

Успехи физиологических наук, 2020, T. 51, № 3, стр. 16-22
Механизмы развития дефицита тетрагидробиоптерина при шизофрении
Т. В. Жиляева a, А. С. Благонравова a, Г. В. Рукавишников b, В. И. Ларионова c, Г. Э. Мазо b, *
a ФГБОУ ВО “Приволжский исследовательский медицинский университет” Минздрава России
Нижний Новгород, Россия
b ФГБУ “Национальный медицинский исследовательский центр психиатрии и неврологии им. В.М. Бехтерева” Минздрава России
Санкт-Петербург, Россия
c ФГБОУ ВО “Северо-западный государственный медицинский университет им. И.И. Мечникова”
Минздрава России
Санкт-Петербург, Россия
* E-mail: galina-mazo@yandex.ru
Поступила в редакцию 24.12.2019
После доработки 27.12.2019
Принята к публикации 29.12.2019
Аннотация
В статье представлен обзор современных исследований о роли тетрагидробиоптерина (ВН4) в качестве одного из управляемых факторов риска развития шизофрении и отдельных кластеров ее симптомов. Рассмотрены возможные механизмы участия ВН4, как ключевого кофактора синтеза центральных нейромедиаторов, в этиопатогенетических процессах при шизофрении. Приведены основные гипотезы, которые могли бы объяснить причины развития дефицита ВН4 при шизофрении (генетические полиморфизмы, нарушения обмена фолатов, воспаление и оксидативный стресс), а также возможные перспективы их подтверждения в специальных исследованиях. Обосновывается актуальность и практическая значимость персонифицированного подхода к коррекции уровня ВН4 при шизофрении в зависимости от механизмов развития его дефицита при этом расстройстве, а также важность трансдиагностического подхода к изучению дефицита ВН4 при психических расстройствах.
ВВЕДЕНИЕ
В последние годы появляется все больше сведений о том, что шизофрения является фенотипом, соответствующим множеству генотипических и биологических вариантов, в каждом из которых ведущую роль играют отдельные молекулярные механизмы [5, 31, 44]. Это открывает возможность разработки принципиально новых патогенетически ориентированных подходов к терапии шизофрении, базирующихся на выделении внутри гетерогенной группы больных шизофренией подтипов с учетом общих молекулярных механизмов патогенеза. Такой подход соответствует принципам персонифицированной медицины [2, 4].
Среди изучаемых молекулярных патогенетических факторов особого внимания заслуживают управляемые факторы риска, своевременное выявление которых у субъекта может способствовать профилактике заболевания или снижению его тяжести при манифестных случаях. К таким факторам, в частности, могут относиться выявляемые лабораторно-биохимические, патофизиологические, иммунологические отклонения, которые потенциально могут быть вовлечены в этиопатогенетические механизмы развития заболевания или отдельных кластеров его симптомов.
К таким факторам в полной мере может относиться дефицит тетрагидробиоптерина (далее ВН4), который, согласно специальным исследованиям, выявляется у 30% пациентов с шизофренией [40, 41]. При своевременном выявлении дефицита ВН4 он может подаваться коррекции (частичной или полной) путем добавления извне синтетического ВН4 (сапроптерина).
Первоначально дефицит ВН4 связывали с атипичной фенилкетонурией (далее ФКУ), поскольку ВН4 является кофактором фермента фенилаланин-гидроксилазы, превращающего фенилаланин в тирозин. Интерес к этому эндогенному веществу резко возрос после публикации Kure и соавт. (1999) [25] о том, что применение ВН4 у четырех пациентов с гиперфенилаланинемией (ГФА) привело к значимому снижению уровня фенилаланина плазмы. После ряда кинических испытаний синтетическая форма ВН4 (препарат сапроптерин – “Куван”, “Эфкурия”) была одобрена для снижения уровня фенилаланина при гиперфенилаланинемии (далее ГФА), зависимой от ВН4 [36].
Однако в последние десятилетия показано, что существует дефицит ВН4, не ассоциированный с ФКУ и ГФА, а проявляющийся целым комплексом нервно-психических расстройств [10]. ВН4 является ключевым кофактором, участвующим в синтезе нескольких нейротрансмиттеров: гидроксилировании ароматических аминокислот ферментами фенилаланин-гидроксилазой (превращение фенилаланина в тирозин), тирозин-3-гидроксилазой (превращение тирозина в L-допа – процесс, лимитирующий скорость синтеза дофамина и норадреналина) и триптофан-5-гидроксилазой (превращение триптофана в серотонин) [8, 22]. Кроме того, было показано, что ВН4 стимулирует высвобождение глутумата, серотонина, а также дофамина независимо от его кофакторной гидроксилазной активности, то есть независимо от участия в синтезе нейромедиаторов [24, 30], а также регулирует экспрессию тирозин-3-гидроксилазы в нервных окончаниях [47]. Таким образом, ВН4 играет ключевую роль в поддержании должного уровня ряда нейротрансмиттеров (серотонина, норадреналина, дофамина и глутамата) посредством нескольких различных механизмов, что позволило отдельным авторам [12] выдвинуть гипотезу о роли ВН4 в этиопатогенетических процессах при шизофрении.
Ключевое участие ВН4 в поддержании синтеза и высвобождения дофамина из нервных окончаний может иметь непосредственное отношение к патогенезу шизофрении. В эксперименте было показано, что у генетически модифицированных мышей с искусственно созданным дефицитом ВН4 выявлялась недостаточность дофамина в головном мозге, а также поведенческие нарушения, характерные для экспериментальной модели шизофрении [26]. Не исключено, что кофакторная и регуляторная роль ВН4 в отношении серотонина тоже может быть ассоциирована с этиопатогенезом шизофрении – нарушения в серотонинергической нейротрансмиссии при этом заболевании в настоящее время активно изучается [17, 20].
ВН4 также является кофактором фермента синтазы оксида азота, которая осуществляет синтез оксида азота (NO) из L-аргинина [39]. NO инициирует сигнальный каскад фосфорилирования белков [46], является регулятором моноаминергической нейротрансмиссии [23], а также глутаматергической системы (вовлечен в функционирование NMDA и других глутаматных рецепторов, в том числе играет роль вторичного мессенджера NMDA-рецепторов) [37]. Теория глутаматергической NMDA-рецепторной гипофункции при шизофрении достаточно активно развивается в течение длительного времени и занимает весомое место в понимании патогенеза шизофрении [15]. В экспериментальной модели на генномодифицированных мышах было показано, что дефицит синтеза ВН4 приводит к поведенческим нарушениям, характерным для шизофрении, и эти нарушения опосредованы снижением NMDA-рецепторной глутаматергической нейротрансмиссии [1]. Нарушения в работе сигнальной системы NO были обнаружены при психических расстройствах, в том числе при шизофрении. У больных шизофренией по сравнению с группами контроля здоровых лиц была выявлена сниженная клеточная экспрессия нейрональной синтазы оксида азота в определенных отделах головного мозга [6], а также сниженный уровень метаболитов оксида азота в цереброспинальной жидкости [38]. Это также может свидетельствовать о возможной ключевой роли ВН4 в этиопатогенезе шизофрении.
При субоптимальном уровне ВН4 незначительное снижение активности фенилаланин-гидроксилазы приводит к накоплению фенилаланина, но не в такой степени, как это наблюдается при генетических мутациях, характерных для ФКУ. В отдельных исследованиях показано, что у больных шизофренией по сравнению со здоровыми субъектами контрольной группы отмечается более высокий уровень фенилаланина плазмы [39, 49]. Кроме того, у пациентов с ФКУ было показано, что гиперфенилаланинемия ассоциирована с развитием психотических симптомов [7].
Проверкой гипотезы о вовлечении ВН4 в этиопатогенез шизофрении был ряд работ, посвященных лабораторному измерению уровня биоптерина в различных биологических жидкостях у больных шизофренией по сравнению с группами контроля. Первые исследования, посвященные измерению уровня биоптерина плазмы, выявили у больных шизофренией (n = 17) по сравнению со здоровым контролем (n =114) значимый дефицит биоптерина [26]. Это подтвердилось и в недавних исследованиях, согласно результатам которых более 30% пациентов с шизофренией имеют дефицит BH4 [40, 41].
ОСНОВНЫЕ МЕХАНИЗМЫ, ВЕДУЩИЕ К ВН4 ПОТЕРЯМ У ПАЦИЕНТОВ С ШИЗОФРЕНИЕЙ
Вполне закономерными являются предположения, что дефицит ВН4 при шизофрении может быть обусловлен генетическими факторами. В генах, контролирующих синтез и ресинтез ВН4 к настоящему времени обнаружено несколько различных мутаций, которые приводят к тяжелым нервно-психическим расстройствам с детства, когда развиваются ФКУ и ГФА. Однако, не исключено существование мягких форм генетических отклонений – полиморфизмов, при которых нарушения синтеза ВН4 не так выражены, как при точечных генетических мутациях и, соответственно, фенотипические проявления не так рано и ярко проявляются, как при ФКУ. Поиск таких полиморфизмов в генах, участвующих в синтезе/ресинтезе BH4, а также их ассоциации с шизофренией в настоящее время осуществляется. Так, в префронтальной коре у больных шизофренией обнаружено снижение уровня фактора транскрипции Nurr1 [49], который помимо прочих функций регулирует экспрессию гена ГТФ циклогидролазы I (GTPCH1) – фермента, участвующего в синтезе ВН4 [18]. Результаты исследования ассоциации другого генетического фактора – полиморфизма гена промоутера сепиаптерин-редуктазы – с шизофренией отрицательны [16]. В еще одном недавнем исследовании обнаружено, что у больных шизофренией чаще, чем в общей популяции выявляется полиморфизм гена ключевого фермента синтеза ВН4 – ГТФ-циклогидролазы-1 (rs10137071), носительство А аллеля которого приводит к снижению экспрессии гена и соответственно уровня ВН4 [13]. Однако у ряда пациентов с нормальным генотипом данного генетического полиморфизма (GG) в приведенном исследовании уровень BH4 плазмы также был низким, что требует поиска других причин, которыми могут быть другие генетические полиморфизмы ферментов синтеза/ресинтеза ВН4, а также негенетические (биохимические и патофизиологические) процессы, которые могут влиять на уровень ВН4.
Дефицит ВН4 может быть обусловлен как нарушением его синтеза из-за недостаточности функции ферментов, участвующих в этом процессе, так и проблемами его ресинтеза из дигидробиоптерина (ВН2) – продукта, образующегося в коферментных реакциях в результате окисления ВН4. ВН4 синтезируется из гуанозинтрифосфата (ГТФ) с помощью трех последовательных ферментативных реакций в которых участвуют: ГТФ гидролаза I (GTPCH1), 6-пирувоилтетрагидроптерин синтетаза (6-PTS) и сепиаптеринредуктаза (SR). Дигидроптеридинредуктаза (DHPR) – фермент, восстанавливающий активную форму BH4 из ВН2. Благодаря постоянному процессу ресинтеза, пул ВН2, формирующийся из ВН4 после коферментной отработки его в соответствующих биохимических реакциях, постоянно превращается обратно в активный ВН4, необходимый для синтеза нейромедиаторов (Схема 1 ).
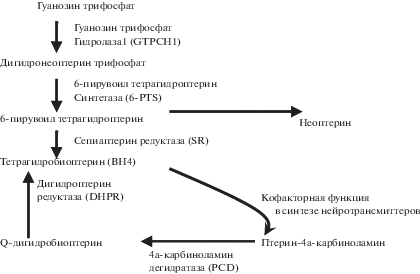
Схема 1 . Основные пути синтеза и ресинтеза ВН4.
При серьезных нарушениях синтеза ВН4 по основному пути, кроме DHPR, в ресинтезе ВН4 из ВН2 может играть роль фермент фолатного цикла дигидрофолатредукатаза (DHFR) [32]. Помимо этого было показано, что 5-метилтетрагидрофолат способен отдавать 2 атома водорода дигидробиоптерину (ВН2), в результате чего образуется ВН4 и 5,10-метилентетрагидрофолат – посредством этого процесса фолаты могут играть важную роль в регулировании всех биохимических реакций, зависимых от ВН4. Особенно это важно в тех ситуациях, когда ресинтез ВН4 по основному пути с помощью фермента DHPR по каким-либо причинам нарушен [32].
Таким образом, дефицит ВН4 может быть ассоциирован с дефицитом фолатов независимо от первоначальной причины в связи с наличием нескольких точек пересечения ресинтеза ВН4 с циклом обмена фолатов. Однако экспериментальные и биохимические исследования о взаимосвязи фолатов и ВН4 подтверждены клиническими наблюдениями о наличии положительной корреляции между уровнем фолатов и ВН4 только у пациентов с депрессией, но не шизофренией [14]. Вероятно, в аналогичных исследованиях при шизофрении эта корреляция также может подтвердиться. Согласно специальным исследованиям, при аффективных расстройствах уровень ВН4 достоверно выше, чем при шизофрении [48], поэтому при шизофрении нельзя исключить механизмы развития дефицита ВН4, отличные от депрессии.
Нарушения обмена фолатов в течение нескольких десятилетий изучаются с позиции этиопатогенетического фактора, играющего роль в развитии шизофрении. Накоплен большой объем данных, позволивший сделать мета-анализы о более частом наличии генетических дефектов и биохимических нарушений в цикле обмена фолатов у больных шизофренией по сравнению со здоровыми субъектами (10 069 пациентов с шизофренией и 13 372 субъектов контрольной группы) [35, 48, 50, 51]. Результаты, полученные в России, согласуются с данными зарубежных авторов о более частом наличии нарушений обмена фолатов при шизофрении и их ассоциации с различными проявлениями этого заболевания [3].
Первые клинические случаи о положительном влиянии фолатов на симптомы шизофрении были описаны в 1970-е [32], а к настоящему времени получены результаты ряда клинических испытаний, согласно которым коррекция витаминами нарушений в цикле обмена фолатов способствует нормализации биохимических параметров (повышению уровня фолатов и кобаламина плазмы, снижению уровня гомоцистеина плазмы до нормального) и при этом частичной редукции негативных и когнитивных симптомов у больных шизофренией, имеющих генетические предпосылки к нарушениям обмена фолатов [3, 11, 42–44]. При этом конкретные механизмы, посредством которых фолаты могут участвовать в патогенезе шизофрении, известны лишь гипотетически и за исключением гипотезы гипометилирования [42] изучались не в связи с шизофренией, а в связи с другими патологическими процессами [1, 21, 28, 29, 45]. Имеется целый ряд гипотез, среди которых в том числе можно назвать нарушение ресинтеза ВН4 из тетрагидрофолата [11]. При этом дефицит ресинтеза ВН4 в связи с нарушениями обмена фолатов также не имеет подтверждения in vivo у пациентов с шизофренией. Кроме того, развивающаяся при дефиците фолатов гипергомоцистеинемия может вызывать оксидативный стресс и эндотелиальную дисфункцию [9, 19, 27]. Это в свою очередь может также приводить к дефициту ВН4 за счет вторичных воспалительных процессов (механизмы описаны ниже), что также остается не изученным до настоящего времени.
Активно изучаемым патогенетическим механизмом шизофрении, который может быть ассоциирован с дефицитом ВН4, является нейровоспаление, маркером которого может служить в частности увеличение уровня С-реактивного белка (индикатора воспаления) [31]. При хроническом воспалении повышенный синтез неоптерина (маркера активации иммунной системы) может реализовываться за счет снижения синтеза ВН4 (так как неоптерин и ВН4 синтезируются из общего предшественника, Схема 1 ) [33]. Кроме того, BH4 относительно нестабильное соединение и в условиях воспаления и оксидативного стресса может подвергаться неэнзиматическому необратимому окислению в дигидроксантоптерин [34]. Потери ВН4 при воспалительных процессах и оксидативном стрессе могут быть весьма ощутимыми за счет дублирующих механизмов.
Таким образом, дефицит ВН4 может выполнять роль связующего звена между нарушениями в дофаминовой, серотониновой и глутаматергической нейротрансмиссии в патогенезе шизофрении, а также объяснять ряд дополнительных патогенетических механизмов (нарушения обмена фолатов, медленное нейровоспаление и окидативный стресс), вероятно, имеющих место при этом психическом расстройстве (Схема 2 ).

Схема 2 . Гипотетические механизмы развития дефицита ВН4 при шизофрении.
ОБСУЖДЕНИЕ
До сих пор, несмотря на существование целого ряда гипотез, не было проведено ни одного исследования, в котором in vivo подтверждалась бы каузальная связь дефицита ВН4 с дефицитом фолатов, параметрами воспаления и оксидативного стресса при шизофрении. Более того, такая каузальная связь не изучена и у здоровых людей, а также у людей с другими заболеваниями, при которых также выявляются нарушения обмена фолатов, воспаление и оксидативный стресс (из психических расстройств – биполярное расстройство, депрессия, расстройства аутистического спектра, ряд неврологических и эндокринно-обменных расстройств). Поэтому полученные результаты могут оказаться важными для лучшего понимания патогенеза как психических, так и соматических расстройств, и данная гипотеза может изучаться с позиции трансдиагностического подхода в психиатрии.
Актуальность проведения исследований механизмов развития ВН4 при шизофрении диктуется не только трудностями понимания молекулярных механизмов развития шизофрении (и других эндогенных психических расстройств), но также необходимостью разработки алгоритмов коррекции известных биохимических нарушений и соответственно патогенетического лечения (профилактики) социально-значимых заболеваний.
Сведения о роли ВН4 в синтезе основных нейромедиаторов, задействованных в развитии дефицитарной симптоматики шизофрении, внушают определенный оптимизм в отношении возможности коррекции тех дименсий симптомов (негативных и когнитивных), которые оказывают максимальное влияние на социальное функционирование пациентов и которые практически резистентны к имеющемуся в настоящее время арсеналу психофармакологических средств. Кроме того, представления о моноаминергическом дефиците при депрессии позволяют предположить, что синтетический ВН4 может оказаться эффективным средством в отношении депрессивного кластера симптомов при шизофрении. Если гипотеза о роли дефицита ВН4 в этиопатогенезе шизофрении верна, то закономерным выводом из нее будет являться фармакологическое вмешательство – назначение синтетического ВН4 больным шизофренией и оценка на этом фоне динамики симптомов заболевания, что уже предлагалось отдельными учеными [11]. К настоящему времени проведено единственное клиническое испытание применения синтетического ВН4 при шизофрении, результаты еще не опубликованы (https://clinicaltrials.gov/ct2/show/results/NCT01706965). Однако гипотетически, применение синтетического ВН4 может оказаться не эффективным, если не будут скомпенсированы механизмы развития его дефицита, такие как нарушение ресинтеза из-за нарушений обмена фолатов и потери при воспалении и оксидативном стрессе. Учитывая высокую коммерческую стоимость сапроптерина и низкую стоимость витаминов В9 и В12, а также противовоспалительных средств и антиоксидантов, раскрытие вклада этих механизмов в потери ВН4 при шизофрении имеет важное практическое значение.
Оптимальными были бы исследования с персонифицированным вмешательством – назначением синтетического биоптерина только тем пациентам с шизофренией, у которых лабораторно подтверждается дефицит ВН4, ассоциированный с генетическими нарушениями ферментов его синтеза/ресинтеза. Представления о взаимосвязи дефицита ВН4 с дефицитом фолатов, воспалением и оксидативным стрессом также могут быть подтверждены в специальных проспективных исследованиях с персонифицированной коррекцией выявленных у пациента биологических маркеров (нарушений обмена фолатов, воспаления, оксидативного стресса) и отслеживанием изменений в уровне ВН4 плазмы у пациентов.
Таким образом, гипотеза многофакторности развития дефицита ВН4 при шизофрении является перспективной для изучения как с точки зрения фундаментального понимания этиопатогенетических механизмов при шизофрении, так и с прикладной точки зрения, так как она может лечь в основу для разработки новых способов коррекции симптомов (профилактики) этого заболевания.
Список литературы
Арзуманян Е.С., Степанова М.С. Механизмы токсического действия гомоцистеиновой кислоты на нейрональные клетки // Нейрохимия. 2010. Т. 27. № 3. С. 251.
Дубинина Е.Е., Щедрина Л.В., Мазо Г.Э. Основные биохимические аспекты патогенеза депрессии. Часть 1 // Успехи физиологических наук. 2018. Т. 49. № 1. С. 28.
Жиляева Т.В., Сергеева А.В., Благонравова А.С., Касимова Л.Н. Психопатологическая характеристика и особенности социального функционирования больных шизофренией с носительством Т-аллеля в полиморфном локусе гена обмена фолатов MTHFR677C> T // Журн. неврологии и психиатрии им. С.С. Корсакова. 2016. Т. 116. № 11. С. 5.
Мазо Г.Э., Кибитов А.О., Рукавишников Г.В., и соавт. Терапевтическая резистентность при депрессии как объект междисциплинарного биомедицинского исследования // Социальная и клиническая психиатрия. 2017. Т. 27. № 4. С. 70.
Рукавишников Г.В., Мазо Г.Э. Депрессия при шизофрении: патофизиологические механизмы и терапевтические подходы // Современная терапия психических расстройств. 2018. № 3. С. 18.
Bernstein H.G., Bogerts B., Keilhoff G. The many faces of nitric oxide in schizophrenia // Schizophr Res. 2005. V. 78. P. 69.
Bilder D.A., Burton B.K., Coon H. et al. Psychiatric symptoms in adults with phenylketonuria. Mol Genet Metab. 2013. V. 108. P. 155.
Blau N., Thony B., Cotton R.G.H. et al. Disorders of tetrahydrobiopterin and related biogenic amines // The metabolic and molecular bases of inherited disease. 2001. McGraw-Hill, New York. P. 1725.
Boldyrev A.A. Molecular mechanisms of homocysteine toxicity // Biochemistry (Moscow). 2009. V. 74. № 6. P. 589. https://doi.org/10.1134/s0006297909060017
Bonafé L., Thöny B., Penzien J.M., Czarnecki B., Blau N. Mutations in the Sepiapterin Reductase Gene Cause a Novel Tetrahydrobiopterin-Dependent Monoamine-Neurotransmitter Deficiency without Hyperphenylalaninemia // The American J. Human Genetics. 2001. V. 69. № 2. P. 269. https://doi.org/10.1086/321970
Brown H.E., Roffman J.L. Vitamin Supplementation in the Treatment of Schizophrenia // CNS drugs. 2014. V. 28. №7. P. 611.
Choi Y.K., Tarazi F.I. Alterations in dopamine and glutamate neurotransmission in tetrahydrobiopterin deficient spr-/- mice: relevance to schizophrenia // BMB Rep. 2010. V. 43. P. 593.
Clelland J.D., Read L.L., Smeed J., Clelland C.L. Regulation of cortical and peripheral GCH1 expression and biopterin levels in schizophrenia-spectrum disorders // Psychiatry Research. 2018. Vol. 262. P. 229. https://doi.org/10.1016/j.psychres.2018.02.020
Coppen A., Swade C., Jones S.A. et al. Depression and tetrahydrobiopterin: The folate connection // J. Affective Disorders. 1989. V. 16. № 2–3. P. 103. https://doi.org/10.1016/0165-0327(89)90062-1
Coyle J.T. Glutamate and schizophrenia: beyond the dopamine hypothesis // Cell Mol Neurobiol. 2006. V. 26. P. 365.
Fu J., Ma G., Mai H. et al. Association study of sepiapterin reductase gene promoter polymorphisms with schizophrenia in a Han Chinese population // Neuropsychiatric Disease and Treatment. 2015. P. 2793. https://doi.org/10.2147/ndt.s92986
Garay R.P., Bourin M., de Paillette E. et al. Potential serotonergic agents for the treatment of schizophrenia // Expert Opinion on Investigational Drugs. 2015. V. 25(2). P. 159.
Gil M., McKinney C., Lee M.K. et al. Regulation of GTP cyclohydrolase I expression by orphan receptor Nurr1 in cell culture and in vivo // J. Neurochemistry. 2006. V. 101. № 1. P. 142. https://doi.org/10.1111/j.1471-4159.2006.04356.x
Goff D.C., Bottiglieri T., Arning E. et al. Folate, Homocysteine, and Negative Symptoms in Schizophrenia // American J. Psychiatry. 2004. V. 161. № 9. P. 1705. https://doi.org/10.1176/appi.ajp.161.9.1705
Halberstadt A.L., Geyer M.A. Serotonergic hallucinogens as translational models relevant to schizophrenia // Int. J. Neuropsychopharmacol. 2013. V. 13. P. 1.
Imamura K., Takeshima T., Nakaso K., Nakashima K. Homocysteine is toxic for dopaminergic neurons in primary mesencephalic culture // Neuroreport. 2007. V. 18. № 13. P. 1319.
Kaufman S. Tetrahydrobiopterin. Basic biochemistry and role in human disease // Johns Hopkins University Press, Baltimore, 1997. V. 26. P. 187.
Kiss J.P. Role of nitric oxide in the regulation of monoaminergic neurotransmission // Brain Res Bull. 2000. V. 52. P. 459.
Koshimura K., Miwa S., Lee K. et al. Enhancement of dopamine release in vivo from the rat striatum by dialytic perfusion of 6R-L-erythro-5,6,7,8-tetrahydrobiopterin // J. Neurochem. 1990. V. 54. P. 1391.
Kure S., Hou D.C., Ohura T. et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency // J. Pediatr. 1999. V. 135. P. 375.
Leeming R.J., Blair J.A., Melikian V. et al. Biopterin derivatives in human body fluids and tissues // J. Clin. Pathol. 1976. V. 29. P. 444.
Levine J., Sela B.A., Osher Y., Belmaker R.H. High homocysteine serum levels in young male schizophrenia and bipolar patients and in an animal model // Prog. Neuropsychopharmacol Biol. Psychiatry. 2005. V. 29. № 7. P. 1181.
Linnebank M., Lutz H., Jarre E. et al. Binding of copper is a mechanism of homocysteine toxicity leading to COX deficiency and apoptosis in primary neurons, PC12 and SHSY-5Y cells // Neurobiol Dis. 2006. V. 23. № 3. P. 725.
Lipton S.A., Kim W.K., Choi Y.B. et al. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor // Proc. Natl. Acad. Sci. USA. 1997. V. 94. P. 5923. https://doi.org/10.1073/pnas.94.11.5923
Mataga N., Imamura K., Watanabe Y. 6R-tetrahydrobiopterin perfusion enhances dopamine, serotonin, and glutamate outputs in dialysate from rat striatum and frontal cortex // Brain Res. 1991. V. 551. P. 64.
Miller B.J., Culpepper N., Rapaport M.H. C-reactive protein levels in schizophrenia // Clin Schizophr Relat Psychoses. 2013. V. 21. P. 1.
Mudd S.H., Freeman J.M. N-5,10-methylenetetrahydrofolate reductase deficiency and schizophrenia: a working hypothesis // J. Psychiatr. Res. 1974. V. 11. P. 259.
Murr C., Widner B., Wirleitner B., Fuchs D. Neopterin as a marker for immune system activation // Curr Drug Metab. 2002. V. 3. P. 175.
Neurauter G., Schrocksnadel K., Scholl-Burgi S. et al. Chronic Immune Stimulation Correlates with Reduced Phenylalanine Turnover // Current Drug Metabolism. 2008. V. 9. №7. P. 622. https://doi.org/10.2174/138920008785821738
Nishi A., Numata S., Tajima A. et al. Meta-analyses of blood homocysteine levels for gender and genetic association studies of the MTHFR C677T polymorphism in schizophrenia //Schizophrenia bulletin. 2014. V. 40. № 5. P. 1154.
Okusaga O.O. 6R-l-erythro-5,6,7,8-tetrahydrobiopterin (BH4): A potential treatment for all symptom domains of schizophrenia // Medical Hypotheses. 2014. V. 82. № 3. P. 395.
Prast H., Philippu A. Nitric oxide as modulator of neuronal function // Prog Neurobiol. 2001. V. 64. P. 51.
Ramirez J., Garnica R., Boll M.-C. et al. Low concentration of nitrite and nitrate in cerebrospinal fluid from schizophrenic patients; a pilot study // Schizophr Res 2004. V. 86. P. 357.
Rao M.L., Gross G., Strebel B. et al. Serum amino acids, central monoamines, and hormones in drug-naive, drug-free, and neuroleptic-treated schizophrenic patients and healthy subjects // Psychiatry Res. 1990. V. 34. P. 243.
Richardson M.A., Read L.L., Taylor Clelland C.L. et al. Evidence for a Tetrahydrobiopterin Deficit in Schizophrenia // Neuropsychobiology. 2005. V. 52. P. 190.
Richardson M.A., Read L.L., Reilly M.A. et al. Analysis of Plasma Biopterin Levels in Psychiatric Disorders Suggests a Common BH4 Deficit in Schizophrenia and Schizoaffective Disorder // Neurochemical Research. 2006. V. 32. № 1. P. 107. https://doi.org/10.1007/s11064-006-9233-5
Roffman J.L., Brohawn D.G., Nitenson A.Z. et al. Genetic Variation Throughout the Folate Metabolic Pathway Influences Negative Symptom Severity in Schizophrenia // Schizophrenia Bulletin. 2011. V. 39. № 2. P. 330. https://doi.org/10.1093/schbul/sbr150
Roffman J.L., Lamberti J.S., Achtyes E. et al. Randomized Multicenter Investigation of Folate Plus Vitamin B 12 Supplementation in Schizophrenia // JAMA Psychiatry. 2013. V. 70. 5. P. 481. https://doi.org/10.1001/jamapsychiatry.2013.900
Roffman J.L., Petruzzi L.J., Tanner A.S. et al. Biochemical, physiological and clinical effects of l-methylfolate in schizophrenia: a randomized controlled trial // Molecular Psychiatry. 2017. V. 23. № 2. P. 316.
Shi Q., Savage J.E., Hufeisen S.J. et al. l-Homocysteine Sulfinic Acid and Other Acidic Homocysteine Derivatives Are Potent and Selective Metabotropic Glutamate Receptor Agonists // J. Pharmacol. Exp. Ther. 2003. V. 305. P. 131.
Snyder S.H., Ferris C.D. Novel neurotransmitters and their neuropsychiatric relevance // Am J Psychiatry. 2000. V. 157. P. 1738.
Sumi-Ichinose C., Urano F., Kuroda R. et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin: a study from 6-pyruvoyltetrahydropterin synthase knockout mice // J Biol Chem. 2001. V. 276. P. 41150.
Wang D., Zhai J.X., Liu D.W. Serum folate levels in schizophrenia: A meta-analysis // Psychiatry Research. 2016. V. 235. P. 83. https://doi.org/10.1016/j.psychres.2015.11.045
Xing G., Zhang L., Russell S., Post R. Reduction of dopamine-related transcription factors Nurr1 and NGFI-B in the prefrontal cortex in schizophrenia and bipolar disorders // Schizophr. Res. 2006. V. 84. P. 36.
Yadav U., Kumar P., Gupta S., Rai V. Role of MTHFR C677T gene polymorphism in the susceptibility of schizophrenia: An updated meta-analysis // Asian J. Psychiatr. 2016. V. 20. P. 41.
Zhang Y., Hodgson N.W., Trivedi M.S. et al. Decreased brain levels of vitamin B12 in aging, autism and schizophrenia // PLoS One. 2016. V. 11. № 1. P. e0146797.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук