

Успехи физиологических наук, 2021, T. 52, № 1, стр. 90-104
Сиртуины: роль в регуляции окислительного стресса и патогенезе нейродегенеративных заболеваний
А. Э. Пухальская a, А. С. Дятлова a, Н. С. Линькова a, b, c, *, И. М. Кветной a, d, e
a Отдел биогеронтологии АНО ВО НИЦ “Санкт-Петербургский институт биорегуляции и геронтологии”
Санкт-Петербург, Россия
b Кафедра терапии, гериатрии и антивозрастной медицины Академии постдипломного образования
ФГБУ ФНКЦ ФМБА России
Москва, Россия
c Кафедра медико-биологических дисциплин, Белгородский национальный исследовательский государственный университет
Белгород, Россия
d Кафедра патологии Санкт-Петербургского государственного университета
Санкт-Петербург, Россия
e Центр молекулярной биомедицины, ФГБУ “Санкт-Петербургский научно-исследовательский институт фтизиопульмонологии” Минздрава РФ
Санкт-Петербург, Россия
* E-mail: miayy@yandex.ru
Поступила в редакцию 07.07.2020
После доработки 23.08.2020
Принята к публикации 28.09.2020
Аннотация
Сиртуины (SIRTs) – семейство гистоновых деацетилаз, эпигенетически регулирующих основные функции клеток. В обзоре проанализирована роль SIRTs в регуляции окислительно-восстановительных реакций в клетке при стрессе. Окислительный стресс и митохондриальная дисфункция являются одной из причин развития нейродегенеративных патологий – болезней Альцгеймера (БА) и Паркинсона (БП). SIRTs, обеспечивающие антиоксидантную защиту нейронов, могут играть важную роль в патогенезе БА и БП. В статье обобщены молекулярные механизмы нейропротекторных свойств SIRT1, 2, 3, 6 при БА и SIRT1, 3 – при БП. Роль других белков семейства SIRTs в патогенезе нейродегенеративных заболеваний требует дальнейшего изучения. SIRTs могут являться потенциальными маркерами диагностики и терапевтическими мишенями при БА и БП.
ВВЕДЕНИЕ
Сиртуины (SIRTs) относятся к III классу гистоновых деацетилаз и являются универсальными регуляторами функций клеток. Гистоновые деацетилазы обеспечивают гипоацетилирование остатков лизина гистоновых белков. Это приводит к сокращению расстояния между нуклеосомой и ДНК и вызывает изменение транскрипции генов. SIRTs в качестве кофактора используют никотинамидадениндинуклеотид (НАД+), а “классические” деацетилазы классов I, II, IV регулируют экспрессию генов без участия НАД+. Кроме того, белки классов I, II и IV гомологичны между собой, но не имеют гомологии с SIRTs [62, 80]. Интересно, что мишенями гистоновых деацетилаз могут являться не только гистоны, но и некоторые другие белки, например, транскрипционный фактор p53 [51].
SIRTs были открыты как регуляторы транскрипции неактивных (“молчащих”) генов дрожжей Saccharomyces cerevisiae, вследствие чего получили название “silent information regulators”. В 1999 году Kaeberlein и McVey показали, что гиперэкспрессия гена sir2 увеличивает продолжительность жизни дрожжей Saccharomyces cerevisiae на 30% [42]. В 2000 году Imai и соавт. идентифицировали белок SIR2 как НАД-зависимую гистоновую деацетилазу (HDAC), которая деацетилирует остатки лизина К9 и К14 гистона H3 и К16 гистона H4. Авторы предположили, что способность белка SIR2 к модификации гистонов может быть связана с увеличением продолжительности жизни у дрожжей и с избыточным количеством копий гена sir2 [36]. Известно, что ацетилирование и деацетилирование гистонов по остаткам лизина являются ключевыми методами регуляции экспрессии генов в определенных областях гистонов [90]. SIRTs, помимо деацетилазной активности, обладают другими ферментативными активностями: АДФ-рибозилированием, демалонилированием, десукцинилированием [26].
Исследования SIRTs, проведенные позднее, показали сходство последовательностей этих белков у прокариот и эукариот, что свидетельствует об их высокой консервативности [28]. Каждый из семи SIRTs млекопитающих имеет консервативный НАД+-связывающий каталитический коровый домен, представленный 250–270 аминокислотными остатками. Коровый домен содержит большой домен, укладку Росманна (участок, необходимый для связывания НАД+), и малый домен – цинковую ленту с гибким спиральным субдоменом. Между этими доменами расположен участок, в котором происходит катализ [22].
Все существующие SIRTs подразделяются на 5 классов (I–IV и U). У млекопитающих выделяют 7 членов семейства сиртуинов (SIRT1–7), являющихся представителями классов I–IV и имеющих разные профили ферментативной активности и субклеточной компартментализации [22]. SIRT1 локализован в ядре, но может циркулировать в пространстве между цитоплазмой и ядром [3]. SIRT2 в основном находится в цитоплазме, но может связываться с хроматином в процессе митоза [66]. SIRT3 локализуется в митохондриях и перемещается в ядро в ответ на стресс (например, при повреждении ДНК) [88]. SIRT4 и SIRT5 локализуются в митохондриях, а SIRT6 и SIRT7 – в гетерохроматических областях и ядрышкe [66]. Основной активностью SIRT4 и SIRT6 является АДФ-рибозилирование, тогда как SIRT5 проявляет демалонилирующую и десукцинилирующую активность [18].
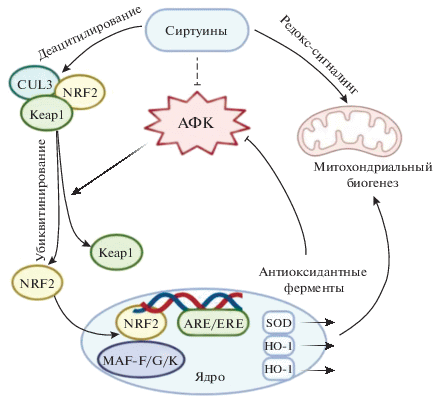
У млекопитающих показано участие SIRTs в образовании гетерохроматина, сайленсинге транскрипции, регуляции ионных каналов и модуляции окислительно-восстановительных процессов. Известно, что SIRTs участвуют в регуляции секреции инсулина, гликолизе, орнитиновом цикле, биогенезе митохондрий, глюкогенезе, окислении жиров [10, 13, 31, 34, 99]. SIRTs вовлечены в патогенез различных ассоциированных с возрастом заболеваний, в том числе нейродегенеративных [30, 97]. Некоторые целевые белки SIRTs, такие как Keap1 и CUL3, являются эффекторами основных путей окислительного стресса (рис. 1) [65, 95, 104, 108]. Тем не менее, механизмы, лежащие в основе воздействия каждого SIRT на эти эффекторы и на антиоксидантную и окислительно-восстановительную передачу сигналов, представляют собой активно развивающуюся область исследований.
Рис. 1.
Схема взаимодействия сиртуинов с NRF2: регуляция экспрессии генов антиоксидантной защиты (по [93]). Обозначения: CUL3 – куллин 3; ARE – элемент антиоксидантного ответа; ERE – элемент электрофильного ответа; GSH – глутатион; HO-1 – гемоксигеназа 1; Keap1 – kelch-подобный ECH-ассоциированный белок 1; NRF2 – ядерный фактор E2; АФК – активные формы кислорода; SOD – супероксиддисмутаза.
Цель обзора – анализ роли SIRTs в окислительно-восстановительной регуляции, окислительном стрессе и патогенезе нейродегенеративных заболеваний.
Роль SIRTs в окислительно-восстановительном балансе. Свободные радикалы (активные формы кислорода (АФК) и азота) генерируются в процессе многочисленных метаболических и биосинтетических путей, включая дыхательную цепь митохондрий, фагоцитоз, синтез простагландина и систему цитохрома P450. Кроме того, неферментативные клеточные реакции также могут служить источником свободных радикалов. Примерами таких реакций являются взаимодействие между кислородом и органическими соединениями и реакции, инициируемые ионизирующим излучением. В норме АФК и азота необходимы для протекания биохимических реакций и стимуляции сигнальных каскадов. Однако избыток свободных радикалов может повреждать ДНК, РНК и другие биологически важные молекулы в клетке.
Антиоксиданты, как правило, действуют по одному из двух путей: либо прерывая каскад образования свободных радикалов, либо предотвращая его возникновение. Основные антиоксидантные ферменты, такие как супероксиддисмутаза (SOD), каталаза и глутатионпероксидаза, предотвращают запуск окислительного каскада, расщепляя молекулы, ответственные за образование свободных радикалов. Другой важный тип антиоксидантных ферментов, пероксиредоксины, контролируют уровень пероксида в клетке.
Идея о том, что SIRTs участвуют в регуляции окислительно-восстановительного баланса, подтверждается их взаимосвязью с несколькими молекулами-элементами антиоксидантного ответа (ARE), которые опосредуют сигнальные каскады регуляции транскрипции генов в клетках, подверженных окислительному стрессу. Молекулы ARE обладают биологическими и структурными особенностями, которые позволяют улавливать изменения окислительно-восстановительного статуса клеток. После этого они активируют транскрипционные ответы, опосредованные главным образом молекулой NRF2 [113, 115]. В условиях отсутствия стресса белок NRF2 локализуется в цитоплазме, где разрушается кластером специализированных белков Keap1 и CUL3 путем убиквитинирования. При окислительном стрессе система убиквитинирования Keap1-CUL3 нарушается, NRF2 накапливается в цитоплазме, транслоцируется в ядро, где инициирует транскрипцию антиоксидантных генов и их белков совместно с одним из белков Maf (MAFF, MAFG, MAFK) [49]. В ядре NRF2 связывается с элементом электрофильного ответа (ERE), который дополнительно модулирует экспрессию генов, участвующих в детоксикации и элиминации электрофильных агентов, что приводит к повышению антиоксидантной активности клеток (рис. 1) [119]. При дисрегуляции ARE/ERE окислительный стресс может привести к развитию нейродегенеративных, аутоиммунных, сердечно-сосудистых заболеваний, канцерогенезу и ускоренному старению организма [87].
Было показано, что SIRT2 деацетилирует NRF2, что приводит к снижению общего и ядерного уровней NRF2 [112]. Кроме того, SIRT2 также может регулировать уровень NRF2 в ядре путем регуляции фосфорилирования Akt-киназы, что приводит к модуляции уровней общего глутатиона и глутамат-цистеинлигазы. Таким образом, SIRT2 может быть ключевым модулятором данного аспекта антиоксидантного ответа [14]. SIRT1, по-видимому, также вовлечен в этот процесс. Нокдаун SIRT1 ингибирует экспрессию генов NRF2, HO-1, SOD1. Антиоксидант ресвератрол, активатор SIRT1, модулирует экспрессию NRF2-зависимых генов, что способствует нейропротекции при церебральных ишемических повреждениях [109].
Установлено, что SIRT6 коактивирует NRF2 для защиты мезенхимальных стволовых клеток человека (hMSCs) от окислительного стресса [79]. Интересно, что сигнальные пути, ассоциированные с окислительным стрессом, по-видимому, участвуют в модуляции активности SIRTs, а также влияют на их экспрессию, посттрансляционные модификации и белок-белковые взаимодействия [86].
Таким образом, большинство SIRTs млекопитающих могут быть связаны с передачей сигналов при окислительном стрессе. Ниже подробно рассмотрены имеющиеся данные об участии каждого сиртуина человека в регуляции окислительно-восстановительного баланса.
SIRT1 является наиболее изученным среди всех сиртуинов млекопитающих. SRT2104, синтетический активатор SIRT1, снижает уровень маркеров перекисного окисления липидов в печени и мышцах, повышает уровень SOD2 в мышечной ткани мышей линии C57BL/6 [64]. По-видимому, такое действие SIRT1 может быть опосредовано ключевыми редокс-чувствительными факторами транскрипции, включая FOXO3a и p53. Семейство транскрипционных факторов FOXO участвует в регуляции широкого спектра генов, связанных с антиоксидантной защитой [48]. Известно, что SIRT1 деацетилирует белок FOXO3a, который индуцирует антиоксидантный ответ посредством активации СОД2 и каталазы. Деацетилирование белка FOXO3a при помощи SIRT1 приводит к его активации. Это способствует повышению антиоксидантной защиты. Кроме того, FOXO3a регулирует экспрессию митохондриальных генов, что приводит к модуляции уровня АФК [20]. р53, обычно рассматриваемый как белок-супрессор опухолей, также является редокс-чувствительным белком и субстратом SIRT1 [61]. В отсутствие клеточного стресса p53 способен снижать уровень внутриклеточных АФК и повышать продукцию антиоксидантных белков, таких как SOD2 и глутатионпероксидаза-1. Нарушение регуляции p53 приводит к повышению внутриклеточных уровней АФК и окислению ДНК [77].
Показано, что SIRT1 регулирует ацетилирование транскрипционного фактора PGC-1α, главного регулятора митохондриального биогенеза [71].
По-видимому, окислительный стресс может вызвать нарушение функционирования SIRT1. В ответ на окислительный стресс SIRT1 перераспределяется на уровне хроматина, вызывая нарушения регуляции транскрипции. Избыток перекиси водорода приводит к активации генов, связанных с SIRT1, включая те, которые участвуют в метаболизме, апоптозе, ионном транспорте, подвижности клеток и передаче сигналов посредством G-белка [74]. Вызванный пероксидом водорода окислительный стресс подавляет синтез SIRT1 в кератиноцитах в зависимости от дозы и времени воздействия. Обработка клеток активатором SIRT1 ресвератролом предотвращает вызванную окислителем гибель клеток и предотвращает их старение [14]. Ингибиторы SIRT1, сиртинол и никотинамид, усиливают апоптоз клеток, вызванный перекисью водорода [35]. Эти данные свидетельствуют о том, что SIRT1 является ключевой молекулой в предотвращении окислительного повреждения клеточных структур.
SIRT2 экспрессируется в головном мозге, почках, поджелудочной железе, яичках, печени и жировой ткани млекопитающих [25]. В цитоплазме SIRT2 выполняет функцию организации цитоскелета, деацетилируя α-тубулин. В ядре SIRT2 деацитилирует остаток лизина K16 в гистоне H4, участвуя в регуляции клеточного цикла [26]. Кроме того, SIRT2 участвует в формировании ядерной мембраны посредством деацетилирования белка ANKLE2 – регулятора сборки ядерной оболочки [44]. Активность и экспрессия SIRT2 изменяются в зависимости от энергетического состояния клетки: активируются в состоянии с низким уровнем энергии и подавляются в состоянии с высоким уровнем энергии [25]. Это предполагает участие SIRT2 в регуляции энергетического метаболизма и клеточного гомеостаза.
SIRT2 также активирует регулятор митохондриального биогенеза PGC-1α, что приводит к повышению экспрессии антиоксидантных ферментов и снижению уровня АФК. Как и SIRT1, SIRT2 деацетилирует FOXO3a в ответ на окислительный стресс [50].
Другими мишенями SIRT2 являются метаболические ферменты: глюкозо-6-фосфатдегидрогеназа (G6PD), фосфоглицератмутаза (PGAM2), транскрипционный фактор NF-κB [25]. В условиях окислительного стресса SIRT2 активирует G6PD, ключевой фермент в пентозофосфатном пути, который продуцирует НАДФ-Н в цитозоле. НАДФ-Н является молекулой, необходимой для противодействия окислительному повреждению, сохраняя глутатион в восстановленной форме. Кроме того, SIRT2 активирует транскрипционный фактор NF-κB. Продемонстрировано, что транскрипция нескольких NF-κB-зависимых генов влияет на уровни АФК в клетке. В свою очередь, активность NF-κB регулируется уровнями АФК. NF-κB регулирует синтез ферментов, способствующих выработке АФК, таких как НАДФН-оксидаза, ксантиноксидоредуктаза, индуцибельная NO-синтаза, циклооксигеназа-2 и цитохром р450. NF-κB также регулирует активность ферментов SOD1, 2, тиоредоксины и глутатион-S-трансферазы, которые способствуют ингибированию АФК. SIRT2, по-видимому, обеспечивает регуляцию NF-κB в зависимости от окислительно-восстановительного состояния клетки. Эти данные свидетельствуют о том, что SIRT2 играет критическую роль в модуляции окислительного стресса, защищая организм от метаболических нарушений.
SIRT3 локализуется в ядре, но в ответ на стрессорные сигналы, такие как повреждение ДНК, транспортируется в митохондрии, где расщепляется при помощи пептидазы до активной формы. SIRT3 модулирует метаболизм митохондрий и повышает продолжительность жизни экспериментальных животных. Для мышей, у которых SIRT3 не синтезируется, характерно снижение потребления кислорода и одновременное увеличение продукции АФК в клетках, а также более высокие показатели окислительного стресса в мышцах [40]. Аналогичные результаты были получены при исследовании культур клеток линий MCF7, T47D и CAMA, не синтезирующих SIRT3. В этих клетках повышалась экспрессия АФК, что могло вызывать повреждение ДНК и активировать молекулу HIF1α, вовлеченную в механизм нарушения васкуляризации и ангиогенеза, энергетического обмена, выживаемости клеток и инвазии опухолей [21]. При ограничении калорийности питания SIRT3 активирует ферменты 3-гидрокси-3-метилглутарил-КoA-синтазу и ацил-КoA-дегидрогеназу, образование кетоновых тел и окисление длинноцепочечных жирных кислот, что связано с увеличением продолжительности жизни [92].
Было показано, что полиморфизм в гене SIRT3 чаще встречается у долгожителей [6, 7]. Переменное число тандемных повторов в энхансерной области 5 интрона гена SIRT3, по-видимому, влияет на активность этого энхансера. Был сделан вывод о том, что люди, несущие аллель с наименее активным энхансером, с меньшей вероятностью доживут до старости. Такой вариант аллеля практически отсутствовал у мужчин старше 90 лет, проживающих в Италии [6]. Тем не менее, более поздние исследования на больших выборках не подтвердили эти выводы. Это позволяет предположить, что влияние SIRT3 на продолжительность жизни менее значительно, чем было заявлено ранее [53].
Как и другие сиртуины, SIRT3 опосредует деацетилирование ферментов, которые ответственны за снижение АФК в клетке, что приводит к защите от окислительного стресса, ассоциированных с ним патологий (нейродегенеративные, сердечно-сосудистые заболевания, канцерогенез) и ускоренного старения организма [2]. SIRT3 активирует изоцитратдегидрогеназу (IDH2), SOD2 и каталазу – ферменты, нейтрализующие АФК [65]. У мышей, нокаутных по SIRT3, находящихся на рационе с высоким содержанием холестерина, повышается уровень АФК в эндотелии сосудов [106]. Повышенные уровни АФК в клетке стимулируют транскрипцию SIRT3 и, следовательно, могут приводить к деацетилированию SOD2 через петлю обратной связи [65].
Пониженные уровни SIRT3 были обнаружены в эпидермальных кератиноцитах человека после воздействия озона, что коррелировало с повреждением ДНК, более высоким уровнем перекиси водорода и сниженной концентрацией SOD2 в клетке [63]. Отсутствие регуляции АФК в кератиноцитах, которые формируют и поддерживают защитный слой кожи, влияет на их дифференцировку. Когда в кератиноцитах происходит дисрегуляция SIRT3, уровни супероксид аниона увеличиваются, способствуя экспрессии маркеров дифференцировки. Противоположное наблюдается в кератиноцитах с индуцированной гиперэкспрессией SIRT3. В этих клетках снижены уровни супероксид аниона и экспрессия маркеров дифференцировки. Таким образом, SIRT3 участвует в подавлении дифференцировки эпидермиса посредством снижения окислительного стресса [4]. SIRT3 модулирует митохондриальную функцию, регулируя уровни НАД+, и может выступать в качестве защитного фактора от острых заболеваний печени и почек [67]. Таким образом, показана роль SIRT3 в защите клеток от окислительного повреждения и генотоксического стресса.
SIRT4 локализован в митохондриях и участвует в рибозилировании аденозиндифосфата (АДФ) [1]. Высокая экспрессия SIRT4 обнаружена в тканях сердца, почек, печени и мозга. Изначально считалось, что SIRT4 не обладает НАД-зависимой деацетилазной активностью. Однако недавно установлено, что SIRT4 обладает способностью деацетилировать лизин, что позволяет ему контролировать секрецию инсулина. У мышей, нокаутных по SIRT4, наблюдается повышенная секреция инсулина [1].
Все сиртуины, кроме SIRT4, играют ключевую роль в снижении митохондриального окислительного стресса при диете с ограничением калорийности [103], однако это не отменяет роль SIRT4 в регуляции окислительного стресса.
SIRT4 участвует в регуляции продукции АФК в митохондриях, хотя неясно, влияет ли он на активацию антиоксидантных ферментов, локализованных в митохондриальном матриксе. При вызванной ангиотензином II (AngII) гипертрофии сердца у мышей гиперэкспрессия SIRT4 способствовала снижению содержания АФК в митохондриях кардиомиоцитов. Нокаут гена SIRT4 в этой модели приводил к повышенному синтезу АФК в митохондриях кардиомиоцитов [59]. Аналогичные результаты наблюдались в кардиомиоцитах крыс, что позволяет предположить, что SIRT4 может контролировать синтез АФК в клетках сердца. Установлено, что SIRT4 ингибирует связывание SOD2 с SIRT3, что приводит к увеличению ацетилирования и снижению активности этого фермента [59]. Эти результаты свидетельствуют о том, что SIRT4 может играть важную роль в управлении сигнальными молекулами, участвующими в антиоксидантной реакции.
Показано, что SIRT4 является неотъемлемым фактором окисления жирных кислот в печени и мышечных клетках. Нокдаун SIRT4 увеличивает окисление жирных кислот и потребление кислорода в гепатоцитах мыши, возможно, посредством регуляции экспрессии SIRT1 [70]. Окисление жирных кислот является ключевым источником митохондриальных АФК, а нарушение регуляции этого процесса связано с повреждением почек при сахарном диабете [83].
SIRT4 исследуется в качестве биомаркера коронарных заболеваний сердца. У пациентов с сердечно-сосудистой патологией, ожирением и жировым гепатозом уровень SIRT4 в крови ниже, чем у здоровых людей [94]. Подобных исследований немного, поэтому неясно, может ли SIRT4 служить в качестве биомаркера ишемической болезни сердца. К тому же, экспрессия SIRT4 различается в зависимости от типа клеток, что необходимо учитывать перед использованием его в качестве биомаркера различных заболеваний.
SIRT5 локализуется в митохондриях, а его функция заключается в деацетилировании, демалонилировании и десукцинировании белков [26]. Экспрессия SIRT5 обнаружена в тканях головного мозга, сердца, печени и в лимфобластах, где он накапливается в межмембранных пространствах митохондрий [58]. SIRT5 участвует в клеточном метаболизме, детоксикации, регуляции окислительного стресса, энергетическом балансе, а также выступает как посредник при апоптозе [56]. Тем не менее, авторы не пришли к консенсусу относительно роли SIRT5 в этих процессах.
SIRT5 известен как регулятор β-окисления жирных кислот в митохондриях, цикла мочевины и клеточного дыхания [110]. SIRT5 деацетилирует и активирует карбамоилфосфатсинтетазу (CPS1), которая катализирует первую стадию цикла мочевины. Показано, что у мышей, нокаутных по SIRT5, во время голодания повышается уровень аммиака в моче. У мышей со сверхэкспрессией SIRT5 наблюдалась повышенная активность CPS1, что способствовало превращению аммиака в менее токсичную мочевину [75]. Аммиак активирует синтез АФК и снижает содержание глутатиона в клетке [11], что свидетельствует о косвенном участии SIRT5 в регуляции окислительного стресса.
Интересно, что SIRT5 защищает кардиомиоциты от апоптоза, индуцированного окислительным стрессом [56]. Подавление окислительного стресса рассматривается как возможный механизм предотвращения апоптоза в клетках нейробластомы линии SH-EP [54]. Эти результаты согласуются с данными, полученными на опухолевых и эпителиальных клетках легких [55, 101]. Показано, что SIRT5 связывается с SOD1 и десукцинирует ее, повышая ее активность. SOD1 опосредованное снижение АФК наблюдалось при коэкспрессии SOD1 и SIRT5. Вероятно, SIRT5 осуществляет посттрансляционную регуляцию SOD1 в опухолевых клетках легких [55].
В клетках, трансфецированных SIRT5, снижается концентрация АФК. Это позволяет предположить, что SIRT5 подавляет развитие окислительного стресса в клетке. Вероятно, функция SIRT5 заключается в обеспечении клеточного ответа на окислительный стресс.
SIRT6 локализован в ядре клетки, а его функцией является НАД+-зависимое деацетилирование лизинов K9 и K56 гистона H3 (H3K9 и H3K56) [96]. SIRT6-опосредованное деацетилирование гистона H3 способствует регуляции экспрессии генов посредством рекрутирования факторов транскрипции, например NF-κB [117]. Помимо того, что SIRT6 играет важную роль в регуляции структуры хроматина и рекрутировании транскрипционных факторов, он участвует в репарации ДНК [38]. SIRT6 регулирует темп старения у млекопитающих [26]. У мышей, нокаутных по SIRT6, снижалась продолжительность жизни и наблюдался фенотип преждевременного старения, включая снижение уровней глюкозы и инсулиноподобного фактора роста (IGF-1) в крови [68]. Учитывая важную роль SIRT6 в клеточном гомеостазе, нарушения синтеза этого фермента, по-видимому, оказывают влияние на развитие различных патологических процессов [5, 84]. SIRT6 считается важным метаболическим сенсором, который обеспечивает связь сигналов от окружающей среды с метаболическим гомеостазом и реакциями на стресс у млекопитающих [5, 100].
Предполагается роль SIRT6 как медиатора окислительного стресса и маркера повреждения миокарда при ишемии-реперфузии. Сверхэкспрессия SIRT6 защищает кардиомиоциты от повреждения при ишемии-реперфузии путем снижения окислительного стресса и активизации эндогенных антиоксидантов через ось AMPK–FOXO3α, обеспечивающую устойчивость к окислительному стрессу [100].
Показано, что SIRT6 защищает hMSC от окислительного стресса путем активации NRF2. В hMSC, не синтезирующих SIRT6, нарушается окислительно-восстановительный метаболизм, что приводит к повышенной чувствительности к окислительному стрессу. Было высказано предположение, что SIRT6 служит коактиватором NRF2, запускающим антиоксидантные пути ответа на окислительный стресс [79].
Кроме того, SIRT6 и NF-κB показали протекторный эффект при ускоренном старении эндотелия, опосредованном высокими уровнями глюкозы. Снижение уровня SIRT6 во время кратковременного воздействия высокого уровня глюкозы приводило к увеличению экспрессии NF-κB, в то время как гиперэкспрессия SIRT6 снижала экспрессию NF-κB. Защитные эффекты антиоксиданта эрготионеина связывают с увеличением количества SIRT1 и SIRT6 в клетках и их негативной регуляцией NF-κB. Это указывает на высокий потенциал обоих SIRTs в отношении регуляции редокс-сигналов [17]. SIRT6 также участвует в контроле воспаления при диабетических атеросклеротических поражениях эндотелия [57]. В ответ на стресс наблюдается репрессия SIRT6, что приводит к ацетилированию гистонов и повышению экспрессии генов [116]. Функции SIRT6, по-видимому, являются антигликолитическими и антиоксидантными, что обеспечивает защиту клеток от АФК [33].
SIRT7 экспрессируется в ядрышке, где осуществляет позитивную регуляцию транскрипции рибосомальной ДНК (рДНК) путем связывания с гистонами [26]. Различный уровень экспрессии мРНК SIRT7 обнаруживается во всех тканях, но более высокий наблюдается в тканях с высокой метаболической активностью. При старении у людей уровень экспрессии SIRT7 снижается [46], а у нокаутных по SIRT7 мышей наблюдается преждевременное старение [98]. При физиологическом старении SIRT7 может транслоцироваться из ядрышка в цитоплазму и хроматин, где ингибирует транскрипцию рДНК [47].
Было показано, что сверхэкспрессия SIRT7 активирует транскрипцию, опосредованную РНК-полимеразой I, тогда как нокдаун или ингибирование SIRT7 снижает ее. SIRT7 играет ключевую роль в энергетическом балансе клетки и в условиях стресса способствует прекращению транскрипции рДНК. Обнаружена роль SIRT7 в регуляции митохондриального гомеостаза посредством деацетилирования белка GABPβ1, одной из субъединиц комплекса, участвующего в регуляции экспрессии митохондриальных генов Clpp, Polrmt, Mfn1, Fars2, Elac2, Nt5m [85].
Таким образом, SIRTs представляют собой особый класс эпигенетических регуляторов транскрипции, что определяет их важную роль в модуляции экспрессии широкого спектра генов. Важнейшей функцией SIRTs является их роль в поддержании окислительно-восстановительного баланса клетки. В настоящее время возрастает интерес к роли SIRTs в развитии нейродегенерации и патогенезе болезней Альцгеймера и Паркинсона.
Роль SIRTs в старении головного мозга и развитии нейродегенеративных заболеваний. Процесс старения характеризуется многочисленными изменениями на организменном, тканевом и клеточном уровнях. С возрастом стареющие клетки накапливаются в тканях, нарушая их нормальное функционирование. Сенесцентные клетки модифицируют микроокружение, секретируя цитокины, хемокины и медиаторы воспаления. Такой секреторный фенотип является одной из причин наблюдаемого у людей пожилого и старческого возраста хронического воспаления (inflamm-aging), а также может ускорять темп репликативного старения соседних клеток. Сенесцентные клетки, кроме секреторного фенотипа, обладают рядом особенностей, таких как повышенный уровень секреции ингибиторов клеточного цикла, активность β-галактозидазы, наличие повреждений ДНК. Увеличение повреждений ДНК с возрастом является результатом нарушения эффективности систем репарации ДНК. Считается, что повреждения ДНК являются основной причиной клеточного старения. Это касается как репликативного, так и стрессового (окислительного, генотоксического) старения. Повреждения ДНК нарушают нормальное функционирование клеток, но в норме эффективности систем репарации достаточно для защиты клеток от накопления повреждений ДНК. Однако возрастное снижение репарации ДНК приводит к накоплению повреждений и, как следствие, старению клеток [72].
Как было сказано выше, основными функциями SIRTs являются репарация ДНК, контроль воспаления и обеспечение антиоксидантной защиты. Поэтому SIRTs рассматриваются в качестве факторов, способных замедлить возрастные изменения организма путем обеспечения физиологического уровня репарации ДНК и регуляции окислительно-восстановительного баланса.
В ряде работ показана роль SIRTs в патогенезе болезни Альцгеймера (БА) и других нейродегенеративных заболеваний. Снижение уровней мРНК и белков SIRT1, SIRT3 в головном мозге пациентов с БА коррелирует со стадией и продолжительностью заболевания [41, 60]. Аналогичные данные о снижении уровня SIRT1 были получены in vitro на линии клеток нейробластомы SH-SY5Y, обработанной нейротоксическим амилоидом Aβ25-35 [52]. В модели БА у мышей наблюдалась повышенная экспрессия мРНК SIRT3, которая соответствовала пространственному и временному профилям накопления амилоида Aβ. У пациентов старческого возраста с БА высокий уровень мРНК SIRT3 наблюдался в височных долях мозга [105]. SIRT5 был идентифицирован в активированной микроглии мозга пациентов с БА [60]. Взаимодействие амилоида Aβ42, сфингозинкиназ и митохондриальных SIRT3–5 может играть важную роль в патогенезе БА [15]. При этом избыточная экспрессия белка-предшественника амилоида (АРР) и пресенилина-1 приводила к снижению экспрессии мРНК и синтеза белка SIRT3 в мозге мышей с БА. Это предполагает более сложные механизмы взаимодействия SIRTs и основных молекул, участвующих в патогенезе БА [111].
Предполагается, что SIRT1 обеспечивает баланс между амилоидогенным и неамилоидогенным процессингом APP, предотвращая развитие БА [82]. Кроме того, SIRT1 может способствовать деградации Aβ через путь LKB1/AMPKα, основной функцией которого является контроль метаболизма и роста нейронов [81].
Также сообщается, что активация или сверхэкспрессия SIRT1 влияют на токсичность Aβ, опосредованную микроглией, благодаря своей способности ингибировать передачу сигналов NF-κB [89]. SIRT1 может защищать нейроны от потери синапсов, характерной для ранних стадий БА [24].
При этом ингибитор SIRT2, молекула AGK2, вызывает смещение баланса между α- и β-секретазами, снижая амилоидную нагрузку на клетки, что приводит к улучшению когнитивных функций в моделях трансгенных мышей с БА [8]. AGK-2 снижает опосредованную амилоидом Aβ42 активацию глии [89]. Таким образом, SIRT1 и SIRT2 регулируют процессинг APP, вероятно, противоположным образом.
Помимо амилоидной гипотезы развития БА существует также τ-гипотеза. Предполагается, что накопление патологически модифицированного τ-протеина, ассоциированного с микротрубочками, приводит к формированию нейрофибриллярных клубков, что становится причиной нарушения аксонального транспорта и разрушения аксонов. По некоторым данным SIRTs опосредуют лептин-зависимое ингибирование фосфорилирования τ-белка [27]. SIRT1 также осуществляет деацетилирование τ-протеина. Поэтому изменения активности SIRTs могут снижать количество нейрофибриллярных клубков [16]. Более того, SIRT1 и τ-белок имеют общий восходящий механизм регуляции, являясь мишенями для микроРНК-132 и AMPK-киназы [32, 81].
SIRT3, по-видимому, также участвует в патогенезе БА путем модуляции фосфорилирования τ-белка. Нокдаун Sirt3 в моделях ex vivo и in vivo вызывал повышение фосфорилированного τ-протеина. Кроме того, в аутопсийном материале коры головного мозга, полученном от пациентов с БА экспрессия Sirt3 была снижена. Вероятно, повышенные уровни Aβ могут снижать экспрессию Sirt3, что приводит к повышению его ацетилирования и формированию нейрофибриллярных клубков [114]. С другой стороны, сообщается, что сверхэкспрессия Sirt3 предотвращает индуцированные Aβ патологические изменения в мозге мышей с БА, поэтому направление взаимодействия SIRT3 и Aβ еще предстоит выяснить [114].
У 4-месячных мышей с дефицитом синтеза SIRT6 наблюдаются нарушения в поведении и обучении. При гистологическом исследовании в головном мозге таких животных обнаруживают большее количество поврежденной ДНК и гиперфосфорилированного τ-белка. Вероятно, SIRT6 регулирует стабильность и фосфорилирование τ-протеина путем активации киназы GSK3α/β [43].
Помимо Aβ и τ-белка, двух важнейших молекулярных факторов патогенеза БА, SIRTs способны влиять на пути, участвующие в нейропротекции. Предполагается, что SIRT1 посредством взаимодействия с рецептором ретиноевой кислоты β (RAR-β) и дальнейшей активации металлопротеазы ADAM10 индуцирует расщепление рецептора Notch. Высвобождение внутриклеточного домена Notch активирует транскрипцию генов, связанных с нейрогенезом и нейрональной дифференцировкой в ответ на патологические повреждения. Кроме того, мишени Notch включают гены, важные для синаптической пластичности, обучения и памяти, а также для генерации синапсов [12]. Таким образом, из всех SIRTs в контексте патогенеза БА SIRT1 представляется наиболее изученным. Нейропротекторное действие SIRT1 при БА, вероятно, является многоуровневым и обусловлено как активацией сигнального пути Notch, так и влиянием на процессинг APP и метаболизм τ-протеина.
Предполагается, что в патогенез болезни Паркинсона (БП), затрагивающей дофаминергические структуры мозга, также вовлечены SIRTs. Как и в случае с БА, SIRT1 в моделях БП проявлял нейропротекторные свойства. Так, ресвератрол, являющийся активатором SIRT1, снижал проявления паркинсонизма у мышей в модели БП, индуцированной МФТП (1-метил-4-фенил-1,2,3,6-тетрагидропиридин) [9]. У мышей с МФТП-индуцированным паркинсонизмом при применении ресвератрола наблюдается опосредованная SIRT1 активация белка PGC-1α, повышающем устойчивость к окислительному стрессу и нейродегенерации [69]. Кроме того, варианты промотора гена SIRT1, связанные с пониженным синтезом белка SIRT1, коррелировали с возникновением спорадической формы БП [118].
Исследования БП в моделях на животных связывают недостаток SIRT1 с агрегацией α-синуклеина. Нейропротекторный эффект ресвератрола в модели БП in vitro, объясняется его способностью вызывать аутофагическую деградацию α-синуклеина через SIRT1 [107].
Мишенью SIRT1 при БП также могут являться шапероны, способствующие правильной укладке белков. Установлено, что шаперон Hsp70 предотвращает агрегацию α-синуклеина. SIRT1 деацетилирует фактор 1 теплового шока (HSF1), что способствует его длительному связыванию с последовательностью-мишенью в гене, кодирующем Hsp70. Это приводит к повышенной экспрессии Hsp70 в стрессовых условиях и, вероятно, предотвращает избыточное накопление α-синуклеина в нейронах [102].
Напротив, применение ингибиторов SIRT2 при МФТП-индуцированном паркинсонизме у мышей снижает потерю дофаминергических нейронов, улучшает неврологический и поведенческий дефицит [29]. Ингибитор SIRT2 AGK2 блокирует токсический эффект α-синуклеина в модели БП in vitro [78].
Согласно данным de Oliveira и соавт. (2017), сверхэкспрессия SIRT2 в нейронах черной субстанции крыс приводит к блокированию ацетилирования α-синуклеина, вызывая его агрегацию и усиливая токсичность. Кроме того, SIRT2, вероятно, облегчает транспорт агрегированного α-синуклеина путем ацетилирования α-тубулина [19]. С этой точки зрения ацетилирование представляется ключевым механизмом, регулирующим агрегацию и токсичность α-синуклеина и облегчающим его аксональный транспорт, демонстрируя потенциальную терапевтическую ценность ингибирования SIRT2 при синуклеинопатиях [76].
SIRT3 и SIRT1 показали нейропротективные свойства при БП, стабилизируя цепь переноса электронов и снижая окислительный стресс в дофаминергических нейронах черной субстанции головного мозга. В работе Gleave и соавт. продемонстрировано, что даже в том случае, когда трансдукцию вектора SIRT3-myc производили после индукции БП и развития клеточного стресса и поведенческих аномалий, усиление синтеза SIRT3 снижало степень дегенерации дофаминергических нейронов путем снижения ацетилирования белков митохондрий [23].
Делеция в гене Sirt3 усиливала окислительный стресс и снижала мембранный потенциал митохондрий в дофаминергических нейронах черной субстанции. Некоторые авторы считают, что связанное с возрастом снижение защитной функции белка SIRT3 является основным фактором, лежащим в основе усиления митохондриального окислительного стресса и апоптоза дофаминергических нейронов черной субстанции при БП [91, 120].
SIRT6 может усиливать нейродегенеративные эффекты при БП. В немногочисленных исследованиях на эту тему продемонстрировано, что уровень белка SIRT6 в мозге пациентов с БП, выше, чем у здоровых людей [37]. У нокаутных по SIRT6 мышей с МФТП-индуцированным паркинсонизмом неврологические и поведенческие изменения были менее выражены по сравнению с животными с нормальной экспрессией SIRT6 [73]. Вероятно, SIRT6 играет провоспалительную роль в патогенезе БП, способствуя выработке и секреции провоспалительных цитокинов [39]. Однако, некоторые авторы считают, что снижение экспрессии SIRT6 в головном мозге является признаком ускоренного старения и связано с прогрессированием нейродегенеративных заболеваний [37, 43, 45], в связи с чем усиление нейродегенерации при БП под действием SIRT6 требует дальнейшего изучения.
ЗАКЛЮЧЕНИЕ
Исходя из имеющихся литературных данных, основной функцией SIRTs млекопитающих является поддержание окислительно-восстановительного баланса клетки и обеспечение антиоксидантной защиты. Во многих работах авторы подчеркивают наличие взаимосвязи между активностью SIRTs и продолжительностью жизни экспериментальных животных и человека.
Не менее интересным и актуальным направлением исследований представляется изучение роли SIRTs в патогенезе нейродегенеративных расстройств. Механизмы патогенеза и способы прижизненной диагностики таких нейродегенеративных заболеваний, как БА и БП, до сих пор остаются дискуссионными. Наиболее распространенными теориями возникновения БА считаются амилоидная и τ-гипотеза, БП – синуклеиновая гипотеза. В целом они объясняют механизмы развития заболеваний, но неясно, что является триггером для накопления патогенных белков Aβ42, τ и α-синуклеина. Нарушения функции SIRTs приводят к дисрегуляции окислительно-восстановительного баланса. Это вызывает нарушения функций нейронов. Наиболее изучены в контексте патогенеза БА и БП SIRT1, 2, 3, 6. При этом SIRT1 и SIRT3, по-видимому, играют роль нейропротекторов, в то время как SIRT2 усугубляет течение БП и БА, а SIRT6 обладает разнонаправленными эффектами. Влияние SIRT4, 5, 7 на возникновение и развитие БА и БП практически не изучено. Перспективным направлением молекулярной медицины является проведение дальнейших исследований влияния SIRTs на развитие нейродегенеративных заболеваний. Это позволит рассматривать SIRTs как потенциальные мишени для фармакотерапии БА и БП.
Список литературы
Anderson K.A., Huynh F.K., Fisher-Wellman K. et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion // Cell Metabolism. 2017. № 4(25). P. 838–855.e15. https://doi.org/10.1016/j.cmet.2017.03.003
Ansari A., Rahman M.S., Saha S.K. et al. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease // Aging Cell. 2017. № 1(16). P. 4–16. https://doi.org/10.1111/acel.12538
Bai W., Zhang X. Nucleus or cytoplasm? The mysterious case of SIRT1’s subcellular localization // Cell Cycle (Georgetown, Tex.). 2016. № 24(15). P. 3337–3338. https://doi.org/10.1080/15384101.2016.1237170
Bause A.S., Matsui M.S., Haigis M.P. The protein deacetylase SIRT3 prevents oxidative stress-induced keratinocyte differentiation // The J. Biological Chemistry. 2013. № 51(288). P. 36484–36491. https://doi.org/10.1074/jbc.M113.472324
Beauharnois J.M., Bolívar B.E., Welch J.T. Sirtuin 6: a review of biological effects and potential therapeutic properties // Molecular BioSystems. 2013. № 7(9). P. 1789–1806. https://doi.org/10.1039/c3mb00001j
Bellizzi D., Dato S., Cavalcante P. et al. Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13 // Genomics. 2007. № 1(89). P. 143–150. https://doi.org/10.1016/j.ygeno.2006.09.004
Bellizzi D., Rose G., Cavalcante P. et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages // Genomics. 2005. № 2(85). P. 258–263. https://doi.org/10.1016/j.ygeno.2004.11.003
Biella G., Fusco F., Nardo E. et al. Sirtuin 2 Inhibition Improves Cognitive Performance and Acts on Amyloid-β Protein Precursor Processing in Two Alzheimer’s Disease Mouse Models // J. Alzheimer’s Disease: JAD. 2016. № 3(53). P. 1193–1207. https://doi.org/10.3233/JAD-151135
Blanchet J., Longpré F., Bureau G. et al. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice // Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2008. № 5(32). P. 1243–1250. https://doi.org/10.1016/j.pnpbp.2008.03.024
Blander G., Guarente L. The Sir2 family of protein deacetylases // Annual Review of Biochemistry. 2004. № 73. P. 417–435. https://doi.org/10.1146/annurev.biochem.73.011303.073651
Bobermin L.D., Wartchow K.M., Flores M.P. et al. Ammonia-induced oxidative damage in neurons is prevented by resveratrol and lipoic acid with participation of heme oxygenase 1 // Neurotoxicology. 2015. № 49. P. 28–35. https://doi.org/10.1016/j.neuro.2015.05.005
Bonda D.J., Lee H.-G., Camins A. et al. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations // The Lancet. Neurology. 2011. № 3(10). P. 275–279. https://doi.org/10.1016/S1474-4422(11)70013-8
Burnett P., Valentini S., Cabreiro F. et al. Absence of effects of Sir2 overexpression on lifespan in P. elegans and Drosophila // Nature. 2011. № 7365(477). P. 482–485. https://doi.org/10.1038/nature10296
Cao W., Hong Y., Chen H. et al. SIRT2 mediates NADH-induced increases in Nrf2, GCL, and glutathione by modulating Akt phosphorylation in PC12 cells // FEBS Letters. 2016. № 14(590). P. 2241–2255. https://doi.org/10.1002/1873-3468.12236
Cieślik M., Czapski G.A., Strosznajder J.B. The Molecular Mechanism of Amyloid β42 Peptide Toxicity: The Role of Sphingosine Kinase-1 and Mitochondrial Sirtuins // PloS One. 2015. № 9(10). P. e0137193. https://doi.org/10.1371/journal.pone.0137193
Corpas R., Revilla S., Ursulet S. et al. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement Through Proteostatic and Neurotrophic Mechanisms // Molecular Neurobiology. 2017. № 7(54). P. 5604–5619. https://doi.org/10.1007/s12035-016-0087-9
D’Onofrio N., Servillo L., Giovane A. et al. Ergothioneine oxidation in the protection against high-glucose induced endothelial senescence: Involvement of SIRT1 and SIRT6 // Free Radical Biology & Medicine. 2016. (96). P. 211–222. https://doi.org/10.1016/j.freeradbiomed.2016.04.013
Du J., Zhou Y., Su X. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase // Science (New York, N.Y.). 2011. № 6057 (334). P. 806–809. https://doi.org/10.1126/science.1207861
Esteves A.R., Arduíno D.M., Silva D.F. et al. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease // Molecular Neurobiology. 2018. № 2(55). P. 1440–1462. https://doi.org/10.1007/s12035-017-0420-y
Ferber E.P., Peck B., Delpuech O. et al. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression // Cell Death and Differentiation. 2012. № 6 (19). P. 968–979. https://doi.org/10.1038/cdd.2011.179
Finley L.W.S., Carracedo A., Lee J. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization // Cancer Cell. 2011. № 3 (19). P. 416–428. https://doi.org/10.1016/j.ccr.2011.02.014
Frye R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins // Biochemical and Biophysical Research Communications. 2000. № 2 (273). P. 793–798. https://doi.org/10.1006/bbrc.2000.3000
Gleave J.A., Arathoon L.R., Trinh D. et al. Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant α-synuclein rat model of parkinsonism // Neurobiology of Disease. 2017. (106). P. 133–146. https://doi.org/10.1016/j.nbd.2017.06.009
Godoy J.A., Zolezzi J.M., Braidy N. et al. Role of Sirt1 during the ageing process: relevance to protection of synapses in the brain // Molecular Neurobiology. 2014. № 3(50). P. 744–756. https://doi.org/10.1007/s12035-014-8645-5
Gomes P., Fleming Outeiro T., Cavadas P. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism // Trends in Pharmacological Sciences. 2015. № 11(36). P. 756–768. https://doi.org/10.1016/j.tips.2015.08.001
Grabowska W., Sikora E., Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process // Biogerontology. 2017. № 4(18). P. 447–476. https://doi.org/10.1007/s10522-017-9685-9
Greco S.J., Hamzelou A., Johnston J.M. et al. Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and β-amyloid in neurons // Biochemical and Biophysical Research Communications. 2011. № 1(414). P. 170–174. https://doi.org/10.1016/j.bbrc.2011.09.050
Greiss S., Gartner A. Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation // Molecules and Cells. 2009. № 5(28). P. 407–415. https://doi.org/10.1007/s10059-009-0169-x
Guan Q., Wang M., Chen H. et al. Aging-related 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurochemial and behavioral deficits and redox dysfunction: improvement by AK-7 // Experimental Gerontology. 2016. № 82. P. 19–29. https://doi.org/10.1016/j.exger.2016.05.011
Guarente L. Sir2 links chromatin silencing, metabolism, and aging // Genes & Development. 2000. № 9 (14). P. 1021–1026.
Haigis M.P., Sinclair D.A. Mammalian sirtuins: biological insights and disease relevance // Annual Review of Pathology. 2010. № 5. P. 253–295. https://doi.org/10.1146/annurev.pathol.4.110807.092250
Hernandez-Rapp J., Rainone S., Goupil P. et al. microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice // Scientific Reports. 2016. № 6. P. 30953. https://doi.org/10.1038/srep30953
Hou K.-L., Lin S.-K., Chao L.-H. et al. Sirtuin 6 suppresses hypoxia-induced inflammatory response in human osteoblasts via inhibition of reactive oxygen species production and glycolysis-A therapeutic implication in inflammatory bone resorption // BioFactors (Oxford, England). 2017. № 2(43). P. 170–180. https://doi.org/10.1002/biof.1320
Houtkooper R.H., Pirinen E., Auwerx J. Sirtuins as regulators of metabolism and healthspan // Nature Reviews. Molecular Cell Biology. 2012. № 4(13). P. 225–238. https://doi.org/10.1038/nrm3293
Ido Y., Duranton A., Lan F. et al. Resveratrol Prevents Oxidative Stress-Induced Senescence and Proliferative Dysfunction by Activating the AMPK-FOXO3 Cascade in Cultured Primary Human Keratinocytes // PLoS One. 2015. № 2(10). https://doi.org/10.1371/journal.pone.0115341
Imai S., Armstrong P.M., Kaeberlein M. et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase // Nature. 2000. № 6771(403). P. 795–800. https://doi.org/10.1038/35001622
Jęśko H., Wencel P., Strosznajder R.P. et al. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders // Neurochemical Research. 2017. № 3(42). P. 876–890. https://doi.org/10.1007/s11064-016-2110-y
Jia G., Su L., Singhal S. et al. Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging // Molecular and Cellular Biochemistry. 2012. № 1–2(364). P. 345–350. https://doi.org/10.1007/s11010-012-1236-8
Jiang H., Khan S., Wang Y. et al. Sirt6 regulates TNFα secretion via hydrolysis of long chain fatty acyl lysine // Nature. 2013. № 7443(496). P. 110–113. https://doi.org/10.1038/nature12038
Jing E., Emanuelli B., Hirschey M.D. et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production // Proceedings of the National Academy of Sciences of the United States of America. 2011. № 35(108). P. 14608–14613. https://doi.org/10.1073/pnas.1111308108
Julien P., Tremblay P., Emond V. et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease // J. Neuropathology and Experimental Neurology. 2009. № 1(68). P. 48–58. https://doi.org/10.1097/NEN.0b013e3181922348
Kaeberlein M., McVey M., Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms // Genes & Development. 1999. № 19(13). P. 2570–2580. https://doi.org/10.1101/gad.13.19.2570
Kaluski S., Portillo M., Besnard A. et al. Neuroprotective Functions for the Histone Deacetylase SIRT6 // Cell Reports. 2017. № 13(18). P. 3052–3062. https://doi.org/10.1016/j.celrep.2017.03.008
Kaufmann T., Kukolj E., Brachner A. et al. SIRT2 regulates nuclear envelope reassembly through ANKLE2 deacetylation // J. Cell Science. 2016. № 24(129). P. 4607–4621. https://doi.org/10.1242/jcs.192633
Khojah S.M., Payne A.P., McGuinness D. et al. Segmental Aging Underlies the Development of a Parkinson Phenotype in the AS/AGU Rat // Cells. 2016. № 4(5). https://doi.org/10.3390/cells5040038
Kiran S., Anwar T., Kiran M. et al. Sirtuin 7 in cell proliferation, stress and disease: Rise of the Seventh Sirtuin! // Cellular Signalling. 2015. № 3(27). P. 673–682. https://doi.org/10.1016/j.cellsig.2014.11.026
Kiran S., Chatterjee N., Singh S. et al. Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal // The FEBS Journal. 2013. № 14(280). P. 3451–3466. https://doi.org/10.1111/febs.12346
Klotz L.-O., Sánchez-Ramos P., Prieto-Arroyo I. et al. Redox regulation of FoxO transcription factors // Redox Biology. 2015. № 6. P. 51–72. https://doi.org/10.1016/j.redox.2015.06.019
Kobayashi A., Kang M.-I., Okawa H. et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2 // Molecular and Cellular Biology. 2004. № 16(24). P. 7130–7139. https://doi.org/10.1128/MCB.24.16.7130-7139.2004
Krishnan J., Danzer P., Simka T. et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system // Genes & Development. 2012. № 3(26). P. 259–270. https://doi.org/10.1101/gad.180406.111
Landry J., Sutton A., Tafrov S.T. et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases // Proceedings of the National Academy of Sciences of the United States of America. 2000. № 11(97). P. 5807–5811. https://doi.org/10.1073/pnas.110148297
Lattanzio F., Carboni L., Carretta D. et al. Treatment with the neurotoxic Aβ (25-35) peptide modulates the expression of neuroprotective factors Pin1, Sirtuin 1, and brain-derived neurotrophic factor in SH-SY5Y human neuroblastoma cells // Experimental and Toxicologic Pathology: Official Journal of the Gesellschaft Fur Toxikologische Pathologie. 2016. № 5(68). P. 271–276. https://doi.org/10.1016/j.etp.2016.02.001
Lescai F., Blanché H., Nebel A. et al. Human longevity and 11p15.5: a study in 1321 centenarians // European Journal of Human Genetics. 2009. № 11(17). P. 1515–1519. https://doi.org/10.1038/ejhg.2009.54
Liang F., Wang X., Ow S.H. et al. Sirtuin 5 is Anti-apoptotic and Anti-oxidative in Cultured SH-EP Neuroblastoma Cells // Neurotoxicity Research. 2017. № 1(31). P. 63–76. https://doi.org/10.1007/s12640-016-9664-y
Lin Z.-F., Xu H.-B., Wang J.-Y. et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS // Biochemical and Biophysical Research Communications. 2013. № 1(441). P. 191–195. https://doi.org/10.1016/j.bbrc.2013.10.033
Liu B., Che W., Zheng P. et al. SIRT5: a safeguard against oxidative stress-induced apoptosis in cardiomyocytes // Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2013. № 4(32). P. 1050–1059. https://doi.org/10.1016/j.bbrc.2013.10.033
Liu R., Liu H., Ha Y. et al. Oxidative stress induces endothelial cell senescence via downregulation of Sirt6 // BioMed Research International. 2014. P. 902842. https://doi.org/10.1155/2014/902842
Lu W., Zuo Y., Feng Y. et al. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer // Tumour Biology: The J. International Society for Oncodevelopmental Biology and Medicine. 2014. № 11(35). P. 10699–10705. https://doi.org/10.1007/s13277-014-2372-4
Luo Y.-X., Tang X., An X.-Z. et al. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity // European Heart Journal. 2017. № 18(38). P. 1389–1398. https://doi.org/10.1093/eurheartj/ehw138
Lutz M.I., Milenkovic I., Regelsberger G. et al. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease // Neuromolecular Medicine. 2014. № 2(16). P. 405–414. https://doi.org/10.1007/s12017-014-8288-8
Maillet A., Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance // Antioxidants & Redox Signaling. 2012. № 11(16). P. 1285–1294. https://doi.org/10.1089/ars.2011.4434
Marks P.A., Xu W.-S. Histone deacetylase inhibitors: Potential in cancer therapy // J. Cellular Biochemistry. 2009. № 4(107). P. 600–608. https://doi.org/10.1002/jcb.22185
McCarthy J.T., Pelle E., Dong K. et al. Effects of ozone in normal human epidermal keratinocytes // Experimental Dermatology. 2013. № 5 (22). P. 360–361. https://doi.org/10.1111/exd.12125
Mercken E.M., Mitchell S.J., Martin-Montalvo A. et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass // Aging Cell. 2014. № 5(13). P. 787–796. https://doi.org/10.1111/acel.12220
Merksamer P.I., Liu Y., He W. et al. The sirtuins, oxidative stress and aging: an emerging link // Aging. 2013. № 3(5). P. 144–150. https://doi.org/10.18632/aging.100544
Michishita E., Park J.Y., Burneskis J.M. et al. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins // Molecular Biology of the Cell. 2005. № 10(16). P. 4623–4635. https://doi.org/10.1091/mbc.e05-01-0033
Morigi M., Perico L., Rota P. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury // The Journal of Clinical Investigation. 2015. № 2(125). P. 715–726. https://doi.org/10.1172/JCI77632
Mostoslavsky R., Chua K.F., Lombard D.B. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6 // Cell. 2006. № 2(124). P. 315–329. https://doi.org/10.1016/j.cell.2005.11.044
Mudò G., Mäkelä J., Liberto V.D. et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease // Cellular and Molecular Life Sciences. 2012. № 7(69). P. 1153–1165. https://doi.org/10.1007/s00018-011-0850-z
Nasrin N., Wu X., Fortier E. et al. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells // The Journal of Biological Chemistry. 2010. № 42(285). P. 31995–32002. https://doi.org/10.1074/jbc.M110.124164
Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} // The Journal of Biological Chemistry. 2005. № 16(280). P. 16456–16460. https://doi.org/10.1074/jbc.M501485200
Ng F., Tang B.L. When is Sirt1 activity bad for dying neurons? // Frontiers in Cellular Neuroscience. 2013. № 7. P. 186. https://doi.org/10.3389/fncel.2013.00186
Nicholatos J.W., Francisco A.B., Bender P.A. et al. Nicotine promotes neuron survival and partially protects from Parkinson’s disease by suppressing SIRT6 // Acta Neuropathologica Communications. 2018. № 1(6). P. 120. https://doi.org/10.1186/s40478-018-0625-y
Oberdoerffer P., Michan S., McVay M. et al. DNA damage-induced alterations in chromatin contribute to genomic integrity and age-related changes in gene expression // Cell. 2008. № 5(135). P. 907–918. https://doi.org/10.1016/j.cell.2008.10.025
Ogura M., Nakamura Y., Tanaka D. et al. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1 // Biochemical and Biophysical Research Communications. 2010. № 1(393). P. 73–78. https://doi.org/10.1016/j.bbrc.2010.01.081
Oliveira R.M. de, Vicente Miranda H., Francelle L. et al. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease // PLoS Biology. 2017. № 3(15). e2000374. https://doi.org/10.1371/journal.pbio.2000374
Ou H.-L., Schumacher B. DNA damage responses and p53 in the aging process // Blood. 2018. № 5(131). P. 488–495. https://doi.org/10.1182/blood-2017-07-746396
Outeiro T.F., Kontopoulos E., Altmann S.M. et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease // Science (New York, N.Y.). 2007. № 5837(317). P. 516–519. https://doi.org/10.1126/science.1143780
Pan H., Guan D., Liu X. et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2 // Cell Research. 2016. № 2(26). P. 190–205. https://doi.org/10.1038/cr.2016.4
Pandey R., Müller A., Napoli P.A. et al. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes // Nucleic Acids Research. 2002. № 23(30). P. 5036–5055. https://doi.org/10.1093/nar/gkf660
Park S.Y., Lee H.R., Lee W.S. et al. Cilostazol Modulates Autophagic Degradation of β-Amyloid Peptide via SIRT1-Coupled LKB1/AMPKα Signaling in Neuronal Cells // PloS One. 2016. № 8(11). e0160620. https://doi.org/10.1371/journal.pone.0160620
Qin W., Yang T., Ho L. et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction // The J. Biological Chemistry. 2006. № 31(281). P. 21745–21754. https://doi.org/10.1074/jbc.M602909200
Rosca M.G., Vazquez E.J., Chen Q. et al. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes // Diabetes. 2012. № 8(61). P. 2074–2083. https://doi.org/10.2337/db11-1437
Roth M., Chen W.Y. Sorting out functions of sirtuins in cancer // Oncogene. 2014. № 13(33). P. 1609–1620. https://doi.org/10.1038/onc.2013.120
Ryu D., Jo Y.S., Lo Sasso G. et al. A SIRT7-dependent acetylation switch of GABPβ1 controls mitochondrial function // Cell Metabolism. 2014. № 5 (20). P. 856–869. https://doi.org/10.1016/j.cmet.2014.08.001
Santos L., Escande P., Denicola A. Potential Modulation of Sirtuins by Oxidative Stress // Oxidative Medicine and Cellular Longevity. 2016. P. 9831825. https://doi.org/10.1155/2016/9831825
Saso L., Firuzi O. Pharmacological applications of antioxidants: lights and shadows // Current Drug Targets. 2014. № 13(15). P. 1177–1199. https://doi.org/10.2174/1389450115666141024113925
Scher M.B., Vaquero A., Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress // Genes & Development. 2007. № 8(21). P. 920–928. https://doi.org/10.1101/gad.1527307
Scuderi P., Stecca P., Bronzuoli M.R. et al. Sirtuin modulators control reactive gliosis in an in vitro model of Alzheimer’s disease // Frontiers in Pharmacology. 2014. № 5. P. 89. https://doi.org/10.3389/fphar.2014.00089
Seto E., Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes // Cold Spring Harbor Perspectives in Biology. 2014. № 4(6). a018713. https://doi.org/10.1101/cshperspect.a018713
Shi H., Deng H.-X., Gius D. et al. Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress // Human Molecular Genetics. 2017. № 10(26). P. 1915–1926. https://doi.org/10.1093/hmg/ddx100
Shimazu T., Hirschey M.D., Hua L. et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production // Cell Metabolism. 2010. № 6 (12). P. 654–661. https://doi.org/10.1093/hmg/ddx100
Singh P.K., Chhabra G., Ndiaye M.A. et al. The Role of Sirtuins in Antioxidant and Redox Signaling // Antioxidants & Redox Signaling. 2018. № 8(28). P. 643–661. https://doi.org/10.1089/ars.2017.7290
Tarantino G., Finelli P., Scopacasa F. et al. Circulating levels of sirtuin 4, a potential marker of oxidative metabolism, related to coronary artery disease in obese patients suffering from NAFLD, with normal or slightly increased liver enzymes // Oxidative Medicine and Cellular Longevity. 2014. e920676. https://doi.org/10.1155/2014/920676
TenNapel M.J., Lynch P.F., Burns T.L. et al. SIRT6 minor allele genotype is associated with >5-year decrease in lifespan in an aged cohort // PloS One. 2014. № 12(9). e115616. https://doi.org/10.1371/journal.pone.0115616
Tennen R.I., Bua D.J., Wright W.E. et al. SIRT6 is required for maintenance of telomere position effect in human cells // Nature Communications. 2011. № 2. P. 433. https://doi.org/10.1038/ncomms1443
Toiber D., Sebastian P., Mostoslavsky R. Characterization of nuclear sirtuins: molecular mechanisms and physiological relevance // Handbook of Experimental Pharmacology. 2011. № 206. P. 189–224. https://doi.org/10.1007/978-3-642-21631-2_9
Vakhrusheva O., Smolka P., Gajawada P. et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice // Circulation Research. 2008. № 6(102). P. 703–710. https://doi.org/10.1161/CIRCRESAHA.107.164558
Vaquero A., Reinberg D. Calorie restriction and the exercise of chromatin // Genes & Development. 2009. № 16(23). P. 1849–1869. https://doi.org/10.1101/gad.1807009
Wang X.-X., Wang X.-L., Tong M. et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms // Basic Research in Cardiology. 2016. № 2(111). P. 13. https://doi.org/10.1007/s00395-016-0531-z
Wang Y., Zhu Y., Xing S. et al. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3 // Cell Stress & Chaperones. 2015. № 5 (20). P. 805–810. https://doi.org/10.1007/s12192-015-0599-7
Watanabe S., Ageta-Ishihara N., Nagatsu S. et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system // Molecular Brain. 2014. № 7. P. 62. https://doi.org/10.1186/s13041-014-0062-1
Wątroba M., Szukiewicz D. The role of sirtuins in aging and age-related diseases // Advances in Medical Sciences. 2016. № 1(61). P. 52–62. https://doi.org/10.1016/j.advms.2015.09.003
Webster B.R., Lu Z., Sack M.N. et al. The role of sirtuins in modulating redox stressors // Free Radical Biology & Medicine. 2012. № 2(52). P. 281–290. https://doi.org/10.1016/j.freeradbiomed.2011.10.484
Weir H.J.M., Murray T.K., Kehoe P.G. et al. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease // PloS One. 2012. № 11(7). P. e48225. https://doi.org/10.1371/journal.pone.0048225
Winnik S., Gaul D.S., Siciliani G. et al. Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: protective role of a novel C/EBP-β-dependent feedback regulation of SOD2 // Basic Research in Cardiology. 2016. № 3(111). P. 33. https://doi.org/10.1007/s00395-016-0552-7
Wu Y., Li X., Zhu J.X. et al. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease // Neuro-Signals. 2011. № 3(19). P. 163–174. https://doi.org/10.1159/000328516
Wu Y.-T., Wu S.-B., Wei Y.-H. Roles of sirtuins in the regulation of antioxidant defense and bioenergetic function of mitochondria under oxidative stress // Free Radical Research. 2014. № 9(48). P. 1070–1084. https://doi.org/10.3109/10715762.2014.920956
Xue F., Huang J.-W., Ding P.-Y. et al. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning // Behavioural Brain Research. 2016. (309). P. 1–8. https://doi.org/10.1016/j.bbr.2016.04.045
Yang L., Ma X., He Y. et al. Sirtuin 5: a review of structure, known inhibitors and clues for developing new inhibitors // Science China. Life Sciences. 2017. № 3 (60). P. 249–256. https://doi.org/10.1007/s11427-016-0060-7
Yang W., Zou Y., Zhang M. et al. Mitochondrial Sirt3 Expression is Decreased in APP/PS1 Double Transgenic Mouse Model of Alzheimer’s Disease // Neurochemical Research. 2015. № 8(40). P. 1576–1582. https://doi.org/10.1007/s11064-015-1630-1
Yang X., Park S.-H., Chang H.-P. et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2 // The J. Clinical Investigation. 2017. № 4(127). P. 1505–1516. https://doi.org/10.1172/JCI88574
Yang Y., Tian T., Wang Y. et al. SIRT6 protects vascular endothelial cells from angiotensin II-induced apoptosis and oxidative stress by promoting the activation of Nrf2/ARE signaling // European J. Pharmacology. 2019. № 859. e172516. https://doi.org/10.1016/j.ejphar.2019.172516
Yin J., Han P., Song M. et al. Amyloid-β Increases Tau by Mediating Sirtuin 3 in Alzheimer’s Disease // Molecular Neurobiology. 2018. № 11(55). P. 8592–8601. https://doi.org/10.1007/s12035-018-0977-0
Yu J., Sun W., Song Y. et al. SIRT6 protects retinal ganglion cells against hydrogen peroxide-induced apoptosis and oxidative stress by promoting Nrf2/ARE signaling via inhibition of Bach1 // Chemico-Biological Interactions. 2019. № 300. P. 151–158. https://doi.org/10.1016/j.cbi.2019.01.018
Yu J., Wu Y., Yang P. High glucose-induced oxidative stress represses sirtuin deacetylase expression and increases histone acetylation leading to neural tube defects // J. Neurochemistry. 2016. № 3 (137). P. 371–383. https://doi.org/10.1111/jnc.13587
Yu S.-S., Cai Y., Ye J.-T. et al. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-κB-dependent transcriptional activity // British Journal of Pharmacology. 2013. № 1(168). P. 117–128. https://doi.org/10.1111/j.1476-5381.2012.01903.x
Zhang A., Wang H., Qin X. et al. Genetic analysis of SIRT1 gene promoter in sporadic Parkinson’s disease // Biochemical and Biophysical Research Communications. 2012. № 4(422). P. 693–696. https://doi.org/10.1016/j.bbrc.2012.05.059
Zhang H., Forman H.J. Reexamination of the electrophile response element sequences and context reveals a lack of consensus in gene function // Biochimica Et Biophysica Acta. 2010. № 7(1799). P. 496–501. https://doi.org/10.1016/j.bbagrm.2010.05.003
Zhou Z.D., Tan E.K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease // Ageing Research Reviews. 2020. e101107. https://doi.org/10.1016/j.arr.2020.101107
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук