

Успехи современной биологии, 2022, T. 142, № 1, стр. 5-24
Роль сестринов в регуляции клеточного ответа на стресс
1 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова
Москва, Россия
2 Trinity College
Dublin 2, Ireland
* E-mail: abudanov@hotmail.com
Поступила в редакцию 25.10.2021
После доработки 20.11.2021
Принята к публикации 20.11.2021
- EDN: GEHUMZ
- DOI: 10.31857/S0042132422010033
Аннотация
Представленная работа посвящена анализу белков семейства сестринов и их роли в ответе клеток на стресс. В работе описаны структура и функции сестринов и их ключевая роль в регуляции киназы mTOR и метаболизма. Также в данном обзоре подробно рассмотрены функции сестринов в регуляции старения и возрастных заболеваний.
ВВЕДЕНИЕ
Адекватный и своевременный ответ на стресс – ключевая функция любого организма, необходимая для сохранения жизнеспособности и нормального функционирования в постоянно меняющихся условиях внешней среды. Механизмы ответа на стресс присущи каждой клетке организма, что придает им устойчивость к стрессу, способствуя поддержанию целостности и функциональности тканей организма. Хотя механизмы ответа на стресс обусловлены конкретным типом стресса, многие стрессовые воздействия имеют общие последствия и активируют сходные механизмы ответа. Например, гипоксия/ишемия, ДНК повреждения и стресс эндоплазматического ретикулума (СЭР) приводят к накоплению активных форм кислорода (АФК), что во многом обусловлено нарушениями нормального функционирования митохондрий. Повреждения в структуре митохондрий приводят к увеличению утечки электронов из цепи окислительного фосфорилирования и, как следствие, продукции радикалов и других высокоактивных молекул, способных повреждать макромолекулы и клеточные структуры, что может привести к необратимым изменениям в клетках и тканях организма. В целях предотвращения повреждений в условиях стресса клетки перестраивают свой метаболизм, который при нормальных условиях задействован в биосинтетических процессах, сопряженных с расходом значительного количества энергии, на катаболические процессы, направленные на производство АТФ посредством деградации различных высокомолекулярных соединений. Также катаболические процессы выполняют другую ключевую функцию – они ответственны за удаление поврежденных органелл и поврежденных белков, способных нарушать нормальные функциональные процессы в клетке. Одним из ключевых механизмов, участвующих в контроле качества клеточных структур и поддерживающих производство АТФ в условиях стресса, является процесс аутофагии, ответственный за деградацию клеточного содержимого в лизосомах. Другим важным следствием активации ответа на стресс является индукция программируемой клеточной смерти (ПКС). Процесс ПКС, тесно сопряженный с работой иммунной системы, приводит к удалению из организма сильно поврежденных клеток, которые могут нарушать функционирование данной ткани и подвергаться злокачественной трансформации. Нарушения в работе и координации механизмов защиты от стресса, таких как аутофагия и ПКС, имеют крайне нежелательные последствия для организма и приводят к развитию множества заболеваний, таких как сердечно-сосудистые заболевания, рак, диабет и нейродегенеративные болезни, являющиеся причинами смерти организма. Старение является естественным процессом ослабления нормальной реакции клеток на стресс, что приводит к значительному накоплению повреждений, приводящих к патологиям и смерти. Также различные воздействия, поддерживающие пути ответа на стресс, могут значительно замедлять процесс старения и развития различных возрастных заболеваний. Понимание роли тех или иных белков в регуляции сигнальных путей, ответственных за реагирование на стресс, может привести к созданию новых лекарств, направленных на усиление или ослабление функции данных белков, либо для поддержания клеточного гомеостаза, как в случае инфаркта или инсульта, либо для удаления поврежденных и вредоносных клеток, как в случае рака. Интересно, что многие белки, активированные стрессом, многофункциональны и играют уникальную роль при разных условиях стресса. Одними из стресс-регуляторных белков являются сестрины.
МЕХАНИЗМЫ КЛЕТОЧНОЙ РЕАКЦИИ НА СТРЕСС
Нарушения в протекании нормальных физиологических процессов посредством внешних воздействий или каких-либо внутренних поломок приводят к состоянию, называемому стрессом. Для сохранения жизнеспособности и функциональности клетка должна своевременно и эффективно реагировать на стресс, чтобы предотвратить накопление повреждений. В то же время в условиях чрезмерного стресса клетка может инициировать ПКС, чтобы не допустить накопление мутаций, основного фактора канцерогенеза, и неконтролируемый некроз, активатор воспалительных процессов (Dalina et al., 2018). Таким образом, клетка постоянно отслеживает любые нарушения посредством контроля экспрессии и активности стресс-респонсивных белков, функция которых состоит в поддержании гомеостаза и в обеспечивании жизнеспособности клеток.
р53 – ключевой регулятор ответа на повреждения ДНК
Поддержание стабильности генома – ключевая функция каждого живого организма, необходимая для сохранения и передачи генетической информации при делении клеток организма, а также в череде поколений (Lane, 1992). Данная функция особенно критична для представителей класса позвоночных, поскольку их организм состоит из большого количества делящихся и подвижных клеток, которые при отсутствии должного контроля могли бы автономно размножаться и распространяться по организму, нарушая нормальную работу органов и тканей.
Опухолевый супрессор ген TP53 кодирует белок р53, являющийся транскрипционным фактором, который постоянно отслеживает различные виды повреждений ДНК, включая двуцепочечные и одноцепочечные разрывы и различные модификации оснований в ДНК (Lane, 1992). Важной особенностью белка р53 в нормальных клетках является его низкая стабильность (около 20 мин), однако генотоксический стресс приводит к стабилизации этого белка (Levine, 1997). Короткая жизнь белка р53 обусловлена его взаимодействием с белком Mdm2/HDM2, Е3-убиквитин лигазой, направляющей р53 в протеасомы (Kastenhuber, Lowe, 2017). Белок Mdm2 сам по себе является транскрипционной мишенью белка р53, что обеспечивает важную обратную связь при поддержании уровня р53 (Kastenhuber, Lowe, 2017). Одноцепочечные и двуцепочечные повреждения ДНК являются сигналами для активации киназ ATR и AТМ, которые ответственны за фосфорилирование N-концевой области р53 напрямую или через активацию киназ Chk1 и Chk2 соответственно. Фосфорилирование определенных N-концевых аминокислот в белке р53 нарушает его связывание с белком Mdm2, предотвращая разрушение р53 в протеасомах (Kastenhuber, Lowe, 2017). Таким образом, высокие уровни белка р53 поддерживаются до тех пор, пока повреждения ДНК не будут подвергнуты репарированию. Стоит однако отметить, что участие киназного каскада ATR/ATM-Chk1/2 не является единственным механизмом регуляции уровня и активности р53. Существует множество других специфических для разных типов стрессов модификаций белка р53, которые включают фосфорилирование, ацетилирование, метилирование, а также другие модификации аминокислот в различных участках белка, что приводит к тонкой настройке активности р53 для адекватного изменения поведения клетки (Kastenhuber, Lowe, 2017).
Ключевой функцией р53 является его роль в регуляции транскрипции. Хотя р53 способен регулировать транскрипцию нескольких тысяч генов, только около сотни генов являются его прямыми мишенями (Andrysik et al., 2017; Fischer, 2017). Данные гены ответственны за несколько ключевых функций в клетке, таких как регуляция клеточного цикла, метаболизма, аутофагии, стабильности р53, а также контроль репарации ДНК (Fischer, 2017). Хотя транскрипционные программы, направленные на поддержание жизнеспособности клеток и активации клеточной смерти, могут противоречить одна другой, выбор той или иной из них зависит от многих факторов, включая интенсивность и продолжительность стресса и тип клеток (Budanov, 2011; Kruiswijk et al., 2015). Таким образом, белок р53 в условиях умеренного стресса способен поддерживать клеточный гомеостаз посредством контроля клеточного цикла, а также процессов репарации и регуляции метаболизма, но, в то же время, активирует ПКС при условиях, когда стресс угрожает появлением необратимых и опасных изменений поведения клетки (Budanov, 2011, 2012; Kruiswijk et al., 2015).
Окислительный стресс
Нарушения баланса производства и разрушения АФК приводят к накоплению в клетке молекул с высоким окислительным потенциалом. Вследствие высокой реакционной способности, АФК могут окислять и повреждать различные компоненты клетки, что является причиной окислительного стресса (D’Autreaux, Toledano, 2007; Budanov, 2011). Эукариотические клетки оснащены большим разнообразием антиоксидантных белков, активность которых направлена на нейтрализацию различных АФК. Так, супероксид дисмутаза ответственна за инактивацию супероксидов, каталаза – за разрушение перекиси водорода. В клетке также содержатся два типа пероксидаз: глютатион пероксидазы (GPX1/2) и пероксиредоксины (Prx1-6), активность которых поддерживается антиоксидантными молекулами: глютатионом и тиоредоксином (Martindale, Holbrook, 2002; Kinnula et al., 2004). Особенностью пероксиредоксинов является их высокий уровень экспрессии и относительная низкая пероксидазная активность, что позволяет им играть важную роль в регуляции сигнальных путей, в которых сигнальными молекулами служат АФК (Wood et al., 2003). Пероксиредоксины представляют собой тиоловые пероксидазы, а их энзиматический цикл включает окисление каталитической –SH-группы цистеина до состояния –SOH, с последующим формированием –S–S-мостика с другим цистеином, что в конечном счете приводит к восстановлению цистеинов с помощью тиоредоксина. Интересной особенностью эукариотических пероксиредоксинов является их способность к избыточному окислению каталитического цистеина до состояния –SO2H, что приводит к временному подавлению пероксидазной активности. Активность гиперокисленного пероксиредоксина может быть восстановлена с помощью белка сульфиредоксина (Srxn1) (Jeong et al., 2012).
Повышение уровней АФК приводит к активации множества сигнальных путей в клетке. Небольшие повышения уровней АФК могут стимулировать пролиферацию клеток и играть важную роль в поддержании гомеостаза (Martindale, Holbrook, 2002). Импульсные повышения уровней АФК могут приводить к окислению и подавлению активных центров редокс-зависимых фосфатаз, например, белковых фосфатаз, участвующих в сигнальных путях, регулируемых рецепторными тирозиновыми киназами или MAPK, а также липидной фосфатазой PTEN (Martindale, Holbrook, 2002; Kamata et al., 2005). Под действием АФК может изменяться конформационное состояние белков через стабилизацию внутрибелковых и межбелковых дисульфидных связей, приводя к усилению или ослаблению энзиматической активности. Это описано для некоторых транскрипционных факторов, например NF-kB (Martindale, Holbrook, 2002). Однако высокие уровни АФК могут приводить и к стимуляции путей, участвующих в индукции программы клеточной смерти, что наблюдается в случае продолжительной активации JNK, представителя семейства МАРК (Kamata et al., 2005). Также АФК, производимые в ответ на стрессовую активацию р53, значительно усиливают ПКС, индуцированную данным белком (Polyak et al., 1997). Напротив, антиоксидантные гены, индуцированные в ответ на активацию транскрипционных факторов NF-kB и семейства FOXO, могут защищать от клеточной смерти (Kamata et al., 2005; D’Autreaux, Toledano, 2007).
Одним из наиболее значимых транскрипционных факторов, активируемых в ответ на окислительный стресс и защищающих клетки от стресса, является белок NRF2. В отсутствии стресса NRF2 взаимодействует с белком KEAP1, который стимулирует убиквитинирование NRF2 и его протеасомную деградацию за счет взаимодействия с компонентами E3-убиквитин-лигазного комплекса – белками Cul3 и RBX1. Напротив, накопление АФК приводит к окислению критических цистеинов в белке КЕАР1. Это приводит к освобождению ионов Zn2+, связанных с белком, и к конформационным изменениям в белке, что предотвращает процесс убиквитинирования и разрушения белка NRF2 (D’Autreaux, Toledano, 2007). В результате NRF2 накапливается в ядре, активируя транскрипцию множества антиоксидантных генов, кодирующих пероксиредоксины, сульфиредоксин, тиоредоксин редуктазу, тиоредоксин и глютатион S-трансферазу (Cuadrado et al., 2019).
Стресс эндоплазматического ретикулума
СЭР вызван накоплением в эндоплазматическом ретикулуме неправильно свернутых белков, что приводит к нарушению множества процессов в клетке, таких как транспорт белков к мембранам, синтез и разрушение белков и контроль ионов Са2+. Данный тип стресса тесно связан с различными стрессовыми воздействиями, такими как нагревание, окислительный стресс, глюкозное голодание и вирусная инфекция (Xu et al., 2005). СЭР стимулирует ответ на неправильно свернутые белки (UPR), который опосредован белком-шапероном Grp78/BiP (Xu et al., 2005). Белок Grp78 в норме взаимодействует с несколькими белками, ответственными за активацию трех основных сигнальных путей СЭР: PERK, Ire1 и ATF6. Данные белки пронизывают мембраны эндоплазматического ретикулума (ЭР), связывая внутренний отсек ЭР с цитоплазмой. При накоплении неправильно свернутых белков в цистернах ЭР данные белки конкурируют за связывание с Grp78, что приводит к освобождению сигнальных белков и их активации (Xu et al., 2005).
Белки, активированные в ответ на СЭР, приводят к активации нескольких транскрипционных программ. Ser/Thr-киназа PERK при высвобождении из комплекса с Grp78 димеризуется на мембранах ЭР, активируется по механизму аутофосфорилирования и фосфорилирует фактор инициации трансляции eIF2α, что приводит к выключению трансляции большинства клеточных мРНК. Однако некоторые мРНК, слабо транслируемые при нормальных условиях, такие как мРНК, кодирующая транскрипционный фактор ATF4, получают значительные преимущества при трансляции, что приводит к накоплению белка в клетке и к активации его транскрипционной программы. Это приводит как к активации белков защиты от стресса (например шаперонов), так и белков, ответственных за индукцию ПКС, например, белка GADD153/CHOP (Xu et al., 2005). Белок NRF2, играющий исключительно важную роль в защите от окислительного стресса, также активируется киназой PERK посредством фосфорилирования белка KEAP1. Белок Ire1, содержащий Ser/Thr-киназный и эндонуклеазный домены, также димеризуется по механизму, сходному с PERK, и способствует вырезанию ингибирующего трансляцию интрона в мРНК транскрипционного фактора XBP1, активирующего транскрипцию генов, участвующих в ретроградном транспорте неправильно свернутых белков из ЭР в цитозоль и далее в системы разрушения белков (Xu et al., 2005). Третий ключевой компонент СЭР, транскрипционный фактор ATF6, в ответ на диссоциацию из комплекса с белком Grp78 транслоцируется в аппарат Гольджи, где часть белка, которая заякоривает белок на мембране, отрезается протеазами и позволяет ATF6 перемещаться в ядро, где он регулирует экспрессию соответствующих генов (Xu et al., 2005).
СЕСТРИНОВЫЕ ГЕНЫ И ИХ БЕЛКОВЫЕ ПРОДУКТЫ
Поиск новых стресс-регулируемых генов привел к обнаружению сестриновых генов (Budanov et al., 2010). Первый представитель данного семейства, первоначально названный PA26 и позже переименованный в SESN1, был обнаружен при поиске новых мишеней транскрипционного фактора р53 (Velasco-Miguel et al., 1999). SESN2, прежде названный Hi95, был обнаружен при поиске генов, активируемых в ответ на продолжительную гипоксию (Budanov et al., 2002). Последний представитель сестринов у млекопитающих, ген SESN3, был впервые обнаружен на основании биоинформатического выявления его гомологии с генами SESN1 и SESN2 при анализе структуры генома человека и плазмидных клонов, содержащих последовательности кДНК (Budanov et al., 2002; Peeters et al., 2003).
Гены, кодирующие сестрины
В геноме человека гены, кодирующие сестрины, расположены на разных хромосомах: ген SESN1, кодирующий Sesn1, – в хромосомном локусе 6q21, SESN2, кодирующий Sesn2, – в локусе 1p35.3 и SESN3, кодирующий белок Sesn3, – в локусе 11p21 (Budanov et al., 2010). Все три гена, вероятно, произошли от общего предка путем мультипликации и последующей дивергенции, хотя и остались высокогомологичными между собой (Budanov et al., 2010). Сестриновые гены кодируют различающиеся изоформы, которые образуются за счет альтернативного сплайсинга и использования альтернативных участков, с которых начинается их транскрипция. SESN2 кодирует единственный белок размером 480 аминокислотных остатков (а. о.) с молекулярной массой 60 кДа. Продуктами гена SESN1 являются три белка размером 551 а. о. (Т1), 491 а. о. (Т2) и 426 а. о. (Т3) с молекулярными массами 68, 55 и 48 кДа соответственно. Все изоформы Sesn1 отличаются по последовательности на N-конце, кодируемой отдельным экзоном, но идентичны в С-концевой части, кодируемой экзонами 4–12 (Velasco-Miguel et al., 1999). В свою очередь, ген SESN3 ответственен за синтез двух белков – размером 492 а. о. (Т1) с молекулярной массой 57.3 кДа и 317 а. о. (T2) с молекулярной массой 36 кДа (Budanov et al., 2010; Chen et al., 2010). Для каждого из трех сестриновых генов характерна одна основная изоформа белка (55 кДа Sesn1, 57.3 КДа Sesn3 и 60 кДа Sesn2), и данные изоформы схожи по размеру и по последовательности. Эти изоформы близки по первичной структуре белковым продуктам уникальных генов сестрина беспозвоночных, таких как dSesn D. melanogaster и сSesn нематоды Caenorhabditis elegans (C. elegans) (Budanov et al., 2002; Lee et al., 2010; Zeltukhin et al., 2018). Ген, кодирующий белок, напоминающий по последовательности сестрины, был обнаружен у некоторых простейших, хотя подобные гены не были найдены у представителей царств растений и грибов, в том числе у дрожжей (Budanov et al., 2010; Wolfson, Sabatini, 2017). Высокое сходство первичных последовательностей сестринов позволяет предположить, что все эти белки имеют сходные функции. Это предположение подкрепляется экспериментальными данными, указывающими на роль сестринов в регуляции метаболизма и внутриклеточных сигнальных путей (Budanov et al., 2010; Lee et al. 2013). Более того, нарушения некоторых физиологических процессов при инактивации гена сестрина у плодовых мушек, например, нарушенная защита от окислительного стресса, устраняютcя при экспрессии человеческого Sesn2 (Lee et al., 2010). Специфические функции изоформ сестринов, например изоформ T1 и T3 для Sesn1 и Т2 для Sesn3, пока неизвестны. Возможно, эти изоформы имеют уникальные функции, которых нет у “основных” изоформ, хотя также не исключено, что данные изоформы влияют на взаимодействия “основных” изоформ с другими белками, оказывая доминантно-негативный эффект. В обзоре будут рассматриваться только основные высокогомологичные друг другу изоформы сестринов: изоформа 55 кДа для Sesn1 и изоформа 57.3 кДа для Sesn3.
Структура сестринов
Анализ гипотетической белковой структуры сестринов показал, что белки данного семейства состоят из 16 α-спиралей, большая часть которых образует три аналогичных кластера (α3–8, α9–10 и α11–16), разделенных неструктурированными участками (Budanov et al., 2002). Анализ первичной структуры продемонстрировал не высокую, но статистически значимую гомологию N-концевой области сестринов (для Sesn2 а. о. 100–175, область содержит спирали α6–10) с бактериальными антиоксидантными белками AhpD бактерии M. tuberculosis и карбоксимуконалактон декарбоксилазой бактерии Methanosarcina acetivorans (M. acetivorans) (Budanov et al., 2004). AhpD играет важную роль в окислительно-восстановительной цепи белков туберкулезной палочки, восстанавливая активность окисленного пероксиредоксина AhpC. Данная окислительно-восстановительная цепь участвует в защите бактерий от летального действия АФК, продуцируемых макрофагами организма-хозяина (Bryk et al., 2002). Хотя для каталитической активности белков данной группы требуются два цистеиновых основания, у всех представителей семейства сестринов обнаруживается только один из гомологичных цистеинов, соответствующий Cys125 в Sesn2 (Budanov et al., 2004). Рентгеноструктурный анализ белка Sesn2 выявил два основных функциональных участка молекулы, N-концевой домен (NКД), охватывающий а. о. 66–220 белка Sesn2, и С-концевой домен (СКД), содержащий а. о. 339–480 данного белка.
Третичная структура обоих доменов практически идентична структуре белка AhpD, хотя по первичной структуре гомология между NКД и AhpD не очень значительная, а гомология между СКД и AhpD или NКД не выявляется вообще (Kim H. et al., 2015; Wolfson et al., 2016). NКД и СКД соединены шарнирным сегментом (ШС) с областью гомологии в районе спиралей α9–10 (Budanov et al., 2002). Важным свойством белков Sesn1 и Sesn2 является наличие участка в С-концевом домене, ответственного за связывание с аминокислотой лейцином и ему подобными аминокислотами – изолейцином и валином (рис. 1). Как будет показано ниже, данный домен содействует регуляции сестринами клеточных сигнальных путей под действием меняющихся уровней лейцина и, возможно, других аминокислот (Wolfson et al., 2016). Активность сестринов, по-видимому, также может регулироваться посредством фосфорилирования, поскольку несколько консервативных сайтов фосфорилирования для СК2, PKА, PKС и ULK1, которые могут участвовать в регуляции активности сестринов, были обнаружены у данных белков (Budanov et al., 2002, 2010; Kimball et al., 2016).
Рис. 1.
Структура Sesn2. Белок состоит из двух основных доменов: N-концевой домен (NКД) и С-концевой домен (СКД), разделенных шарнирным сегментом (ШC). NКД содержит оксидоредуктазный сегмент (ОC), участвующий в антиокислительной функции сестринов и содержащий гомологичный и консервативный Cys125. СКД содержит участок связывания лейцина (УСЛ).

АКТИВАЦИЯ ЭКСПРЕССИИ СЕСТРИНОВ В ОТВЕТ НА СТРЕССЫ
Важным свойством всех сестриновых генов млекопитающих является активация их экспрессии в ответ на стресс (Budanov et al., 2010). Экспрессия SESN1 индуцируется двумя основными типами транскрипционных факторов. Одним из ключевым регуляторов гена SESN1 является белок р53, активирующийся различными видами стресса, включая повреждения ДНК в ответ на облучение радиацией и ультрафиолетом (УФ), а также обработку хемотерапевтическими препаратами и в ответ на окислительный стресс, нарушения сборки цитоскелета и биогенеза рибосом (Levine, 1997; Velasco-Miguel et al., 1999; Budanov et al., 2002; Budanov, 2011; Kruiswijk et al., 2015). Белок р53 взаимодействует с последовательностями в 1-м и 2-м интронах гена SESN1, активируя его транскрипцию (Velasco-Miguel et al., 1999; Wei et al., 2006). Другими регуляторами гена SESN1 являются транскрипционные факторы семейства FOXO, FOXO1 и FOXO3, стимулирующие экспрессию регулируемых ими генов в ответ на нехватку ростовых факторов и при окислительном стрессе (рис. 2) (Velasco-Miguel et al., 1999; Tran et al., 2002; Chen et al., 2010).
Рис. 2.
Регуляция транскрипции cестринов млекопитающих (а) и дрозофилы (б). (а) р53 играет ключевую роль в регуляции Sesn1 (Т2) и Sesn2 в ответ на повреждения ДНК, в то время как белки FOXO участвуют в активации Sesn1 и Sesn3 в ответ на окислительный стресс. Sesn2 также активируется транскрипционными факторами NRF2, ATF4, ATF6, XBP1, C/EBPβ, HIF1 и c-Jun в ответ на СЭР и окислительный стресс. (б) Сестрин дрозофилы активируется транскрипционными факторами р53 и FOXO.

Ген SESN2 также активируется в ответ на различные стрессовые воздействия, такие как гипоксия, метаболические нарушения, повреждения ДНК, окислительный стресс и СЭР (Budanov et al., 2002, 2004; Ben-Sahra et al., 2013; Ding et al., 2016; Parmigiani, Budanov, 2016). Генотоксический и окислительный стрессы индуцируют экспрессию гена SESN2 посредством активации р53, связывающегося с участком, расположенным на расстоянии 9600 п.н. ниже от конца транскрипта (Budanov et al., 2002; Wei et al., 2006). Однако многие воздействия, такие как нарушения метаболизма и СЭР, индуцируют SESN2 через механизм, опосредованный активацией транскрипционных факторов ATF4 и NRF2 (Parmigiani, Budanov, 2016). Важно отметить, что индукция SESN2 в ответ на продолжительное аминокислотное голодание также опосредована белком ATF4, трансляция которого, в свою очередь, индуцируется в ответ на активацию киназы GCN2 (Ye et al., 2015). Помимо вышеописанных факторов, транскрипция гена SESN2 также активируется транскрипционными факторами C/EBPβ, HIF1, c-Jun, ATF6, и XBP1 при определенных условиях (Parmigiani, Budanov, 2016; Jegal et al., 2017; Byun et al., 2017). В то время как NRF2, C/EBP1β, HIF1 и c-Jun играют важную роль в защите от окислительного стресса, ATF6, XBP1 и ATF4 ответственны за три основные пути регуляции генной экспрессии при стрессе эндоплазматического ретикулума (Walter, Ron, 2011; Parmigiani, Budanov, 2016). Тем не менее, преимущественный вклад перечисленных транскрипционных факторов в активацию гена SESN2 зависит от тканевой специфичности клеток и других не до конца понятных условий. Некоторые типы стресса индуцируют SESN2 за счет кооперации нескольких транскрипционных факторов. Например, индукция SESN2 при глюкозном голодании опосредована одновременной активацией транскрипционных факторов ATF4 и NRF2 (рис. 2) (Ding et al., 2016).
Основными транскрипционными факторами, участвующими в индукции гена SESN3, являются представители семейства транскрипционных факторов FOXO, FOXO1 и FOXO3 (Nogueira et al., 2008; Chen et al., 2010; Hagenbuchner et al., 2012). АФК активируют белки семейства FOXO (Eijkelenboom, Burgering, 2013), и было показано, что SESN3 индуцируется в ответ на накопление АФК посредством FOXO-зависимого механизма (рис. 2) (Nogueira et al., 2008; Hagenbuchner et al., 2012).
Механизмы активации cестринов высококонсервативны в эволюции. Так, было показано, что экспрессия гена dSesn у мух индуцируется в ответ на окислительный стресс через dFOXO-зависимый механизм. Аналогично клеткам млекопитающих у дрозофил повышенная экспрессия гена р53 приводит к активации экспрессии сестрина (Lee et al., 2010).
mTOR – ОСНОВНАЯ МИШЕНЬ СЕСТРИНОВ
Структура mTOR
Эктопическая экспрессия Sesn2 ингибирует образование колоний клеток человека в культуре (Budanov et al., 2002), что позволило предположить роль белков данного семейства в регуляции сигнальных путей, ответственных за рост и пролиферацию клеток. Ключевым регулятором данных процессов является Ser/Thr-киназа mTOR, принадлежащая к семейству фосфатидилинозитол-3-киназа-подобных киназ, PI3KK (Saxton, Sabatini, 2017). mTOR входит в состав двух различных белковых комплексов, названных mTORC1 и mTORC2 (Saxton, Sabatini, 2017). Данные комплексы, помимо киназы mTOR, содержат белки mLST8, иначе называемый GβL и DEPTOR, но различаются по нескольким уникальным субъединицам (Saxton, Sabatini, 2017). В состав комплекса mTORC1 входят белки Raptor и PRAS40. Более того mTORC1 в присутствии антибиотика-макролида рапамицина связывается с белком FKBP12, который, в свою очередь, подавляет киназную активность mTORC1 (Saxton, Sabatini, 2017). Комплекс mTORC2 содержит белки Rictor, mSIN1 и Protor1/2 (Saxton, Sabatini, 2017). Белки Raptor в комплексе mTORC1 и Rictor в комплексе mTORC2 определяют специфичность связывания данных комплексов с субстратами и, как следствие, определяют функциональные особенности каждого из этих комплексов (Saxton, Sabatini, 2017).
Функции mTORC1 и mTORC2
mTORC1 отвечает за рост клетки, поддерживая многие анаболические процессы, такие как синтез белков, липидов и нуклеотидов, осуществляемые посредством фосфорилирования и активации киназы p70S6K (Saxton, Sabatini, 2017). Кроме того, mTORC1 стимулирует 5'-CAP-зависимую трансляцию, фосфорилируя белок 4EBP1. 4EBP1 в норме ингибирует инициацию белкового синтеза, взаимодействуя с ключевым фактором инициации трансляции – белком eIF-4E, являющимся компонентом пре-инициаторного комплекса eIF-4F (Wullschleger et al., 2006). Фосфорилирование по нескольким сайтам белка 4EBP1 киназой mTORC1 приводит к нарушению взаимодействий между 4EBP1 и eIF-4E и предотвращает подавляющее действие 4EBP1 в отношении eIF-4E. Наконец, mTORC1 исключительно важен для подавления макроаутофагии (далее – просто аутофагии), процесса лизосомного переваривания участков цитоплазмы через их помещение внутрь двухслойных мембранных структур, называемых аутофагосомами, которые затем подвергаются слиянию с лизосомами (Mizushima, 2007). Ингибирование аутофагии киназой mTORC1 опосредуется ингибиторным фосфорилированием киназы ULK1, участвующей в инициации аутофагии, а также путем фосфорилирования некоторых других белков, таких как ATG14L/UVRAG (Saxton, Sabatini, 2017). Другим механизмом супрессии аутофагии киназой mTORC1 является фосфорилирование и удерживание в цитоплазме транс-крипционного фактора ТFEB, фосфорилированная форма которого взаимодействует с белком 14-3-3. ТFEB является важным регулятором экспрессии генов, участвующих в механизмах функционирования лизосом и аутофагосом (Settembre et al., 2013). В свою очередь, киназа mTORC2 играет ключевую роль в контроле метаболизма за счет фосфорилирования киназы АКТ и родственных белков – PKC и SGK (Saxton, Sabatini, 2017). Регулируя активность данных киназ, mTORC2 стимулирует транспорт и метаболизм глюкозы, миграцию клеток, преобразования цитоскелета, ионный транспорт и может ингибировать клеточную смерть (Saxton, Sabatini, 2017).
Регуляция комплексов mTORC1 и mTORC2
Комплексы mTORC1 и mTORC2 регулируются через различные, хотя и связанные между собой, механизмы. Ключевую роль в активации комплекса mTORC1 играет белок Rheb, представитель семейства малых ГТФаз. Rheb взаимодействует с mTORC1 и, будучи связанным с ГТФ, активирует киназную активность данного комплекса (Wullschleger et al., 2006). Поскольку Rheb расположен на лизосомах, транслокация mTORC1 на поверхность лизосом исключительно важна для активации этого комплекса (Saxton, Sabatini, 2017). Активность белка Rheb ингибируется белковым комплексом TSC, который состоит из белков TSC1, TSC2 и TBC1D7 и функционирует в качестве белка-активатора ГТФаз (GAP) для Rheb (Manning, Toker, 2017) (рис. 3). Активность TSC негативно регулируется посредством фосфорилирования киназами AKT и ERK, активирующимися в ответ на инсулин и ростовые факторы (Wullschleger et al., 2006). Ключевым активатором киназы АКТ является липидная киназа PI3K, которая ответственна за выработку фосфатидилинозитол-(3,4,5)-трифосфата (PI3P). PI3P, являющийся компонентом клеточных мембран, связывает и стимулирует киназы PDK1 и mTORC2, которые, в свою очередь, фосфорилируют АКТ по Thr308 и Ser473 соответственно, что требуется для активации данной киназы (Sarbassov et al., 2005; Gan et al., 2011; Manning, Toker, 2017). PI3K активируется инсулином и IGF1 при связывании c соответствующими рецепторами, что вызывает фосфорилирование самих рецепторов и некоторых компонентов рецепторного комплекса по Tyr. Это приводит к связыванию рецептора с белками IRS, которые, в свою очередь, взаимодействует с PI3K, индуцируя синтез PI3P и активацию PI3P-зависимых сигнальных путей (Manning, Toker, 2017). Данный процесс негативно регулируется фосфатазой PTEN, которая гидролизует PI3Р и ингибирует сигнальный каскад, опосредованный киназой PI3K.
Рис. 3.
Регуляция киназы mTORС1. Инсулин и IGF1 взаимодействуют с рецептором I/IGFR на плазматической мембране, что при участии фактора IRS1 приводит к активации киназ PI3K и АКТ. АКТ фосфорилирует и ингибирует TSC, что стимулирует Rheb и приводит к активации mTORC1. Параллельно этому аминокислоты (АК) активируют mTORC1 через активацию GATOR2 и подавление GATOR1, которые взаимодействуют при участии белкового комплекса KINGSTOR. Это приводит к активации белков RagA/B и транслокации mTORC1 к лизосомам, где активность RagA/B и mTORC1 поддерживается комплексом Ragulator. Активация mTORC1 приводит к активации анаболических процессов, подавлению аутофагии и увеличению размера клеток.
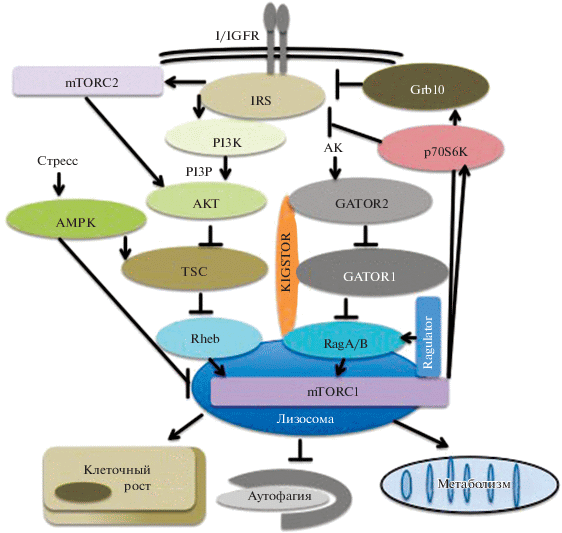
Несмотря на ключевой вклад, который инсулин и ростовые факторы вносят в регуляцию mTORC1, аминокислоты играют доминантную роль в активации mTORC1 посредством механизма, опосредованного локализацией mTORC1 на лизосомах. За лизосомную локализацию mTORC1 отвечают белки Rag, представители семейства малых ГТФаз. Данные белки работают в виде гетеродимеров RagA/B:RagC/D, где активная форма RagA/B связана с ГТФ, а активная форма RagC/D связана с ГДФ (Sancak et al., 2008).
Активность белков RagA/B позитивно регулируется белковым комплексом Ragulator, являющимся фактором ГДФ–ГТФ обмена (GEF), и негативно регулируется белковым комплексом GATOR1. GATOR1, состоящий из белков Nprl2, Nprl3 и DEPDC5, функционирует как GAP для RagA/B (Bar-Peled et al., 2013). Комплекс GATOR1, в свою очередь, ингибируется посредством белок-белковых взаимодействий белковым комплексом GATOR2, состоящим из белков Mios, WDR24, WDR59, Seh1L и Sec13 (Bar-Peled et al., 2013). В то время, как локализация GATOR2 в клетке неизвестна, GATOR1 ассоциирован c белковым комплексом KICSTOR, который расположен на лизосомах, где он осуществляет контроль активности mTORC1 в ответ на аминокислоты и некоторые другие питательные вещества (рис. 3) (Bar-Peled et al., 2013; Peng et al., 2017; Wolfson et al., 2017).
Ингибирование активности mTORC1 идет по механизму обратной связи. Так, mTORC1 активирует киназу p70S6K, которая, в свою очередь, фосфорилирует белок IRS1 по нескольким Ser/Thr основаниям, индуцируя его деградацию и, как следствие, блокирует активацию сигнального пути PI3K–AKT. Другой механизм негативной обратной связи, осуществляемый киназой mTORC1, опосредован фосфорилированием белка Grb10 и является сигналом к его стабилизации и ингибированию сигнала от I/IGFR к AKT (Saxton, Sabatini, 2017).
Ингибирование mTORC1 сестринами опосредовано двумя параллельными механизмами (Parmigiani, Budanov, 2016). Так, было показано, что сестрины препятствуют связыванию ГТФ белком Rheb, предотвращая активацию mTORC1 (рис. 4). Данный механизм опосредован киназой AMPK, состоящей из трех субъединиц: α, β и γ (Budanov, Karin, 2008). Сестрин может работать как каркас, способствующий образованию комплекса между AMPK и ее активатором – киназой LKB1, ответственной за фосфорилирование каталитической α-субъединицы AMPK в ответ на накопление в клетке AМФ и некоторые другие воздействия (Morrison et al., 2015). В последующих работах было показано, что сестрины напрямую взаимодействуют с комплексом GATOR2 и ослабляют ингибиторование киназы mTORC1 комплексом GATOR1 (рис. 4) (Parmigiani et al., 2014; Chantranupong et al., 2014). Следствием ингибирования GATOR1 сестринами является предотвращение связывания комплекса mTORC1 с лизосомами и его активация белком Rheb. Комплексы RagA/B:RagC/D опосредуют ингибирование mTORC1 сестринами, хотя действие сестринов не приводит к изменению ГТФазной активности белков Rag, что могло бы объяснить их ингибирование сестринами (Parmigiani et al., 2014; Chantranupong et al., 2014; Peng et al., 2014). Хотя в одной из работ было заявлено, что сестрины могут напрямую взаимодействовать с комплексами Rag (Peng et al., 2014), по-видимому, это взаимодействвие опосредовано комплексами GATOR1 и GATOR2 (Budanov, 2015).
Рис. 4.
Регуляция киназы mTOR сестринами. Сестрины ингибируют mTORC1 посредством 2-х параллельных механизмов: через стимуляцию киназы AMPK и активацию комплекса TSC, а также через взаимодействие с комплексом GATOR2. Сестрины также активируют mTORC2, что приводит к стимулированию киназы АКТ, ключевого регулятора метаболизма и ингибитора клеточной смерти.

Активность киназы mTORC1 зависит от наличия в среде аминокислот, таких как лейцин, а представители семейства сестринов Sesn1 и Sesn2 являются сенсорами лейцина и других родственных лейцину аминокислот (Wolfson et al., 2016; Saxton et al., 2016). Лейцин взаимодействует с участком связывания лейцина (УСЛ) в молекуле сестрина. УСЛ образует карман, который окружает молекулу лейцина. Связывание лейцина с УСЛ изменяет конформацию молекулы cестрина, переводя ее в позицию так называемого закрытого замка (lid-latch), что препятствует взаимодействию сестрина и комплекса GATOR2 (Saxton et al., 2016). Хотя УСЛ находится в СКД, оба NКД и СКД белка Sesn2 требуются для взаимодействия между молекулой сестрина и комплексом GATOR2 (Parmigiani et al., 2014; Saxton et al., 2016).
Ингибируя mTORC1, сестрины в то же время могут стимулировать киназу АКТ, которая является основным активатором mTORC1. Сестрины регулируют mTORC1 посредством нескольких механизмов (Parmigiani, Budanov, 2016). Во-первых, сестрины могут осуществлять активацию АКТ, ингибируя негативную петлю обратной связи от mTORC1, что опосредуется ингибированием p70S6K, стабилизацией IRS1 и последующей активацией киназ PDK1 и mTORC2, ответственных за активацию АКТ (Lee et al., 2012; Manning, Toker, 2017). Во-вторых, при ультрафиолетовом облучении Sesn2 способствует фосфорилированию AKT, задерживая перемещение липидной фосфатазы PTEN к цитоплазматической мембране и, таким образом, предотвращая подавление передачи сигнала в пути PI3K–АКТ белком PTEN (Zhao et al., 2014). В-третьих, было продемонстрировано, что белки Sesn2 и Sesn3 способны напрямую взаимодействовать с киназой mTORC2, поддерживая фосфорилирование АКТ киназой mTORC2 и предотвращая ингибирование белка Rictor, ключевого компонента mTORC2, белком 14-3-3 (рис. 4) (Tao et al., 2015; Byun et al., 2017).
АНТИОКСИДАНТНАЯ И РЕГУЛЯТОРНАЯ ФУНКЦИИ СЕСТРИНОВ
Антиоксидантная функция
Анализ первичной структуры сестринов показывает их родство с бактериальным антиоксидантным белком AhpD, участвующим в восстановлении окисленных форм пероксиредоксина AhpC, чей каталитический цистеин окисляется во время каталитического цикла до состояния Cys-SOH (Budanov et al., 2004). В клетках эукариот восстановление окисленного Cys осуществляется белком тиоредоксином, однако, в отличие от прокариотических пероксиредоксинов, эукариотические гомологи часто инактивируются путем гиперокисления их каталитического цистеина до сульфинильной Cys-SO2 формы (Wood et al., 2003). Сестрины участвуют в процессе восстановления гиперокисленных форм пероксиредоксинов, хотя, по-видимому, сами не обладают сульфинилредуктазной активностью и действуют через механизм, опосредованный активацией экспрессии сульфиредоксина (Budanov et al., 2004; Woo et al., 2009). Помимо роли сестринов в регуляции экспрессии антиоксидантных генов, N-концевой домен сестринов обладает активностью алкилпероксидазы. Было показано, что высококонсервативный Cys125 в Sesn2 необходим для этого процесса, хотя клеточный субстрат, восстанавливаемый сестринами, не был охарактеризован (рис. 5) (Kim H. et al., 2015).
Рис. 5.
Роль сестринов в регуляции АФК. Сестрины ингибируют накопление внутриклеточных АФК, способствуя прямой деградации некоторых форм АФК, а также поддерживая активность пероксиредоксинов, контролируя аутофагию и нормализуя функционирование митохондрий. В некоторых случаях сестрины супрессируют АФК, подавляя экспрессию Nox4.

Регуляция аутофагии
Сестрины также препятствуют накоплению АФК в клетках и предотвращают окислительный стресс посредством поддержания аутофагии. Аутофагия исключительно важна для снабжения клетки питательными веществами в условиях голодания и контроля за целостностью и функциональностью органелл (Mizushima, 2007). Повреждения в работе органелл, таких как митохондрия и пероксисомы могут приводить к накоплению АФК, которые, в свою очередь, повреждают клеточные структуры, способствуя дальнейшему увеличению уровней АФК и окислительному стрессу (Budanov, 2011; Green et al., 2011). Недостаточная активация аутофагии является причиной нарушения нормальной работы клеток и может привести к старению клеток (senescence) или клеточной гибели. Таким образом, аутофагия контролирует целостность и работоспособность органелл, предотвращая накопление повреждений в клетке и окислительный стресс (Green et al., 2011). Сигнальный путь AMPK–mTORC1 исключительно важен для регуляции аутофагии. В то время как AMPK активирует, mTORC1 подавляет аутофагию через механизм, опосредованный фосфорилированием киназы ULK1 по сайтам, специфическим для каждой из этих киназ (Saxton, Sabatini, 2017). Сестрины участвуют в регуляции обеих киназ и могут активировать аутофагию в ответ на различные стрессы, включая ДНК-повреждения и CЭР (рис. 5) (Li et al., 2012; Saveljeva et al., 2016). Поскольку повреждения митохондрий приводят к значительному накоплению АФК, в клетке существуют механизмы, контролирующие целостность митохондрий. Удаление поврежденных митохондрий с помощью аутофагии, которая в данном случае носит название митофагии, исключительно важна для защиты клеток от окислительного стресса и нарушений метаболизма (Scheibye-Knudsen et al., 2015). Sesn2 играет важную роль в регуляции митофагии при воспалении и сепсисе. Так, было установлено, что Sesn2 контролирует узнавание и доставку поврежденных фрагментов митохондрий в аутофагосомы, взаимодействуя с белком р62/SQSTM1, который является переносчиком субстрата для переваривания в аутофагосомах (Kim et al., 2016).
Регуляция специфической деградации белков
Помимо регуляции общего процесса аутофагии, Sesn2 также контролирует специфическое расщепление белков в лизосомах. Так, антиоксидантная активность сестринов опосредована активацией транскрипционного фактора NRF2. Посредством взаимодействия с белками KEAP1, p62 и RBX1 сестрины вызывают деградацию KEAP1, что приводит к последующей активации белка NRF2 (Bae et al., 2013). NRF2 ингибирует накопление АФК путем индукции экспрессии антиооксидантных генов, таких как сульфиредоксин, который восстанавливает переокисленные формы пероксиредоксинов (Bae et al., 2013). Помимо регуляции белковой стабильности NRF2, Sesn2 также может связывается с белком NRF2, поддерживая его транскрипционную активность в ядре (Tomasovic et al., 2015). Важно отметить, что регуляция стабильности белков специфична для определенного типа клеток. Например, было продемонстрировано, что Sesn2 способствует перевариванию рецептора ростового фактора PDGF-β в клетках глиобластомы, потенциально играя важную роль в контроле сигнальных путей от PDGFR-β, которые участвуют в регуляции роста клетки и пролиферации (Liu et al., 2011).
В некоторых типах клеток, например, в клетках почечных клубочков, Sesn2 может осуществлять свою антиоксидантную функцию через понижение уровня экспрессии Nox4, поддерживаемого активацией киназы АМРК (Eid et al., 2013), однако играет ли данный механизм какую-либо роль в регуляции АФК в других типах клеток на сегодняшний день не установлено (рис. 5).
Регуляция метаболизма и клеточной смерти
Cестрины поддерживают внутриклеточный гомеостаз, регулируя метаболизм посредством киназ AMPK и mTOR, а также других, не до конца выясненных, механизмов. Так, было продемонстрировано, что инактивация Sesn2 снижает производство АТФ (Ben-Sahra et al., 2013; Seo et al., 2015; Ding et al., 2016), вызывая замедление как окислительного фосфорилирования, так и гликолиза (Ding et al., 2016). Таким образом, активация сестринов в ответ на стресс, вероятно, играет важную роль в поддержании производства энергии. Повреждения ДНК стимулируют производство АТФ, поддерживая работу цепи окислительного фосфорилирования, что может быть необходимо для процессов репарации (Brace et al., 2016). Сестрины могут участвовать в этом процессе, поскольку данные белки активируются в ответ на повреждения ДНК (Budanov, 2011). Нарушения метаболизма при сбоях в работе митохондрий, подавлении гликолиза или при недостаточном снабжении клетки необходимыми питательными веществами, такими как глюкоза или аминокислоты, приводят к СЭР и индукции Sesn2, вызванными активацией СЭР-зависимых транскрипционных факторов ATF4 и NRF2 (Ye et al., 2015; Seo et al., 2015; Ding et al., 2016; Garaeva et al., 2016). Sesn2, в свою очередь, поддерживает жизнеспособность клеток в условиях ишемии, а также глюкозного и аминокислотного голодания (Budanov et al., 2002; Ye et al., 2015; Seo et al., 2015; Ding et al., 2016; Byun et al., 2017). Также, Sesn2 защищает от клеточной смерти при обработке ингибитором окислительного фосфорилирования ротеноном или ингибитором гликолиза 2-дезоксиглюкозой (2ДГ) (Ben-Sahra et al., 2013; Hou et al., 2015). Помимо этого, Sesn2 защищает от клеточной смерти при индукции СЭР в ответ на обработку тапсигаргином, бортезомибом и нелфинавиром (Bruning et al., 2013; Saveljeva et al., 2016).
Хотя вклад сестринов в защиту от клеточной смерти во многих случаях связан с регуляцией пути AMPК–mTORC1, что приводит к ингибированию биосинтетических процессов в клетке и поддерживает катаболические процессы, направленные на выработку энергии и восстановление клеточных структур, при глюкозном голодании Sesn2 защищает от некротической гибели через механизм, не связанный с регуляцией киназ AMPK и mTOR, но посредством контроля работы митохондрии и поддержания митохондриального дыхания в условиях стресса (Ding et al., 2016).
Несмотря на защитную роль сестринов в условиях недостатка питательных веществ или при других видах стресса, Sesn2 также поддерживает клеточную смерть при определенных условиях. В частности, Sesn2 стимулирует клеточную смерть при генотоксическом стрессе (Budanov et al., 2002; Sanli et al., 2012), регулируя экспрессию и активность киназы AMPK (Sanli et al., 2012). Sesn2 также поддерживает клеточную смерть в ответ на активацию рецепторов смерти цитокинами, способствуя лизосомной деградации антиапоптических белков семейства IAP, таких как IAP1, IAP2 и XIAP (Ding et al., 2015).
СЕСТРИНЫ В СТАРЕНИИ И ЗАЩИТЕ ОТ ЗАБОЛЕВАНИЙ
Сестрины – супрессоры старения
Старение – процесс, при котором в клетках различных тканей организма накапливаются повреждения, приводящие к функциональным нарушениям в работе органов и, как следствие, к ослаблению жизнеспособности организма и его гибели. Старение тесно связано с развитием различных возрастных патологий, таких как рак, диабет 2-го типа, а также сердечно-сосудистые и нейродегенеративные заболевания (Lopez-Otin et al., 2013). Киназа mTORC1 играет ключевую роль в регуляции старения, поскольку ее генетическое или фармакологическое ингибирование увеличивает продолжительность жизни большинства изученных эукариотических организмов, включая дрожжей, плоских червей, мух и мышей (Cornu et al., 2013; Saxton, Sabatini, 2017). Ингибирование активности mTORC1 также приводит к увеличению продолжительности жизненного интервала, свободного от различных хронических заболеваний (ИСРХЗ) (Johnson et al., 2013). Функция mTORC1 в контроле старения не до конца ясна, однако регуляция аутофагии, сворачивания белков и метаболизма киназой mTORC1, вероятно, важны для данной активности (Johnson et al., 2013). Хотя продолжительный и интенсивный стресс может ускорять процесс старения, слабые стрессовые воздействия могут также способствовать увеличению продолжительности жизни в процессе, названном гормезис, за счет различных путей ответа на стресс, приводящих к активации аутофагии, что может быть опосредовано индукцией сестринов (Kourtis, Tavernarakis, 2011).
Сестрины, будучи ингибиторами mTORC1 и стресс-респонсивными белками, участвуют в контроле старения. На модели C. elegans было показано, что эктопическая экспрессия сестрина способствует увеличению продолжительности жизни, а инактивация эндогенного сестрина – к ее укорочению (Yang et al., 2013). Сестрин также защищает организм червя от различных стрессовых воздействий, включая нагревание, обработку перекисью водорода и воздействие тяжелых металлов (Yang et al., 2013). Инактивация сестрина у D. melanogaster не вызывает уменьшение продолжительности жизни, но укорачивает ИСРХЗ (Lee et al., 2010). Так, мухи с нокаутом по сестрину в раннем возрасте (2–3-х нед.) приобретают черты стареющих особей дикого типа. Также у мух, дефицитных по сестрину, наблюдается дегенерация мышц сердца и груди, сопровождающаяся окислительным стрессом и нарушением метаболизма липидов и углеводов (Lee et al., 2010, 2012). Мухи, дефицитные по сестрину, по фенотипическим характеристикам похожи на мух, лишенных гена аутофагии dATG1, показывая наличие потенциальной связи между индукцией экспрессии сестрина, активацией аутофагии и защитой клеток организма от повреждений, что может замедлить процесс старения (Lee et al., 2010).
Сестрины млекопитающих также защищают от различных заболеваний, связанных со старением и стрессом (рис. 6). Такие болезни, как диабет 2-го типа, сердечно-сосудистые заболевания, ишемия и онкологические заболевания являются основными причинами смерти. Преждевременная смерть от подобных заболеваний вызвана нездоровым и малоподвижным образом жизни в современном мире, и сестрины, активируемые в ответ на физические тренировки, антидиабетические препараты и пищевые добавки, такие как метформин и ресвератрол, могут замедлять развитие возрастных заболеваний (Lee et al., 2013; Parmigiani, Budanov, 2016).
Рис. 6.
Роль сестринов в развитии различных заболеваний. В то время как сестрины защищают от многих болезней, включая метаболический синдром, сердечно-сосудистые и нейродегенеративные заболевания, глухоту и сепсис, они поддерживают эмфизему и способствуют старению клеток иммунной системы. Также сестрины могут супрессировать канцерогенез в некоторых тканях, но поддерживать опухолевый рост у определенных типов рака, таких как злокачественные заболевания кожи.

Метаболический синдром и диабет
Метаболический синдром – группа заболеваний, вызванных нарушением обмена веществ и характеризующихся ожирением, устойчивостью к инсулину и нарушением толерантности к глюкозе, что приводит к развитию диабета 2-го типа (Lee et al., 2013). Инсулин, вырабатываемый клетками поджелудочной железы в ответ на повышение концентрации глюкозы в крови, понижает уровни глюкозы посредством стимулирования потребления глюкозы тканями (особенно такими, как мышцы и жировая ткань) и супрессии производства глюкозы в печени. Одним из ключевых процессов, приводящих к устойчивости к инсулину, является пониженная активации киназы АКТ в клетках в ответ на инсулин (Lee et al., 2013). Причиной данного нарушения является повышенная активность киназы mTORC1, которая поддерживает высокий уровень активности киназы p70S6K (Lee et al., 2013). Активация p70S6K приводит к деградации белка IRS1, необходимого для передачи сигнала от инсулинового рецептора к PI3K с последующей активацией киназ PDK1 и mTORC2, ответственных за фосфорилирование АКТ. Ингибирование обеих киназ ослабляет активность АКТ, следствием чего является подавление внутриклеточного транспорта глюкозы клетками различных тканей и повышенный уровень продукции глюкозы в печени, несмотря на высокий уровень инсулина в крови (Lee et al., 2013; Manning, Toker, 2017).
Среди различных членов семейства сестринов только экспрессия Sesn2 повышена в печени мышей с ожирением, что, по-видимому, опосредовано активацией СЭР, вызванного увеличением концентрации насыщенных жирных кислот в данном органе (Lee et al., 2012; Park et al., 2014). У человека Sesn2 также препятствует развитию инсулиновой устойчивости и неалкогольной жировой болезни печени (НЖБП), характеристикой которой является накопление жировых капель в печени (рис. 6) (Lee et al., 2012). Повышенная экспрессия Sesn2 стимулирует активацию AMPK и ингибирование mTORC1, поддерживая высокую активность АКТ. Данный процесс в результате способствует супрессии глюконеогенеза в печени и, как следствие, понижению уровня сахара в крови (Lee et al., 2012). На модели ожирения у мышей с нокаутом по Sesn2 продемонстрировано, что активация АКТ в ответ на инсулин значительно ослаблена, что является причиной инсулиновой устойчивости. Фенотип данных мышей выражается в неэффективном подавлении глюконеогенеза в печени мышей, что приводит к повышенному производству глюкозы (Lee et al., 2012). Белок Sesn3 также препятствует развитию инсулиновой устойчивости у мышей, лишенных каких-либо признаков ожирения, за счет поддержания активности AKT (Lee et al., 2012; Tao et al., 2015). Механизм активации АКТ сестринами опосредован ингибированием сигнального пути mTORC1–p70S6K, хотя также было показано, что сестрины способны взаимодействовать с киназой mTORC2, повышая ее активность (Tao et al., 2015; Byun et al., 2017).
Сердечно-сосудистые заболевания
Распространенными заболеваниями преклонного возраста являются сердечно-сосудистые, где Sesn2 также играет защитную функцию (рис. 6). Было показано, что мыши, лишенные Sesn2, обладают значительно более выраженной чувствительностью к инфаркту миокарда, вызванному ишемией с последующей реоксигенацией сердца, по сравнению с мышами дикого типа (Morrison et al., 2015). В ответ на ишемию, Sesn2 способствует активации киназы AMPK, что приводит к защите сердечной мышцы от клеточной гибели путем некроза (Morrison et al., 2015). Экспрессия Sesn2 в сердечной мышце мышей с возрастом снижается, увеличивая восприимчивость к инфаркту миокарда у стареющих особей (Quan et al., 2017). Также, Sesn2 защищает кровеносные сосуды и предотвращает развитие атеросклероза посредством механизма, опосредованного активацией AMPK в эндотелиальных клетках (Hwang et al., 2017). Хронические воспаления являются частой причиной атеросклероза и инфаркта, и было показано, что Sesn2 может подавлять развитие данных заболеваний посредством ингибирования сигнальных путей, которые регулируют воспалительные процессы в макрофагах и других клетках иммунной системы (Yang et al., 2015; Kim M.J. et al., 2015; Kim et al., 2016; Yang et al., 2017). Потенциальным механизмом супрессии воспаления белком Sesn2 является активация митофагии, препятствующей накоплению повреждений в митохондриях, которые, в свою очередь, могут приводить к активации провоспалительных сигнальных путей (Kim et al., 2016).
Нейродегенеративные заболевания
Sesn2 может играть защитную роль при нейродегенеративных заболеваниях, включая болезни Альцгеймера (БА) и Паркинсона (БП), которые характеризуются накоплением белковых включений и окислительным стрессом в клетках нейрональной ткани, а также гибелью нейронов (рис. 6) (Majd et al., 2015). В результате это приводит к нейродегенерации и сопутствующим нарушениям функций нервной системы. Было продемонстрировано, что бета-амилоидные пептиды длиной 36–43 а. о., накапливающиеся в нейрональной ткани больных БА, способны индуцировать экспрессию Sesn2, который защищает клетки от клеточной смерти через активацию аутофагии (Kim et al., 2003; Chen et al., 2014). Sesn2 также поддерживает гомеостаз в клетках, способствуя активации транскрипционного фактора TFEB, ответственного за экспрессию группы генов CLEAR. Гены CLEAR играют важную роль в аутофагии и лизосомальном переваривании белковых агрегатов, связанных с развитием БА (Reddy et al., 2016). Особенностью БП является накопление белка синуклеина в нейронах центральной нервной системы (Majd et al., 2015). Повышенная экспрессия Sesn2 наблюдается в среднем мозге пациентов, страдающих БП, и было продемонстрировано, что Sesn2 препятствует накоплению синуклеина в нейрональных клетках (Hou et al., 2015). В мышиной модели БП было также показано, что экспрессия Sesn2 снижает нейротоксичность препарата 1-метил-4-фенилпиридиниума (МРР+), используемого при моделировании БП в мышиных моделях и культурах клеток (Zhou et al., 2013). Сестрины защищают нейроны и клетки нейронального происхождения от окислительного стресса, приводящего к клеточной гибели, что также может объяснять защитный вклад сестринов против патологий данного типа (Papadia et al., 2008; Doonan et al., 2009; Hagenbuchner et al., 2012; Kallenborn-Gerhardt et al., 2013). Помимо защитной функции от нейродегенеративных заболеваний, было также продемонстрировано, что наличие определенных полиморфных аллелей гена SESN3 коррелирует с развитием эпилепсии, и Sesn3 участвует в защите от эпилепсии через подавление воспалительных процессов в мозге (Johnson et al., 2015).
Раковые заболевания
Будучи мишенями р53 и негативными регуляторами киназы mTORC1, активность которой часто повышена при различных видах рака, сестрины являются потенциальными опухолевыми супрессорами (рис. 6). Генетический локус 1p35, содержащий ген SESN2, часто делетирован при нейробластоме, раке толстой кишки, печени и раке груди (Leister et al., 1990; White et al., 1993; Yeh et al., 1994; Nagai et al., 1995), а локус 6q21, содержащий SESN1, часто делетирован при лейкемиях и лимфомах, раке шейки матки, почки, желудка и поджелудочной железы (Abe et al., 1999; Hatano et al., 2001; Carvalho et al., 2002; Lehmann et al., 2008; Thelander et al., 2008). Также было показано, что уровень экспрессии Sesn2 обратно коррелирует с тяжестью болезни при раке легкого и толстого кишечника (Wei et al., 2015; Chen et al., 2016). Интересно, что повышенный уровень белка Sesn2 обнаруживается в плевральной жидкости некоторых пациентов с раком легкого, что, возможно, является следствием гибели клеток с повышенной экспрессией Sesn2 (Tsilioni et al., 2016). Инактивация Sesn2 способствует злокачественной трансформации клеток MEF (Budanov, Karin, 2008) и поддерживает рост привитых опухолей рака легкого (Sablina et al., 2005) и рака толстого кишечника (Ro et al., 2016). Sesn1 супрессирует развитие фолликулярных лимфом (ФЛ), в которых часто наблюдаются хромосомные абберации в области 6q21 и активирующие мутации в гене EZH2, кодирующем компонент гистоновой лизилметилтрансферазы, чья экспрессия приводит к подавлению транскрипции SESN1 (Oricchio et al., 2017). Промотор гена SESN3 метилирован в 20% эндометриальных видов рака матки (Zighelboim et al., 2007), что свидетельствует о возможной роли этого гена в супрессии данного рака. Также возможная роль сестринов в опухолевой супрессии подтверждается тем фактом, что мутантный белок Ras, участвующий в развитии многих типов рака, негативно регулирует экспрессию генов SESN1 и SESN3 (Kopnin et al., 2007).
Однако в некоторых видах рака сестрины могут играть проонкогенную роль, предотвращая гибель поврежденных клеток. Например, в меланомах и карциномах кожи Sesn2 способствует выживанию клеток в условиях стресса, вызванного ультрафиолетом и химиотерапевтическим препаратом 5-фторурацилом, способствуя росту опухолей. Данная активность сестринов объясняет тот факт, что экспрессия Sesn2 повышена во многих злокачественных опухолях кожи (Zhao et al., 2014; Zhao et al., 2017). Потенциальный механизм действия Sesn2 связан с активацией киназы АКТ, ответственной за защиту от клеточной смерти и контроль роста и пролиферации клеток (Zhao et al., 2014; Manning, Toker, 2017).
Хроническая обструктивная болезнь легких
Хотя Sesn2 играет важную защитную роль при многих заболеваниях, в условиях хронического стресса активация Sesn2 может негативно влиять на течение болезни, препятствуя тканевой регенерации. Было показано, что экспрессия Sesn2 повышена у хронических курильщиков и пациентов, страдающих эмфиземой (Heidler et al., 2013). В экспериментальных мышиных моделях эмфиземы инактивация Sesn2 препятствовала развитию этого состояния (рис. 6) (Wempe et al., 2010; Heidler et al., 2013). Одним из выраженных фенотипов эмфиземы является повышенная гибель эпителиальных клеток дыхательных путей и альвеол, что связано со снижением активности киназы mTORC1 и неконтролируемой митофагией, являющейся причиной некротической гибели клеток и изменений тканевой структуры (Yoshida et al., 2010; Mizumura et al., 2014). Продолжительная активация Sesn2 в процессе развитии данной болезни может способствовать клеточной гибели посредством подавления mTORC1 и активации аутофагии/митофагии.
Глухота
Сестрины также ответственны за поддержание слуха. Так, было показано, что уровень экспрессии Sesn2 падает в клетках улитки уха у стареющих мышей, и у мышей, нокаутных по Sesn2, наблюдается повышенная дегенерация волосковых сенсорных клеток внутреннего уха, приводящая к глухоте (Zhang et al., 2017). Инактивация Sesn2 способствует воспалительным процессам и клеточной гибели в улитке уха. В работе из другой лаборатории было также продемонстрировано, что Sesn2 способствует выживанию клеток улитки уха в ответ на действие антибиотика гентамицина, обладающего ототоксическим эффектом (рис. 6) (Ebnoether et al., 2017).
Роль сестринов в иммунном ответе
Как было показано в нескольких работах, сестрины участвуют в иммунном ответе и негативно регулируют воспалительные процессы в различных тканях (Yang et al., 2015; Johnson et al., 2015; Ro et al., 2016; Hwang et al., 2017; Yang et al., 2017; Zhang et al., 2017). Также, на нескольких мышиных моделях было показано, что Sesn2 играет защитную роль от сепсиса (Kim et al., 2016). Однако, в соответствии с данными недавней работы, сестрины могут способствовать износу иммунной системы, ослабляя иммунитет (Lanna et al., 2017). Экспрессия сестринов повышена в Т-клетках пожилых людей и стареющих мышей, и сестрины могут способствовать клеточному старению компонентов иммунной системы, что приводит к ослаблению иммунного ответа при вакцинации в мышиных моделях (Lanna et al., 2017). Объяснением данному феномену может служить вклад сестринов в активацию белковых киназ семейства MAPK, которые, в свою очередь, способствуют процессам клеточного старения (sene-scence) и приводят к снижению пролиферации и активации Т-клеток в ответ на стимуляцию антигеном (рис. 6) (Lanna et al., 2017).
ЗАКЛЮЧЕНИЕ
Стресс активирует экспрессию множества генов с различной активностью, направленной как на поддержание жизнеспособности и функции клеток, так и на индукцию клеточной гибели и/или клеточного старения. Таким образом, конечный результат ответа на стресс зависит от наличия или отсутствия определенных регуляторов физиологических процессов в клетке, способных направлять развитие событий по тому или иному сценарию, либо позволяющему организму преодолевать ситуацию стресса, либо приводящему к гибели организма. Возможность модулировать экспрессию и активность белков, реагирующих на стрессы, имеет исключительную важность для предотвращения и лечения патологий.
В частности, модуляторами ответа клеток на стресс являются белки сестрины, регулирующие метаболизм и клеточную смерть, причем результат их активации зависит от многих факторов, таких как тип клеток, тип стресса и наличие или отсутствие тех или иных дополнительных модуляторов. Знание механизмов регуляции экспрессии сестринов и роли сестринов в контроле ответа на стресс в физиологических и патофизиологических процессах позволит разработать подходы к регуляции их экспрессии и функции для лечения различных заболеваний. Большинство болезней человека и проблемы старения связаны с неспособностью тканей и органов своевременно и контролируемо отвечать на стресс, что имеет разрушительные последствия для организма. Это приводит к хроническим заболеваниям, таким как сердечно-сосудистые и нейродегенеративные патологии, диабет, рак и многие другие, тесно связанные с процессом старения организма (Dalina et al., 2018).
При этом сестрины представляют удобную мишень для лекарств, направленных против диабета, инфаркта и рака, поскольку их экспрессия может быть активирована многими известными фармакологическими препаратами, включая метформин и ресвератрол (Budanov et al., 2010; Parmigiani, Budanov, 2016). Более того, активность сестринов может модулироваться малыми молекулами, которые способны взаимодействовать с участком связывания лейцина в молекуле сестрина и вызывать структурные и функциональные изменения в белке (Dalina et al., 2018).
Иными словами, сестрины имеют уникальные функции в разных клетках и тканях организма при различных воздействиях и участвуют во множестве жизненно важных процессов, требующих дальнейшего изучения.
Список литературы
Abe T., Makino N., Furukawa T. et al. Identification of three commonly deleted regions on chromosome arm 6q in human pancreatic cancer // Gen. Chrom. Cancer. 1999. V. 25 (1). P. 60–64.
Andrysik Z., Galbraith M.D., Guarnieri A.L. et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity // Gen. Res. 2017. V. 27 (10). P. 1645–1657.
Bae S.H., Sung S.H., Oh S.Y. et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage // Cell Metab. 2013. V. 17 (1). P. 73–84.
Bar-Peled L., Chantranupong L., Cherniack A.D. et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1 // Science. 2013. V. 340 (6136). P. 1100–1106.
Ben-Sahra I., Dirat B., K Laurent K. et al., Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death // Cell Death Differ. 2013. V. 20 (4). P. 611–619.
Brace L.E., Vose S.C., Stanya K. et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage // N.P.J. Aging Mech. Dis. 2016. V. 2. P. 16022.
Bruning A., Rahmeh M., Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation // Mol. Oncol. 2013. V. 7 (6). P. 1012–1018.
Bryk R., Lima C. D., Erdjument-Bromage H. et al. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein // Science. 2002. V. 295 (5557). P. 1073–1077.
Budanov A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling // Antioxid. Redox. Signal. 2011. V. 15 (6). P. 1679–1690.
Budanov A.V. Sestrins link tumor suppressors with the AMPK–mTOR signaling network // Protein phosphorylation in human health / Ed. C. Huang. Nvi Sad, Croatia: InTech, 2012. 51–96.
Budanov A.V. SESTRINs regulate mTORC1 via RRAGs: the riddle of GATOR // Mol. Cell. Oncol. 2015. V. 2 (3). P. e997113.
Budanov A.V., Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling // Cell. 2008. V. 134 (3). P. 451–460.
Budanov A.V., Lee J.H., M. Karin M. Stressin’ sestrins take an aging fight // EMBO Mol. Med. 2010. V. 2 (10). P. 388–400.
Budanov A.V., Shoshani T., Faerman A. et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability // Oncogene. 2002. V. 21 (39). P. 6017–6031.
Budanov A.V., Sablina A.A., Feinstein E. et al. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD // Science. 2004. V. 304 (5670). P. 596–600.
Byun J.K., Choi Y.-K., Kim J.-H. et al. A positive feedback loop between Sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells // Cell Rep. 2017. V. 20 (3). P. 586–599.
Carvalho B., Seruca R., Buys C.H.C.M., Kok K. Novel expressed sequences obtained by means of a suppression subtractive hybridisation analysis from the 6q21 region that is frequently deleted in gastric cancer // Eur. J. Cancer. 2002. V. 38 (8). P. 1126–1132.
Chantranupong L., Wolfson R.L., Orozco J.M. et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1 // Cell Rep. 2014. V. 9 (1). P. 1–8.
Chen C.C., Jeon S.M., Bhaskar P.T. et al. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor // Dev. Cell. 2010. V. 18 (4). P. 592–604.
Chen K.B., Xuan Y., Shi W.-J. et al. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer // Am. J. Transl. Res. 2016. V. 8 (4). P. 1903–1909.
Chen Y.-S., Chen S.-D., Wu C.-L. et al. Induction of sestrin2 as an endogenous protective mechanism against amyloid beta-peptide neurotoxicity in primary cortical culture // Exp. Neurol. 2014. V. 253. P. 63–71.
Cornu M., Albert V., Hall M.N. mTOR in aging, metabolism, and cancer // Curr. Opin. Genet. Dev. 2013. V. 23 (1). P. 53–62.
Cuadrado A., Rojo A.I., Wells G. et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases // Nat. Rev. Drug. Discov. 2019. V. 18 (4). P. 295–317.
Dalina A.A., Kovaleva I.E., Budanov A.V. Sestrins are gatekeepers in the way from stress to aging and disease // Mol. Biol. (Mosk.). 2018. V. 52 (6). P. 948–962.
D’Autreaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis // Nat. Rev. Mol. Cell Biol. 2007. V. 8 (10). P. 813–824.
Ding B., Parmigiani A., Yang C., Budanov A.V. Sestrin2 facilitates death receptor-induced apoptosis in lung adenocarcinoma cells through regulation of XIAP degradation // Cell Cycle. 2015. V. 14 (20). P. 3231–3241.
Ding B., Parmigiani A., Divakaruni A.S. et al. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death // Sci. Rep. 2016. V. 6. P. 22538.
Doonan F., Wallace D.M., O’Driscoll C., Cotter T.G. Rosiglitazone acts as a neuroprotectant in retinal cells via up-regulation of sestrin-1 and SOD-2 // J. Neurochem. 2009. V. 109 (2). P. 631–643.
Ebnoether E., Ramseier A., Cortada M. et al. Sesn2 gene ablation enhances susceptibility to gentamicin-induced hair cell death via modulation of AMPK/mTOR signaling // Cell Death Discov. 2017. V. 3. P. 17024.
Eid A.A., Lee D.-Y., Roman L.J. et al. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression // Mol. Cell. Biol. 2013. V. 33 (17). P. 3439–3460.
Eijkelenboom A., Burgering B.M. FOXOs: signalling integrators for homeostasis maintenance // Nat. Rev. Mol. Cell Biol. 2013. V. 14.(2). P. 83–97.
Fischer M. Census and evaluation of p53 target genes // Oncogene. 2017. V. 36 (28). P. 3943–3956.
Gan X., Wang J., Su B., Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate // J. Biol. Chem. 2011. V. 286 (13). P. 10998–11002.
Garaeva A.A., Kovaleva I.E., Chumakov P.M., Evstafieva A.G. Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4 // Cell. Cycle. 2016. V. 15 (1). P. 64–71.
Green D.R., Galluzzi L., Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging // Science. 2011. V. 333 (6046). P. 1109–1112.
Hagenbuchner J., Kuznetsov A., Hermann M. et al. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3 // J. Cell Sci. 2012. V. 125. (Pt 5). P. 1191–1203.
Hatano N., Nishikawa N.S., McElgunn C. et al. A comprehensive analysis of loss of heterozygosity caused by hemizygous deletions in renal cell carcinoma using a subtraction library // Mol. Carcinog. 2001. V. 31 (3). P. 161–170.
Heidler J., Fysikopoulos A., Wempe F. et al. Sestrin-2, a repressor of PDGFRbeta signalling, promotes cigarette-smoke-induced pulmonary emphysema in mice and is upregulated in individuals with COPD // Dis. Model. Mech. 2013. V. 6 (6). P. 1378–1387.
Hou Y.S., Guan J.-J., Xu H.-D. et al. Sestrin2 protects dopaminergic cells against rotenone toxicity through AMPK-dependent autophagy activation // Mol. Cell Biol. 2015. V. 35 (16). P. 2740–2751.
Hwang H.-J., Jung T.W., Choi J.-H. et al. Knockdown of sestrin2 increases pro-inflammatory reactions and ER stress in the endothelium via an AMPK dependent mechanism // Biochim. Biophys. Acta. 2017. V. 1863 (6). P. 1436–1444.
Jegal K.H., Park W., Cho S.S. et al. Activating transcription factor 6-dependent sestrin2 induction ameliorates ER stress-mediated liver injury // Biochim. Biophys. Acta. 2017. V. 1864 (7). P. 1295–1307.
Jeong W., Bae S.H., Toledano M.B., Rhee S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression // Free Radic. Biol. Med. 2012. V. 53 (3). P. 447–456.
Johnson M.R., Behmoaras J., Bottolo L. et al. Systems genetics identifies Sestrin3 as a regulator of a proconvulsant gene network in human epileptic hippocampus // Nat. Commun. 2015. V. 6. P. 6031.
Johnson S.C., Rabinovitch P.S., Kaeberlein M. mTOR is a key modulator of ageing and age-related disease // Nature. 2013. V. 493 (7432). P. 338–345.
Kallenborn-Gerhardt W., Lu R., Syhr K.M.J. et al. Antioxidant activity of sestrin2 controls neuropathic pain after peripheral nerve injury // Antioxid. Redox. Signal. 2013. V. 19 (17). P. 2013–2023.
Kamata H., Honda S.-I., Maeda S. et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases // Cell. 2005. V. 120 (5). P. 649–661.
Kastenhuber E.R., Lowe S.W. Putting p53 in context // Cell. 2017. V. 170 (6). P. 1062–1078.
Kim H., An S., Ro S.H. et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains // Nat. Commun. 2015. V. 6 (10025). P. 1–11.
Kim J.-R., Lee S.-R., Chung H.L. et al. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: involvement of RTP801 in amyloid beta-peptide toxicity // Exp. Mol. Med. 2003. V. 35 (5). P. 403–411.
Kim M.G., Yang J.H., Kim K.M. et al. Regulation of Toll-like receptor-mediated Sestrin2 induction by AP-1, Nrf2, and the ubiquitin-proteasome system in macrophages // Toxicol. Sci. 2015. V. 144 (2). P. 425–435.
Kim M.J., Bae S.H., Ryu J.-C. et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages // Autophagy. 2016. V. 12 (8). P. 1272–1291.
Kimball S.R., Gordon, B. S., Moyer J.E. et al. Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation // Cell Signal. 2016. V. 28 (8). P. 896–906.
Kourtis N. Tavernarakis N. Cellular stress response pathways and ageing: intricate molecular relationships // EMBO J. 2011. V. 30 (13). P. 2520–2531.
Kruiswijk F., Labuschagne C.F., Vousden K.H. p53 in survival, death and metabolic health: a lifeguard with a licence to kill // Nat. Rev. Mol. Cell Biol. 2015. V. 16 (7). P. 393–405.
Kinnula V.L., Paakko P., Soini Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung // FEBS Lett. 2004. V. 569 (1–3). P. 1–6.
Kopnin P.B., Agapova L.S., Kopnin B.P., Chumakov P.M. Repression of sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability // Cancer Res. 2007. V. 67 (10). P. 4671–4678.
Lane D.P. Cancer. p53, guardian of the genome // Nature. 1992. V. 358 (6381). P. 15–16.
Lanna A., Gomes D.C.O., Muller-Durovic B. et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging // Nat. Immunol. 2017. V. 18. P. 354–363.
Lee J.H., Budanov A.V., Park E.J. et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies // Science. 2010. V. 327 (5970). P. 1223–1228.
Lee J.H., Budanov A.V., Talukdar S. et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3 // Cell Metab. 2012. V. 16 (3). P. 311–321.
Lee J.H., Budanov A.V., Karin M. Sestrins orchestrate cellular metabolism to attenuate aging // Cell Metab. 2013. V. 18 (6). P. 792–801.
Lehmann S., Ogawa S., Raynaud S.D. et al. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia // Cancer. 2008. V. 112 (6). P. 1296–1305.
Leister I., Weith A., Brüderlein S. et al. Human colorectal cancer: high frequency of deletions at chromosome 1p35 // Cancer Res. 1990. V. 50 (22). P. 7232–7235.
Levine A.J. p53, the cellular gatekeeper for growth and division // Cell. 1997. V. 88 (3). P. 323–331.
Li D.D., Sun T., Wu X.-Q. et al. The inhibition of autophagy sensitises colon cancer cells with wild-type p53 but not mutant p53 to topotecan treatment // PLoS One. 2012. V. 7 (9). P. e45058.
Liu S.Y., Lee Y.J., Lee T.C. Association of platelet-derived growth factor receptor beta accumulation with increased oxidative stress and cellular injury in sestrin2 silenced human glioblastoma cells // FEBS Lett. 2011. V. 585 (12). P. 1853–1858.
Lopez-Otin C., Blasco M.A., Partridge L. et al. The hallmarks of aging // Cell. 2013. V. 153 (6). P. 1194–1217.
Majd S., Power J.H., Grantham H.J. Neuronal response in Alzheimer’s and Parkinson’s disease: the effect of toxic proteins on intracellular pathways // BMC Neurosci. 2015. V. 16. P. 69.
Manning B.D., Toker A. AKT/PKB signaling: navigating the network //Cell. 2017. V. 169 (3). P. 381–405.
Martindale J.L., Holbrook N.J. Cellular response to oxidative stress: signaling for suicide and survival // J. Cell Physiol. 2002. V. 192 (1). P. 1–15.
Mizushima N. Autophagy: process and function // Gen. Dev. 2007. V. 21 (22). P. 2861–2873.
Mizumura K., Cloonan S.M., Nakahira K. et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD // J. Clin. Invest. 2014. V. 124 (9). P. 3987–4003.
Morrison A., Chen L., Wang J. et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart // FASEB J. 2015. V. 29 (2). P. 408–417.
Nagai H., Negrini M., Carter S.L. et al. Detection and cloning of a common region of loss of heterozygosity at chromosome 1p in breast cancer // Cancer Res. 1995. V. 55 (8). P. 1752–1757.
Nogueira V., Park Y., Chen C.C. et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis // Cancer Cell. 2008. V. 14 (6). P. 458–470.
Oricchio E., Katanayeva N., Donaldson M.C. et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma // Sci. Transl. Med. 2017. V. 9 (396). P. eaak9969.
Papadia S., Soriano F.X., Léveillé F. et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses // Nat. Neurosci. 2008. V. 11 (4). P. 476–487.
Park H.-W., Park H., Ro S.-H. et al. Hepatoprotective role of Sestrin2 against chronic ER stress // Nat. Commun. 2014. V. 5. P. 4233.
Parmigiani A., Budanov A.V. Sensing the environment through sestrins: implications for cellular metabolism // Int. Rev. Cell Mol. Biol. 2016. V. 327. P. 1–42.
Parmigiani A., Nourbakhsh A., Ding B. et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex // Cell Rep. 2014. V. 9 (4). P. 1281–1291.
Peeters H., Debeer P., Bairoch A. et al. PA26 is a candidate gene for heterotaxia in humans: identification of a novel PA26-related gene family in human and mouse // Hum. Genet. 2003. V. 112 (5–6). P. 573–580.
Peng M., Yin N., Li M.O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling // Cell. 2014. V. 159 (1). P. 122–133.
Peng M., Yin N., Li M.O. SZT2 dictates GATOR control of mTORC1 signalling // Nature. 2017. V. 543 (7645). P. 433–437.
Polyak K., Xia Y., Zweier J.L. et al. A model for p53-induced apoptosis // Nature. 1997. V. 389 (6648). P. 300–305.
Quan N., Sun W., Wang L. et al. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism // FASEB J. 2017. V. 31. P. 4153–4167.
Reddy K., Cusak C.L., Nnah I. et al. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency // Cell. Rep. 2016. V. 14 (9). P. 2166–2179.
Ro S.-H., Xue X., Ramakrishnan S.K. et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis // Elife. 2016. V. 5. P. e12204.
Sablina A.A., Budanov A.V., Ilyinskaya G.V. et al. The antioxidant function of the p53 tumor suppressor // Nat. Med. 2005. V. 11 (12). P. 1306–1313.
Sancak Y., Peterson T.R., Shaul Y.D. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1 // Science. 2008. V. 320 (5882). P. 1496–1501.
Sanli T., Linher-Melville K., Tsakiridis T., Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells // PLoS One. 2012. V. 7 (2). P. e32035.
Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the Rictor–mTOR complex // Science. 2005. V. 307 (5712). P. 1098–1101.
Saveljeva S., Cleary P., Mnich K. et al. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival // Oncotarget. 2016. V. 7 (11). P. 12254–12266.
Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease // Cell. 2017. V. 169 (2). P. 361–371.
Saxton R.A., Knockenhauer K.E., Wolfson R.L. et al. Structural basis for leucine sensing by the Sestrin2–mTORC1 pathway // Science. 2016. V. 351 (6268). P. 53–58.
Scheibye-Knudsen M., Fang E.F., Croteau D.L. et al. Protecting the mitochondrial powerhouse // Tr. Cell. Biol. 2015. V. 25 (3). P. 158–170.
Seo K., Ki S.H., Shin S.M. Sestrin2-AMPK activation protects mitochondrial function against glucose deprivation-induced cytotoxicity // Cell Signal. 2015. V. 27 (7). P. 1533–1543.
Settembre C., Fraldi A., Medina D.L., Ballabio A. et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism // Nat. Rev. Mol. Cell Biol. 2013. V. 14 (5). P. 283–296.
Tao R., Xiong X., Liangpunsakul S., Dong X.C. et al. Sestrin3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2–Akt signaling // Diabetes. 2015. V. 64 (4). P. 1211–1223.
Thelander E.F., Ichimura K., Corcoran M. et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia // Leuk. Lymph. 2008. V. 49 (3). P. 477–487.
Tomasovic A., Kurrle N., Sürün D. et al. Sestrin2 protein regulates platelet-derived growth factor receptor β (Pdgfrβ) expression by modulating proteasomal and Nrf2 transcription factor functions // J. Biol. Chem. 2015. V. 290 (15). P. 9738–9752.
Tran H., Brunet A., Grenier J.M. et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein // Science. 2002. V. 296 (5567). P. 530–534.
Tsilioni I., Filippidis A.S., Kerenidi T. et al. Sestrin-2 is significantly increased in malignant pleural effusions due to lung cancer and is potentially secreted by pleural mesothelial cells // Clin. Biochem. 2016. V. 49 (9). P. 726–728.
Velasco-Miguel S., Buckbinder L., Jean P. et al. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes // Oncogene. 1999. V. 18 (1). P. 127–137.
Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation // Science. 2011. V. 334 (6059). P. 1081–1086.
Wei C.L., Wu Q., Vega V.B. et al. A global map of p53 transcription-factor binding sites in the human genome // Cell. 2006. V. 124 (1). P. 207–219.
Wei J.L. et al. Decreased expression of sestrin 2 predicts unfavorable outcome in colorectal cancer // Oncol. Rep. 2015. V. 33 (3). P. 1349–1357.
Wempe F., De-Zolt S., Koli K. et al. Inactivation of sestrin 2 induces TGF-beta signaling and partially rescues pulmonary emphysema in a mouse model of COPD // Dis. Model. Mech. 2010. V. 3 (3–4). P. 246–253.
White P.S., Kaufman B.A., Marshall H.N., Brodeur G.M. Use of the single-strand conformation polymorphism technique to detect loss of heterozygosity in neuroblastoma // Gen. Chrom. Cancer. 1993. V. 7 (2). P. 102–108.
Wolfson R.L., Chantranupong L., Saxton R.A. et al. Sestrin2 is a leucine sensor for the mTORC1 pathway // Science. 2016. V. 351 (6268). P. 43–48.
Wolfson R.L, Sabatini D.M. The dawn of the age of amino acid sensors for the mTORC1 pathway // Cell Metab. 2017. V. 26 (2). P. 301–309.
Wolfson R.L., Chantranupong L., Wyant G.A. et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1 // Nature. 2017. V. 543 (7645). P. 438–442.
Woo H.A., Bae S.H., Park S., Rhee S.G. Sestrin2 is not a reductase for cysteine sulfinic acid of peroxiredoxins // Antioxid. Redox. Signal. 2009. V. 11 (4). P. 739–745.
Wood Z.A., Poole L.B, Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling // Science. 2003. V. 300 (5619). P. 650–653.
Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism // Cell. 2006. V. 124 (3). P. 471–484.
Xu C., Bailly-Maitre B., Reed J.C. Endoplasmic reticulum stress: cell life and death decisions // J. Clin. Invest. 2005. V. 115 (10). P. 2656–2664.
Yang J.H., Kim K.M., Kim M.G. et al. Role of sestrin2 in the regulation of proinflammatory signaling in macrophages // Free Radic. Biol. Med. 2015. V. 78. P. 156–167.
Yang K., Xu C., Zhang Y. et al. Sestrin2 suppresses classically activated macrophages-mediated inflammatory response in myocardial infarction through inhibition of mTORC1 signaling // Front. Immunol. 2017. V. 8. P. 728.
Yang Y.-L., Loh K.-S., Liou B.-Yu. et al. SESN-1 is a positive regulator of lifespan in Caenorhabditis elegans // Exp. Gerontol. 2013. V. 48 (3). P. 371–379.
Ye J., Palm W., Peng M. et al. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2 // Gen. Dev. 2015. V. 29 (22). P. 2331–2336.
Yeh S.H., Chen P.J., Chen H.L. et al. Frequent genetic alterations at the distal region of chromosome 1p in human hepatocellular carcinomas // Cancer Res. 1994. V. 54 (15). P. 4188–4192.
Yoshida T., Mett I., Bhunia A.K. et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema // Nat. Med. 2010. V. 16 (7). P. 767–773.
Zhang C., Sun W., Li J. et al. Loss of sestrin 2 potentiates the early onset of age-related sensory cell degeneration in the cochlea // Neuroscience. 2017. V. 361. P. 179–191.
Zhao B., Shah P., Budanov A.V. et al. Sestrin2 protein positively regulates AKT enzyme signaling and survival in human squamous cell carcinoma and melanoma cells // J. Biol. Chem. 2014. V. 289 (52). P. 35806–35814.
Zhao B., Shah P., Qiang L. et al. Distinct role of Sesn2 in response to UVB-induced DNA damage and UVA-induced oxidative stress in melanocytes // Photochem. Photobiol. 2017. V. 93 (1). P. 375–381.
Zeltukhin A.O., Ilyinskaya G.V., Budanov A.V., Chumakov P.M. Some phenotypic characteristics of nematode Caenorhabditis elegans strain with defective functions of the sestrin (cSesn) gene // Biom. Pharmacol. J. 2018. V. 11 (2). P. 759–767.
Zhou D., Zhan C., Zhong Q., Li S. Upregulation of sestrin-2 expression via P53 protects against 1-methyl-4-phenyl-pyridinium (MPP+)neurotoxicity // J. Mol. Neurosci. 2013. V. 51 (3). P. 967–975.
Zighelboim I., Goodfellow P.J., Schmidt A.P. et al. Differential methylation hybridization array of endometrial cancers reveals two novel cancer-specific methylation markers // Clin. Cancer Res. 2007. V. 13 (10). P. 2882–2889.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии