консервативны, чем последовательности белков, степень ортологии между наборами соответствующих lncRNA неожиданно высока, а именно, от 60 до 70% генов lncRNA являются общими для человека и мыши (Managadze et al., 2013).
Большинство lncRNA демонстрируют специфическую субклеточную локализацию и являются процессированными (полиаденилированными и сплайсированными); это наблюдение позволяет утверждать, что функциональной является, вероятно, их зрелая форма (Kapusta, Feschotte, 2014; Vance, Ponting, 2014). Другим признаком функциональности продуктов lncRNA может быть то, что значительная часть “эволюционного ограничения” на последовательность lncRNA, вероятно, локализована в регуляторных элементах сплайсинга (Chodroff et al., 2010; Schuler et al., 2014). Это подразумевает, что корректный сплайсинг интронов важен для работы lncRNA. Действительно, подавляющее большинство lncRNA с определеенной клеточной функцией, по-видимому, действуют в процессированной форме (Kapusta, Feschotte, 2014; Vance, Ponting, 2014). Сравнительный анализ более 3000 генов lncRNA мыши позволил предположить, что сохранение экзон-интронной структуры может быть общим свойством lncRNA (Ponjavic et al., 2007). Было обнаружено, что 65 и 40% |GT–AG| сайтов сплайсинга lncRNA мыши сохраняются у крысы и человека соответственно. Эти числа значительно превышают количество консервативных интронных GT и AG динуклеотидов, которые не участвуют в сплайсинге, что указывает на эволюционную консервативность сигналов сплайсинга lncRNA (Ponjavic et al., 2007).
Среди транскриптов существуют многочисленные длинные межгенные некодирующие РНК (lincRNA), то есть молекулы РНК длиной более 200 нуклеотидов, которые кодируются вне других идентифицированных генов. Одной из наиболее изученных к настоящему времени lincRNA является Xist, которая участвует в инактивации Х-хромосомы самок плацентарных млекопитающих (Brockdorff et al., 1992; Chang et al., 2006). РНК Xist эволюционно происходит от белок-кодирующего гена Lnx3, который потерял свою способность кодировать белок и стал псевдогеном у ранних плацентарных. Затем последовала интеграция мобильных элементов (Duret et al., 2006; Elisaphenko et al., 2008). Четыре из десяти экзонов Xist, обнаруженных у плацентарных, показывают значительное сходство последовательности с экзонами гена Lnx3, тогда как остальные шесть экзонов Xist схожи с разными транспозонами. Таким образом, некоторые интроны Xist были унаследованы от гена Lnx3, а некоторые, по-видимому, были приобретены в ходе эволюции гена Xist (Elisaphenko et al., 2008). Анализ Xist у нескольких видов млекопитающих показал общую консервативность ее экзон-интронной структуры (Elisaphenko et al., 2008).
В данной работе мы сделали попытку провести крупномасштабные реконструкции эволюции интронов в генах lincRNA, используя множественные геномные выравнивания. Сравнительный анализ предполагаемых позиций интронов в генах lincRNA в различных геномах млекопитающих свидетельствует о том, что некоторые интроны lincRNA сохраняются более 100 млн лет, и, следовательно, первичная/вторичная структура этих молекул, вероятно, функционально важна.
МАТЕРИАЛЫ И МЕТОДЫ
Гены lincRNA человека и мыши, соответствующие геномные выравнивания и данные экспрессии были взяты из ранее опубликованной работы (Managadze et al., 2011), где подробно описаны процедуры обработки данных. Набор данных из 5444 только некодирующих наборов проб мыши был загружен из базы данных NRED (Dinger et al., 2009), составленной на основе трех следующих экспрессионных массивов: 1) Custom noncoding microarray, мышь – 5000 проб (Dinger et al., 2009); 2) GNF SymAtlas, человек – 1200 проб, мышь – 6000 проб; 3) Allen Brain Atlas, мышь – 1300 проб. После фильтрации наборов проб, которые не попали в межгенные области, и установления взаимнооднозначных отношений между идентификаторами РНК и их соответствующими идентификаторами набора проб, мы получили окончательный набор из 2390 lincRNA мыши (NCBI GenBank Accession IDs of RNAs), из которых 977 содержали интроны. После фильтрации наборов зондов с очень низкими медианными уровнями экспрессии, а также с неточным картированием в геноме, был получен окончательный набор из 2013 lincRNA мыши, включая 918 lincRNA, содержащих интроны. Для человека были скачаны данные для 917 наборов проб, и такая же процедура удаления слабо экспрессирующихся или неточно картированных lincRNA дала окончательный набор из 519 lincRNA, включая 211 генов, содержащих интроны. Геномные координаты и последовательности экзонов и интронов генов lincRNA человека и мыши были загружены из UCSC Table Browser (Karolchik et al., 2004) из таблиц all_mrna сборок геномов мыши mm8 и человека hg18. Множественные выравнивания этих областей были получены в системе Galaxy (Goecks et al., 2010). Использовались два разных варианта множественного выравнивания с референсными геномами человека (hg18) и мыши (mm8) из UCSC genome database (Haeussler et al., 2019). Для нашего анализа были использованы следующие виды: человек (Homo sapiens; hg18), шимпанзе (Pan troglodytes; panTro1), корова (Bos taurus; bosTau2), макака (Rhesus macaque; rheMac2), мышь (Mus musculus; mm8), крыса (Rattus norvegicus; rn4), собака (Canis familiaris; canFam2), тенрек (Echinops telfair; echTel1), слон (Loxodonta africana; loxAfr1), кролик (Oryctolagus cuniculus; orCun1), рыба данио (Danio rerio; danRer3), опоссум (Monodelphis domestica; monDom4), броненосец (Dasypus novemcinctus; dasNov1), курица (Gallus gallus; galGal2), фугу (Takifugu rubripes; fr1), иглобрюх (Tetraodon nigroviridis; tetNig1) и лягушка (Xenopus tropicals; xenTro1) (Managadze et al., 2011).
Анализ парсимонии был выполнен с использованием программ DNAPARS, Dollop из пакета PHYLIP. Для того, чтобы проверить значение эволюционной консервативности сигналов сплайсинга (динуклеотиды GT или GC – начало интронов и AG – конец интрона) и позиций интронов, мы оценили долю консервативных сигналов сплайсинга (Freal). После этого мы случайно выбрали динуклеотиды GT/GC (или AG) из выравниваний интронных последовательностей и оценили долю консервативных динуклеотидов GT/GC (или AG) (Fsampled). Мы повторили процедуру выборки 10 000 раз, распределение Fsampled использовалось для вычисления вероятности P (Freal ≤ ≤ Fsampled). Эта вероятность равна доле выбранных сигналов сплайсинга (GT/GC или AG), в которых Fsampled равен или больше, чем Freal. Малые значения вероятности P (Freal ≤ Fsampled) ≤ 0.05 указывают на значительную консервативность сигналов сплайсинга. Та же процедура была повторена для интронов, и в этом случае одновременно изучалось сохранение динуклеотидов GT/GC и AG. Расстояние между GT/GC и AG должно было быть больше 39 нуклеотидов, как было предложено (Deutsch, Long, 1999). Наблюдаемые распределения длин интронов у человека и мыши (табл. 1) и частоты динуклеотидов GT/GC моделировались во время процедуры отбора проб. Для расчета вероятности P (Freal ≤ Fsampled) использовалась доля сохраненных сигналов сплайсинга GT–AG и GC–AG (донорные–акцепторные сайты сплайсинга).
Таблица 1.
Статистика набора данных lincRNA
| Особенности генов lincRNA | Мышь | Человек |
|---|---|---|
| Количество всех lincRNA | 2390 | 589 |
| Количество интрон-содержащих lincRNA | 979 | 245 |
| Количество экзонов | 3439 | 1194 |
| Количество интронов | 2462 | 949 |
| Количество экзонов короче, чем 15 nt | 41 | 7 |
| Количество интронов на lincRNA | 2.52 | 3.86 |
| Средняя длина гена, nt | 11 775 (712) | 17 192 (1921) |
| Медиана длин генов, nt | 2535 | 2626 |
| Средняя длина экзонов, nt | 524 (21) | 409 (48) |
| Медиана длин экзонов, nt | 464 | 356 |
| Средняя длина интронов, nt | 9621 (1631) | 10 562 (4539) |
| Медиана длин интронов, nt | 2615 | 216 |
РЕЗУЛЬТАТЫ
Исследование гена Xist
Подавляющее большинство исследований, направленных на реконструкцию эволюции архитектуры генов эукариот, были сосредоточены на интронах в консервативных частях белок-кодирующих областей. Например, вывод о том, что существенного приобретения интронов у млекопитающих не происходило (Roy et al., 2003), был основан на данных этого типа. Однако эволюция малоконсервативных сегментов белок-кодирующих последовательностей, нетранслируемых областей белок-кодирующих генов, районов альтернативного сплайсинга и генов, происходящих из мобильных элементов, по-видимому, является намного более быстрой и динамичной, с многочисленными приобретениями интронов у млекопитающих (Cordaux et al., 2006; Hong et al., 2006; Zhang, Chasin, 2006; Zhuo et al., 2007; Szcześniak et al., 2011). В целом, из-за отсутствия эволюционной консервативности в районах таких генов, реконструкция событий приобретения и потерь интронов в их эволюции является сложной, а иногда и неточной (особенно без экспериментальной проверки). Соответственно, эволюционные работы сконцентрированы на высококонсервативных генах. Таким образом, выводы о дефиците приобретения интронов в некоторых группах эукариот, таких как млекопитающие, частично возникают из-за смещенной в сторону консервативности выборки, тогда как общая динамика интронов может быть гораздо более интенсивной, чем было принято считать ранее (Roy et al., 2003).
Эта же проблема относится к генам некодирующих РНК. Например, геномы млекопитающих содержат множество (>10 000) генов lncRNA, которые содержат многочисленные интроны (Ponting et al., 2009). В недавнем подробном исследовании было выявлено более 8000 генов lncRNA со средней плотностью интронов ~1.9 шт./т.п.н., и обнаружен частый альтернативный сплайсинг этих некодирующих РНК с ~2.3 изоформами РНК на ген (Cabili et al., 2011). Одной из наиболее изученных молекул lncRNA является Xist, которая участвует в инактивации Х-хромосомы самок плацентарных млекопитающих (Chang et al., 2006). По-видимому, РНК Xist возникла в результате псевдогенизации белок-кодирующего гена Lnx3 у ранних плацентарных с последующей интеграцией мобильных элементов (Elisaphenko et al., 2008). Анализ Xist у нескольких видов млекопитающих показал полную консервативность структуры гена Xist (рис. 1). Четыре из 10 экзонов Xist, обнаруженных у плацентарных, показывают значительное сходство последовательности с экзонами гена Lnx3 (рис. 1), тогда как остальные 6 экзонов Xist гомологичны различным мобильным элементам. Таким образом, некоторые интроны Xist были унаследованы от гена Lnx3, а некоторые, по-видимому, были получены в ходе эволюции этого гена (Elisaphenko et al., 2008). Сравнительный анализ >3000 генов lncRNA мыши дал основания предполагать, что сохранение экзон-интронной структуры может быть общим свойством lncRNA (Ponjavic et al., 2007). Было обнаружено, что 65 и 40% |GT–AG| сайтов сплайсинга lncRNA мыши сохраняются у человека и крысы соответственно. Эти значения существенно выше, чем количество консервативных интронных динуклеотидов GT и AG, которые не участвуют в сплайсинге, что указывает на эволюционную консервативность сигналов сплайсинга lncRNA (Ponjavic et al., 2007). Для детальной реконструкции происхождения и эволюции интронов lncRNA требуются дальнейшие сравнительные геномные исследования.
Рис. 1.
Ген Xist эволюционировал из белок-кодирующего гена и ряда мобильных элементов. Черные прямоугольники показывают экзоны, происходящие из гена Lnx3; темно-серые прямоугольники показывают экзоны, происходящие из мобильных элементов; светло-серые прямоугольники соответствуют псевдогенизированному экзону у паралогичного Lnx3 гена в предке всех рассмотренных видов; заштрихованные прямоугольники соответствуют остаткам белок-кодирующих экзонов. Данные взяты из (Elisaphenko et al., 2008).

Эволюционная консервативность и содержание мобильных элементов в экзонах и интронах lincRNA
Эволюционная консервативность показывает функциональную значимость молекул lincRNA. Эволюционная консервативность может быть исследована при помощи традиционного критерия отбора в белок-кодирующих генах. Критерий определяется отношением несинонимических (Ka) к синонимическим (Ks) заменам. Предполагается, что положительный отбор наблюдается при Ka/Ks > 1, в то время как отрицательный отбор может наблюдаться при Ks/Ka > 1 (Hurst, 2002). При рассмотрении генов lincRNA скорость замен в экзонах (Ke) можно считать аналогичной Ka, а в интронах (Ki), соответственно, – Ks (Louie et al., 2003; Hoffman, Birney, 2007; Resch et al., 2007). Селективный отбор в экзонах lincRNA потенциально может быть определен при условии Ke/Ki < 1.
Показано (Managadze et al., 2011), что скорость замен в экзонах lincRNA мыши значительно ниже скорости замен в интронах (Ke/Ki < 1) (рис. 2а,б). Результаты исследования говорят о том, что селективный отбор действует на экзоны генов lincRNA, и согласуются с более ранними наблюдениями (Ponjavic et al., 2007; Guttman et al., 2009). Также авторы показали, что распределение скоростей замен значительно шире для выборки экзонов lincRNA мыши и человека, чем для выборки интронов (рис. 2а,б). Это указывает на меняющуюся интенсивность селективного отбора на гены lincRNA.
Рис. 2.
Дивергенция между экзонами (а) и интронами (б) lincRNA человека и мыши. По оси абсцисс – эволюционная дистанция. По оси ординат – численность единиц (экзонов/интронов); количество сравненных интронов меньше, чем экзонов, в силу особенностей их выравнивания.
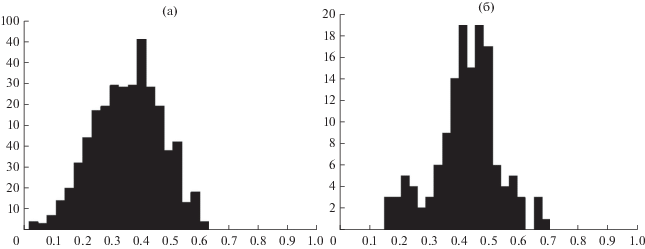
В работе (Managadze et al., 2013) измерили уровень экспрессии lincRNA с использованием микрочипов и данных EST-последовательностей (expressed sequence tag). Исследователи получили высокую корреляцию между уровнями экспрессии данных микрочипов и EST (Pearson CC = 0.39, P < 10–62). В экзонах lincRNA мыши наблюдалась статистически значимая отрицательная корреляция между скоростью эволюции последовательности и ее уровнем экспрессии. Коэффициенты корреляции в основном находятся в диапазоне 0.1–0.16. Напротив, для интронов коэффициенты корреляции были очень низкими и статистически не значимыми. То есть была показана отрицательная корреляция между скоростью эволюции и уровнем экспрессии lincRNA.
Последовательности мобильных элементов TEs (transposable elements) составляют значительную часть геномов у млекопитающих и, в частности, входят в состав генов lincRNA. На рис. 3а показано распределение TEs в предполагаемых областях промотора, экзонах и интронах lincRNA. Наименьшее количество TEs было обнаружено в области промоторов, среднее количество TEs было найдено в экзонах, а наибольшая доля TEs находилась в интронах. Это распределение TEs совместимо с ранее описанной тенденцией отсутствия TEs в функционально важных районах белок-кодирующих генов (рис. 3б; Jordan et al., 2003).
Рис. 3.
Процент содержания TEs в lincRNA (а) и mRNA (б) в геномах человека и мыши. Для гистограммы mRNA используются следующие сокращения: 5'UTR – 5'-нетранслируемый регион; 3'UTR – 3'-нетранслируемый регион; CDS – белок-кодирующая последовательность.
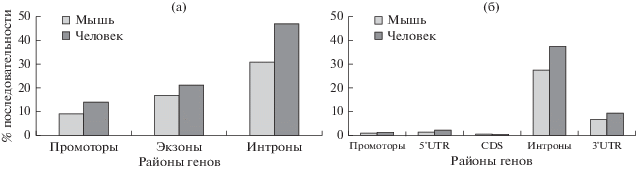
Аналогично белок-кодирующим генам плотность TEs в расширенных районах промотора оказалась значительно выше, чем плотность в коровой области промотора (Jordan et al., 2003). Доли TEs в интронах lincRNA и белок-кодирующих генов практически идентичны, что указывает на сопоставимые функциональные ограничения. В экзонах и в коровой области промотора генов lincRNA доля TEs статистически достоверно выше (P < 10–5 по тесту Фишера), чем в соответствующих белок-кодирующих генах. Эти результаты согласуются с результатами предыдущего исследования, в котором использовались разные наборы генов lincRNA (Kapusta et al., 2013). Таким образом, распределение TEs в генах lincRNA является надежной характеристикой.
Интроны lincRNA в масштабе полных геномов млекопитающих
Мы стремились проанализировать эволюцию интрон-экзонной структуры генов lincRNA млекопитающих в масштабе полных геномов. Такой анализ требует тщательной идентификации ортологичных наборов генов (наборов генов, произошедших от одного гена у последнего общего предка сравниваемых видов), а также идентификации ортологичных интронов в каждом из этих наборов генов. Чтобы избежать потенциальных осложнений, вызванных координированной экспрессией кодирующих белок генов и lincRNA, мы решили проанализировать только наборы lincRNA млекопитающих. Мы использовали наборы данных для человека и мыши, поскольку эти аннотированные наборы lincRNA имеют известные эволюционные свойства и свойства экспрессии генов (Managadze et al., 2011, 2013). Маловероятно, что этот набор данных содержит белок-кодирующие гены, и этот же вывод был сделан для других наборов данных генов lincRNA (Banfai et al., 2012; Guttman et al., 2013). Меньший размер выборки генов lincRNA человека по сравнению с генами lincRNA мыши (табл. 1) не оказал заметного влияния на выводы нескольких предыдущих исследований (Managadze et al., 2011, 2013; Kannan et al., 2015). Характеристики проанализированных наборов lincRNA мыши и человека приведены в таблице 1. Около 40% lincRNA человека и мыши содержат интроны (табл. 1). На содержащие интроны гены lincRNA приходится более 2 интронов со средней длиной более 9000 нуклеотидов, хотя медианные значения намного меньше (табл. 1). Интересно отметить, что, несмотря на более длинные экзоны у мыши, чем у человека, и аналогичные размеры интронов у обоих видов, средняя длина lincRNA мыши значительно короче, чем средняя длина lincRNA человека (Р < 0.0001, согласно двустороннему Т-тесту Стьюдента). Это, по-видимому, связано с бо́льшим количеством интронов, присутствующих в lincRNA человека, по сравнению с lincRNA мыши (3.86 по сравнению с 2.52 в среднем) (табл. 1). Этот результат может отражать различия в процедурах выборки lincRNA, хотя не следует исключать и биологические факторы.
Эволюционная консервативность сигналов сплайсинга
Мы проанализировали эволюционную консервативность GT/GC (начало интрона) и AG (конец интрона), используя попарное сравнение генов lincRNA мыши/человека и 15 других видов (табл. 2). В соответствии с исследованием (Ponjavic et al., 2007), мы обнаружили значительную консервативность сигналов сплайсинга (табл. 2). Попарные сравнения с сигналами сплайсинга мыши позволили сделать вывод, что доля консервативных динуклеотидов GT/GC и AG у крыс составляет 73 и 68% соответственно. Для большинства сравнений доля консервативных GT/GC и AG составляла около 50–60%.
Таблица 2.
Консервативные сигналы сплайсинга
| Вид | Попарное сравнение сайтов сплайсинга, где геном мыши – референсный | ||||||
| название (число ортологов) |
донорный сайт (GT/GC) | акцепторный сайт (AG) | |||||
| совпадений | несовпадений | совпадений, % | совпадений | несовпадений | совпадений, % | ||
| Rattuефs norvegicus | Крыса (2285) |
1555 | 569 | 73 | 1448 | 669 | 68 |
| Oryctolagus cuniculus | Кролик (1522) |
518 | 258 | 67 | 419 | 306 | 58 |
| Homo sapiens | Человек (2091) |
902 | 619 | 59 | 746 | 715 | 51 |
| Pan troglodytes | Шимпанзе (2068) |
826 | 606 | 58 | 703 | 692 | 50 |
| Macaca mulatta | Макака (1971) |
807 | 543 | 60 | 682 | 647 | 51 |
| Bos taurus | Бык (1815) |
694 | 402 | 63 | 560 | 498 | 53 |
| Canis lupus familiaris | Собака (1897) |
714 | 512 | 58 | 627 | 581 | 52 |
| Loxodonta africana | Слон (1485) |
499 | 247 | 67 | 428 | 312 | 58 |
| Echinops telfairi | Тенрек (1256) |
368 | 179 | 67 | 283 | 193 | 59 |
| Takifugu rubripes | Фугу (203) |
36 | 28 | 56 | 24 | 28 | 46 |
| Monodelphis domestica | Опоссум (1068) |
249 | 169 | 60 | 162 | 150 | 52 |
| Dasypus novemcinctus | Броненосец (1426) |
469 | 260 | 64 | 382 | 322 | 54 |
| Gallus gallus | Цыпленок (472) |
113 | 36 | 76 | 75 | 43 | 64 |
| Danio rerio | Данио (207) |
44 | 27 | 62 | 26 | 32 | 45 |
| Tetraodon nigroviridis | Иглобрюх (226) |
46 | 24 | 66 | 29 | 28 | 51 |
| Xenopus tropicalis | Лягушка (312) |
74 | 37 | 67 | 51 | 40 | 56 |
| Вид | Попарное сравнение сайтов сплайсинга, где геном человека – референсный | ||||||
| название (число ортологов) |
донорный сайт (GT/GC) | акцепторный сайт (AG) | |||||
| совпадений | несовпадений | совпадений, % | совпадений | несовпадений | совпадений, % | ||
| Pan troglodytes | Шимпанзе (575) |
870 | 19 | 98 | 867 | 15 | 98 |
| Macaca mulatta | Макака (564) |
800 | 53 | 94 | 828 | 42 | 95 |
| Mus musculus | Мышь (488) |
368 | 120 | 75 | 364 | 05 | 78 |
| Rattus norvegicus | Крыса (476) |
369 | 112 | 77 | 342 | 102 | 77 |
| Oryctolagus cuniculus | Кролик (463) |
445 | 86 | 84 | 415 | 114 | 78 |
| Bos taurus | Бык (527) |
531 | 122 | 81 | 484 | 144 | 77 |
| Canis lupus familiaris |
Собака (476) |
546 | 121 | 82 | 543 | 118 | 82 |
| Loxodonta africana | Слон (458) |
364 | 82 | 82 | 341 | 83 | 80 |
| Echinops telfairi | Тенрек (419) |
196 | 59 | 77 | 175 | 68 | 72 |
| Dasypus novemcinctus | Броненосец (468) |
362 | 95 | 79 | 320 | 122 | 72 |
| Monodelphis domestica | Опоссум (287) |
213 | 35 | 86 | 189 | 62 | 75 |
| Gallus gallus | Цыпленок (131) |
33 | 10 | 77 | 23 | 18 | 56 |
| Takifugu rubripes | Фугу (80) |
48 | 7 | 87 | 51 | 11 | 82 |
| Danio rerio | Данио (79) |
43 | 7 | 86 | 44 | 9 | 83 |
| Tetraodon nigroviridis | Иглобрюх (87) |
49 | 16 | 75 | 52 | 18 | 74 |
| Xenopus tropicalis | Лягушка (89) |
29 | 4 | 88 | 29 | 10 | 74 |
Эти числа значительно превышают количество консервативных интронных GT/GC- и AG-динуклеотидов, которые не участвуют в сплайсинге, что указывает на эволюционную консервативность сигналов сплайсинга в lincRNA (P (Freal ≤ Fsampled) < 0.001). Этот результат свидетельствует о том, что гены lincRNA мыши содержат эволюционно консервативные сигналы сплайсинга. Однако доля консервативных динуклеотидов GT/GC и AG намного больше (около 70–80%) для сравнений интронов lincRNA человека и ортологичных позиций у других видов (табл. 2), уровень консервативности имеет высокую значимость (P (Freal ≤ Fsampled) < 0.001).
Эволюционная консервативность экзон-интронной структуры
Традиционно анализ позиций интронов в белок-кодирующих генах основывался на ортологичных положениях интронов. Для того чтобы пара интронов считалась ортологичной, они должны находиться точно в одном и том же положении в выравненных последовательностях ортологичных кодирующих белок генов. В этом исследовании мы использовали менее строгое определение ортологичных интронов на основе полногеномных выравниваний: для того чтобы пара интронов считалась ортологичной, один интрон должен быть расположен внутри известного гена lincRNA человека или мыши (табл. 1), а другой должен иметь ортологичные GT/GC (начало интрона) и AG (конец интрона) динуклеотиды в ортологичных положениях, по меньшей мере, в одной последовательности из геномных выравниваний. Таким образом, мы использовали позиции интронов мыши или человека в качестве референсной экзон-интронной структуры гена. Эта процедура может приводить к ложноположительным результатам, поскольку некоторые динуклеотиды могут сохраняться, но не служить при этом в качестве сигналов сплайсинга. Такая же проблема существует для сигналов сплайсинга (см. выше), мы использовали статистические тесты для подтверждения значимости консервативности. Мы применили ту же методологию для того, чтобы сделать вывод о консервативности позиций интронов в генах lincRNA.
Чтобы более подробно проанализировать эволюционную динамику интронов, мы обратились к филогенетическому анализу. С этой целью позиции интронов могут быть представлены в виде матрицы данных отсутствия/присутствия интрона (закодированы “0/1”, отсутствующие данные кодируются “?”). Пример такой матрицы для местоположений интронов показан на рис. 4. Мы использовали для анализа геномы трех видов приматов и трех видов грызунов (которые разошлись менее 100 млн лет назад (Elisaphenko et al., 2008). Остальные 11 видов мы использовали для определения консенсуса аутгруппы. Консенсусные последовательности аутгрупп были реконструированы с использованием следующих трех правил. 1) Если в видах аутгруппы была, по крайней мере, одна “1”, состоянию аутгруппы была присвоена “1”. 2) Если в видах аутгруппы были только “0”, состоянию аутгруппы был присвоен “0”. 3) Если в видах аутгруппы не было состояний “1” или “0”, состоянию аутгруппы был присвоен “?” (рис. 4), и эта позиция была удалена из дальнейшего анализа. Пример фрагмента консенсусной последовательности аутгруппы показан на рис. 4. Мы изучали интроны, которые присутствовали в генах lincRNA человека и, по крайней мере, в одном из любых других видов. Мы использовали тот же фильтр для генов lincRNA мыши (ортологичные интроны в генах lincRNA мыши и, по крайней мере, в одном из любых других видов).

Данные об отсутствии/присутствии интрона были подвергнуты анализу методом эволюционной парсимонии (экономии). В существующих подходах невзвешенная парсимония представляется наиболее подходящей в этом случае (Rogozin et al., 2003), поскольку мы не обладаем моделью возникновения/потери интронов в генах lincRNA. Мы применили принцип парсимонии следующим образом: учитывая топологию филогенетического дерева, строили наиболее экономный сценарий эволюции интронов при распределении событий возникновения и потери интронов по ветвям деревьев. Наиболее экономным сценарием будет сценарий с минимальным количеством возникновений и потерь (рис. 5, 6).
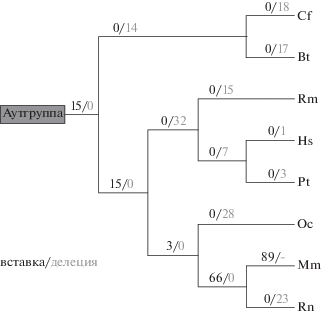
Рис. 5.
Дерево распределения вставок/делеций интронов, полученное при помощи программы DNAPARS, с выравниванием на интроны мыши. На рис. 5–8 для обозначения видов используются сокращения латинских названий, приведенных в таблице 2: Сf – собака; Bt – бык; Rm – макака – rhesus macaque (Macaca mulatta); Hs – человек; Pt – шимпанзе; Oc – кролик; Mm – мышь; Rn – крыса.
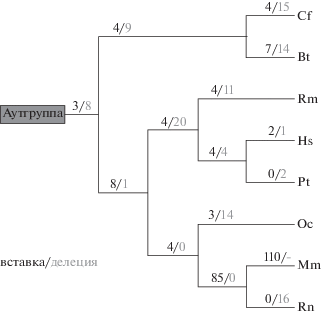
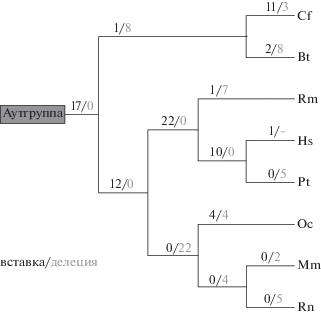
Рис. 6.
Дерево распределения вставок/делеций интронов, полученное при помощи программы DNAPARS, с выравниванием на интроны человека.
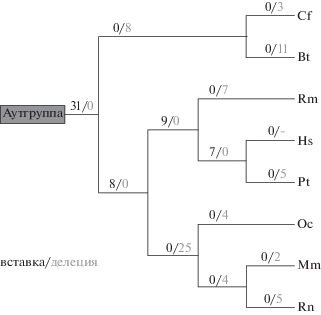
Анализ позиций интронов с использованием программы DNAPARS показал, что многие позиции интронов оставались сохраненными; например, на рисунке 4 имеется пять 100%-но консервативных позиций интронов (для 100%-ной консервативности интрона требуется его присутствие во всех шести видах приматов/грызунов и в консенсусной последовательности аутгруппы). 362 (55%) положения интронов человека сохраняются на 100%, консервативность значима (P (Freal ≤ Fsampled) < 0.001). Количество 100%-но консервативных положений интронов мыши менее впечатляющее (68 интронов, 19%), но все еще очень значимое (P (Freal ≤ Fsampled) < 0.001). Однако значительная часть интронов мыши и человека не консервативны (см., например, рис. 4). Для интронов мыши существует масштабная динамика интронов в ветви, ведущей к кластеру мыши и крысы, как и в последующей ветви, ведущей к мыши (рис. 5). Аналогичная тенденция наблюдалась, когда мы использовали позиции интронов человека в качестве референсной структуры генов (рис. 6), хотя потери в этом сценарии доминировали над возникновениями.
Для реконструкции эволюции интронов мы также применили метод Долло-парсимонии. Закон Долло, также известный как “the law of irreversible evolution”, был сформулирован в 1893 г. (Dollo, 1893). Этот закон утверждает, что сложная биологическая система, которая была потеряна организмом в ходе эволюции, не может снова появиться в своем исходном виде. Другими словами, одна и та же последовательность мутационных событий, которая привела к появлению биологической системы, не может повториться дважды в силу стохастичности эволюционных процессов. Метод Долло-парсимонии как метод филогенетического анализа был впервые формализован в 1977 г. (Farris, 1977). В простейшем виде этот метод рассматривает два состояния в каждом исследуемом сайте: примитивное (0) и производное (1). Возникновение производного состояния 1 из примитивного состояния 0 (0 => 1) разрешается только один раз в рассматриваемом филогенетическом дереве, в то время как число переходов 1 => 0 не ограничено. У мыши обнаружена существенная динамика интронов в ветви, ведущей к кластеру мыши и крысы, как и в последующей ветви, ведущей к мыши (рис. 7). Аналогичная тенденция наблюдалась для ветви, ведущей к кролику, мыши и крысе, когда мы использовали позиции интронов человека в качестве референсной структуры генов (рис. 8). Потери интронов в этом сценарии доминировали над возникновениями для многих ветвей дерева (рис. 8).
ОБСУЖДЕНИЕ
Мы построили сценарии потерь/приобретений интронов с использованием методов парсимонии и Долло-парсимонии. Метод Долло-парсимонии был успешно применен ранее для исследования эволюции интронов в белок-кодирующих генах (Rogozin et al., 2003; Babenko et al., 2004). Следует отметить, что в данной работе критерии наличия/отсутствия интронов в lincRNA являются менее точными по сравнению с такими критериями для интронов в белок-кодирующих генах (Rogozin et al., 2003; Babenko et al., 2004), поэтому модель Долло-парсимонии может быть нереалистичной. Тем не менее, парсимония и Долло-парсимония дают схожие результаты. Анализ при помощи программы DOLLOP с использованием выравнивания на интроны человека выявил существенную динамику потерь/приобретений интронов; в целом приобретения интронов сдвигались ближе к корню дерева. Мы можем сделать вывод, что существенная динамика интронов отличает lincRNA от белок-кодирующих генов (в которых наблюдается меньшая динамика у плацентарных млекопитающих) (Rogozin et al., 2012; Chernikova et al., 2016). Тем не менее, большое число консервативных интронов (предсказанное обоими методами) является общей чертой интронов в двух типах генов вне зависимости от условий реконструкции сценария эволюции интронов.
Очевидный парадокс меньшего количества консервативных интронов мыши по сравнению с интронами человека (хотя набор данных lincRNA мыши намного больше, табл. 1) может быть результатом высокой скорости эволюции состава генов/интронов lincRNA в отряде грызунов (рис. 2, 3). Это согласуется с низкой консервативностью сигналов сплайсинга мыши по сравнению с сигналами сплайсинга человека (табл. 2). Эти результаты могут отражать наблюдаемую быструю динамику генов lincRNA грызунов: было показано, что почти половина локусов lincRNA возникли или были потеряны со времени последнего общего предка мыши и крысы (Kutter et al., 2012). Было высказано предположение, что такая быстрая эволюция lincRNA способствует эволюции экспрессии генов, специфичных для тканей и линий (Kutter et al., 2012). Частые потери интронов наблюдались в нескольких эволюционно консервативных генах lincRNA (Chodroff et al., 2010), поэтому экзон-интронная структура генов lincRNA показала значительно больше возникновений и потерь во время эволюции, тогда как сравнительный анализ позиций интронов в белок-кодирующих генах позвоночных выявил всего несколько потерь, но не выявил видимых возникновений интронов в генах млекопитающих (Roy et al., 2003; Csuros et al., 2011). Более крупные наборы надежных генов lincRNA млекопитающих могут помочь в разработке надежных статистических моделей процесса возникновения/потери интронов и в проверке любых специфических особенностей данного процесса в различных группах позвоночных.
Существенная динамика позиций интронов в генах lincRNA млекопитающих не отменяет наблюдение, что многие интроны lincRNA высоко консервативны (19–55%). Это наблюдение согласуется с предыдущими исследованиями гена Xist и нескольких других эволюционно консервативных генов lincRNA (Elisaphenko et al., 2008; Chodroff et al., 2010). Данный анализ датирует происхождение многочисленных интронов сплайсеосомных lincRNA временем распространения плацентарных млекопитающих около 100 млн лет назад (Elisaphenko et al., 2008). Этот результат свидетельствует о том, что первичная/вторичная структура этих молекул функционально важна, а консервативные интроны могут использоваться в качестве отличительных признаков функциональных генов lincRNA.
Было высказано предположение, что наборы данных по генам lincRNA не содержат многих белок-кодирующих генов (Managadze et al., 2011, 2013; Banfai et al., 2012; Guttman et al., 2013; Calviello et al., 2016); однако мы не можем исключить наличие функциональных коротких открытых рамок считывания (Carvunis et al., 2012; Andrews, Rothnagel, 2014). Одним из возможных признаков того, что lincRNA не содержат много белок-кодирующих областей, является высокая доля мобильных элементов, наблюдаемых в генах lincRNA (Kannan et al., 2015). lincRNA имеют в два раза больше мобильных элементов, чем 3'-концы кодирующих белок генов. Фактически, доля мобильных элементов ближе к интронным областям, чем к любым другим районам белок-кодирующих генов (Kannan et al., 2015). Более низкие скорости замещения экзонов по сравнению с интронами наблюдались для генов lincRNA человека и мыши (Managadze et al., 2011). Однако селективный отбор экзонов в lincRNA намного слабее, чем на несинонимичных позициях в кодирующих белок генах (Managadze et al., 2011). Как сила, так и форма распределения скоростей замещения в экзонах lincRNA более похожи на синонимичные, чем несинонимичные замены в белок-кодирующих генах (Managadze et al., 2011). Это наблюдение также согласуется с идеей, что lincRNA не кодируют белки. Однако нельзя исключать, что присутствие высоко консервативных интронов может быть связано с короткими (и редкими) открытыми рамками считывания. В этом случае интроны могут использоваться в качестве признака функциональных коротких открытых рамок считывания. Окончательный ответ на этот вопрос может быть получен с помощью комбинации экспериментальных и вычислительных методов, включая профилирование рибосом, анализ использования кодонов и консервативности кодонов (Calviello et al., 2016). Альтернативный сплайсинг – еще один фактор, который может влиять на выводы этого исследования. В недавнем подробном исследовании было обнаружено более 8000 генов lincRNA человека со средней плотностью интронов ~1.9 шт. на килобазу, а также обнаружен обширный альтернативный сплайсинг этих некодирующих РНК с ~2.3 изоформами РНК на ген (Cabili et al., 2011). Такие альтернативно сплайсированные lincRNA, вероятно, увеличивают скорость возникновений/потерь интронов.
ЗАКЛЮЧЕНИЕ
Мы представляем крупномасштабные реконструкции эволюции экзонов и интронов генов lincRNA с использованием множественных геномных выравниваний 17 видов позвоночных. Хотя консервативность первичных последовательностей экзонов lincRNA не столь выражена, как у белок-кодирующих генов, но, тем не менее, она существенно выше, чем у интронов генов lincRNA. Сравнительный анализ предполагаемых позиций интронов в генах lincRNA этих геномов позвоночных указывает на то, что некоторые интроны lincRNA сохранялись более 100 млн лет, что позволяет предположить, что эти молекулы, вероятно, являются функционально важными.
Список литературы
Amaral P.P., Dinger M.E., Mattick J.S. Non-coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective // Brief. Funct. Genomics. 2013. V. 12. P. 254–278.
Andrews S.J., Rothnagel J.A. Emerging evidence for functional peptides encoded by short open reading frames // Nat. Rev. Genet. 2014. V. 15. P. 193–204.
Babenko V.N., Rogozin I.B., Mekhedov S.L., Koonin E.V. Prevalence of intron gain over intron loss in the evolution of paralogous gene families // Nucl. Acids Res. 2004. V. 32. P. 3724–3733.
Banfai B., Jia H., Khatun J. et al. Long noncoding RNAs are rarely translated in two human cell lines // Genome Res. 2012. V. 22. P. 1646–1657.
Bertone P., Stolc V., Royce T.E. et al. Global identification of human transcribed sequences with genome tiling arrays // Science. 2004. V. 306. P. 2242–2246.
Brockdorff N., Ashworth A., Kay G.F. et al. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus // Cell. 1992. V. 71. P. 515–526.
Cabili M.N., Trapnell C., Goff L. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses // Genes Dev. 2011. V. 25. P. 1915–1927.
Calviello L., Mukherjee N., Wyler E. et al. Detecting actively translated open reading frames in ribosome profiling data // Nat. Methods. 2016. V. 13. P. 165–170.
Carvunis A.R., Rolland T., Wapinski I. et al. Proto-genes and de novo gene birth // Nature. 2012. V. 487. P. 370–374.
Chang S.C., Tucker T., Thorogood N.P., Brown C.J. Mechanisms of X-chromosome inactivation // Front. Biosci. 2006. V. 11. P. 852–866.
Chernikova D., Managadze D., Glazko G.V. et al. Conservation of the exon-intron structure of long intergenic non-coding RNA genes in eutherian mammals // Life. 2016. V. 6. P. e27.
Chodroff R.A., Goodstadt L., Sirey T.M. et al. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes // Genome Biol. 2010. V. 11. P. R72.
Cordaux R., Udit S., Batzer M.A., Feschotte C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element // PNAS USA. 2006. V. 103. P. 8101–8106.
Csuros M., Rogozin I.B., Koonin E.V. A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes // PLoS Comput. Biol. 2011. V. 7. P. e1002150.
Deutsch M., Long M. Intron-exon structures of eukaryotic model organisms // Nucl. Acids Res. 1999. V. 27. P. 3219–3228.
Dinger M.E., Pang K.C., Mercer T.R. et al. NRED: a database of long noncoding RNA expression // Nucl. Acids Res. 2009. V. 37. P. D122–126.
Dollo L. Le lois de l’evolution // Bulletin de la Societe Belge de Geologie, de Paleontologie et d’Hydrologie. 1893. V. 7. P. 164–167.
Duret L., Chureau C., Samain S. et al. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene // Science. 2006. V. 312. P. 1653–1655.
Elisaphenko E.A., Kolesnikov N.N., Shevchenko A.I. et al. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements // PLoS One. 2008. V. 3. P. e2521.
Farris J.S. Phylogenetic analysis under Dollo’s Law // Syst. Zool. 1977. V. 26. P. 77–88.
Goecks J., Nekrutenko A.,Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences // Genome Biol. 2010. V. 11. P. R86.
Goodrich J.A., Kugel J.F. Non-coding-RNA regulators of RNA polymerase II transcription // Nat. Rev. Mol. Cell. Biol. 2006. V. 7. P. 612–616.
Guttman M., Rinn J.L. Modular regulatory principles of large non-coding RNAs // Nature. 2012. V. 482. P. 339–346.
Guttman M., Amit I., Garber M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals // Nature. 2009. V. 458. P. 223–227.
Guttman M., Russell P., Ingolia N.T. et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins // Cell. 2013. V. 154. P. 240–251.
Haeussler M., Zweig A.S., Tyner C. et al. The UCSC genome browser database: 2019 update // Nucl. Acids Res. 2019.
Hoffman M.M., Birney E. Estimating the neutral rate of nucleotide substitution using introns // Mol. Biol. Evol. 2007. V. 24. P. 522–531.
Hong X., Scofield D.G., Lynch M. Intron size, abundance, and distribution within untranslated regions of genes // Mol. Biol. Evol. 2006. V. 23. P. 2392–2404.
Hurst L.D. The Ka/Ks ratio: diagnosing the form of sequence evolution // Trends Genet. 2002. V. 18. P. 486.
Jordan I.K., Rogozin I.B., Glazko G.V., Koonin E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements // Trends Genet. 2003. V. 19. P. 68–72.
Kannan S., Chernikova D., Rogozin I.B. et al. Transposable element insertions in long intergenic non-coding RNA genes // Front. Bioeng. Biotechnol. 2015. V. 3. P. 71.
Kapusta A., Feschotte C. Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications // Trends Genet. 2014. V. 30. P. 439–452.
Kapusta A., Kronenberg Z., Lynch V.J. et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs // PLoS Genet. 2013. V. 9. P. e1003470.
Karolchik D., Hinrichs A.S., Furey T.S. et al. The UCSC table browser data retrieval tool // Nucl. Acids Res. 2004. V. 32. P. D493–D496.
Kutter C., Watt S., Stefflova K. et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression // PLoS Genet. 2012. V. 8. P. e1002841.
Liu J., Gough J., Rost B. Distinguishing protein-coding from non-coding RNAs through support vector machines // PLoS Genet. 2006. V. 2. P. e29.
Louie E., Ott J., Majewski J. Nucleotide frequency variation across human genes // Genome Res. 2003. V. 13. P. 2594–2601.
Managadze D., Rogozin I.B., Chernikova D. et al. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs // Genome Biol. Evol. 2011. V. 3. P. 1390–1404.
Managadze D., Lobkovsky A.E., Wolf Y.I. et al. The vast, conserved mammalian lincRNome // PLoS Comput. Biol. 2013. V. 9. P. e1002917.
Marques A.C., Ponting C.P. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness // Genome Biol. 2009. V. 10. P. R124.
Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions // Nat. Rev. Genet. 2009. V. 10. P. 155–159.
Ng S.Y., Lin L., Soh B.S., Stanton L.W. Long noncoding RNAs in development and disease of the central nervous system // Trends Genet. 2013. V. 29. P. 461–468.
Ponjavic J., Ponting C.P., Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs // Genome Res. 2007. V. 17. P. 556–565.
Ponting C.P., Belgard T.G. Transcribed dark matter: meaning or myth? // Hum. Mol. Genet. 2011. V. 19. P. R162–R168.
Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs // Cell. 2009. V. 136. P. 629–641.
Resch A.M., Carmel L., Mariño-Ramírez L. et al. Widespread positive selection in synonymous sites of mammalian genes // Mol. Biol. Evol. 2007. V. 24. P. 1821–1831.
Robinson R. Dark matter transcripts: sound and fury, signifying nothing? // PLoS Biol. 2010. V. 8. P. e1000370.
Rogozin I.B., Wolf Y.I., Sorokin A.V. et al. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution // Curr. Biol. 2003. V. 13. P. 1512–1517.
Rogozin I.B., Carmel L., Csuros M., Koonin E.V. Origin and evolution of spliceosomal introns // Biol. Direct. 2012. V. 7. P. 11.
Roy S.W., Fedorov A., Gilbert W. Large-scale comparison of intron positions in mammalian genes shows intron loss but no gain // PNAS USA. 2003. V. 100. P. 7158–7162.
Schuler A., Ghanbarian A.T., Hurst L.D. Purifying selection on splice-related motifs, not expression level nor RNA folding, explains nearly all constraint on human lincRNAs // Mol. Biol. Evol. 2014. V. 31. P. 3164–3183.
Siepel A., Bejerano G., Pedersen J.S. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes // Genome Res. 2005. V. 15. P. 1034–1050.
Szcześniak M.W., Ciomborowska J., Nowak W. et al. Primate and rodent specific intron gains and the origin of retrogenes with splice variants // Mol. Biol. Evol. 2011. V. 28. P. 33–37.
van Bakel H., Hughes T.R. Establishing legitimacy and function in the new transcriptome // Brief. Funct. Genomic Proteomic. 2009. V. 8. P. 424–436.
Vance K.W., Ponting C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs // Trends Genet. 2014. V. 30. P. 348–355.
Zhang X.H., Chasin L.A. Comparison of multiple vertebrate genomes reveals the birth and evolution of human exons // PNAS USA. 2006. V. 103. P. 13427–13432.
Zhuo D., Madden R., Elela S.A., Chabot B. Modern origin of numerous alternatively spliced human introns from tandem arrays // PNAS USA. 2007. V. 104. P. 882–886.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии