

Успехи физиологических наук, 2022, T. 53, № 3, стр. 3-14
Механизмы влияния цитокинового шторма на функцию внешнего дыхания
Н. П. Александрова *
Институт физиологии им. И.П. Павлова РАН
199034 Санкт-Петербург, Россия
* E-mail: aleks@infran.ru
Поступила в редакцию 25.03.2022
После доработки 27.03.2022
Принята к публикации 30.03.2022
- EDN: TNXOFW
- DOI: 10.31857/S0301179822030043
Аннотация
В обзоре на основе современных экспериментальных и клинических данных рассматриваются последствия чрезмерного увеличения системного уровня провоспалительных цитокинов, так называемого цитокинового шторма, вызывающего развитие системного воспаления, характерного для тяжелого течения новой вирусной инфекции SARS COV-19. Обсуждаются физиологические механизмы влияния провоспалительных цитокинов на систему внешнего дыхания на тканевом, органном и системном уровнях. Приводятся собственные экспериментальные данные, согласно которым цитокиновый шторм может усиливать дисфункцию дыхательной системы посредством действия провоспалительных цитокинов на рефлекторные механизмы регуляции вентиляционной функции легких, ослабляя тем самым компенсаторные возможности системы внешнего дыхания. Подчеркивается, что проведение исследований в этом направлении открывает новые перспективы в изучении регуляторных процессов, происходящих в центральной нервной системе, способствует раскрытию тонких механизмов межсистемных взаимодействий, участвующих в центральной регуляции висцеральных функций.
ВВЕДЕНИЕ
Цитокины, эндогенные полипептиды, обладающие мощной плейотропной (многофункциональной) активностью и выполняющие роль сигнальных молекул в межклеточных взаимодействиях, изначально рассматривались как медиаторы, обеспечивающие только локальное взаимодействие между клетками иммунной системы [9]. К настоящему времени установлено, что экспрессия цитокинов, а также и их рецепторов не ограничена клетками только иммунной системы. Они могут продуцироваться и во многих других органах и тканях. Кроме того, было установлено, что цитокины могут оказывать не только местное, паракринное или аутокринное, но и гормоноподобное действие, т.е. влиять на клетки-мишени, находящиеся в различных органах в отдалении от того места, где в данный момент они продуцируются, образуя, таким образом, в организме единую сигнальную сеть. Поэтому в настоящее время цитокины выделяются в самостоятельную систему регуляции защитных реакций организма и нормальных физиологических функций, тесно связанную с нервной и эндокринной системами регуляции [4].
Характерной чертой цитокинов является многофункциональность. Они действуют и на тканевом, и на системном уровне. Экспрессия провоспалительных цитокинов в очаге воспаления активирует лимфоциты, вызывает экспрессию молекул адгезии на эндотелиоцитах, увеличивая тем самым сосудистую проницаемость, активирует фагоциты, усиливает NO-синтазную активность и метаболизм арахидоновой кислоты, способствуя синтезу оксида азота и простагландинов. К системным эффектам цитокинов относится их действие на терморегуляторный центр гипоталамуса, вызывающее подъем температуры тела, влияние на синтез большинства гормонов, индукция в печени синтеза острофазовых белков и компонентов системы комплемента, влияние на кроветворную систему, вызывающее активацию гемопоэза, увеличение количества лейкоцитов за счет ускорения их выхода из костного мозга и депо, повышение свертываемости крови. Все эти биологические эффекты цитокинов имеют единую цель – они направлены на борьбу с патогеном и формирование единой защитной реакции организма, посредством осуществления связи между иммунной, нервной, эндокринной и кроветворной системами. Однако иммунный ответ не всегда бывает адекватным. Он может быть как недостаточным, так и гипертрофированным. В организме существуют эффективные механизмы, предотвращающие гиперпродукцию цитокинов: это экспрессия противовоспалительных цитокинов, связывающие цитокины белки плазмы крови, ингибиторы протеаз, растворимые рецепторы цитокинов, усиление синтеза стероидных гормонов [23, 83]. Несмотря на существование таких механизмов, в ряде случаев контроль оказывается недостаточно эффективным и уровни цитокинов достигают патологически высоких значений. Установлено, что неуправляемый и избыточный иммунный ответ, так называемый “цитокиновый шторм”, может причинить огромный вред организму человека [25, 35, 79].
Цитокиновый шторм или гиперцитокинемия – это потенциально летальная реакция иммунной системы, характеризующаяся быстрой пролиферацией и повышенной активностью Т-клеток, макрофагов и естественных киллеров с высвобождением защитными клетками различных воспалительных цитокинов и химических медиаторов [90]. Выработка большого количества медиаторов воспаления приводит к активации иммунных клеток и высвобождению последними новой порции медиаторов вследствие наличия неконтролируемой положительной обратной связи между этими процессами [19]. Возникает порочный круг, который вызывает разрушение тканей очага воспаления, распространение реакции на соседние ткани, выход провоспалительных цитокинов в кровеносное русло. Воспаление приобретает системный, генерализованный характер, охватывая весь организм в целом и вызывая полиорганное повреждение, ведущее к дыхательной, сердечной, печеночной и почечной недостаточности [28, 80, 82, 86].
Впервые термин “цитокиновый шторм” был введен в научный лексикон американским ученым Джеймсом Феррара в 1993 г. при описании реакции “трансплантат против хозяина”, которая является основным осложнением аллогенной трансплантации костного мозга [41]. В настоящее время исследование патофизиологических механизмов и последствий развития цитокинового шторма приобрело особую актуальность в связи c возникновением и широким распространением коронавирусной инфекции COVID-19. Установлено, что степень повреждения органов дыхания у больных COVID-19 зависит не столько от прямого действия вируса и интенсивности вирусной репродукции, сколько от неконтролируемой выработки провоспалительных цитокинов и развития системной воспалительной реакции. Именно с развитием цитокинового шторма связаны осложнения и летальные случаи этого заболевания [31, 60, 82]. На прямую корреляцию между смертностью и уровнем гиперцитокинемии указывалось и в более ранних исследованиях при изучении патофизиологии тяжелых вирусных заболеваний и сепсиса [20, 24, 25, 30, 73, 90]. Этиологический фактор, запускающий каскады цитокинового шторма, пока не установлен. Существенную роль в этом плане может играть генетическая предрасположенность, а также состояние иммунной системы человека. Установлено, что при заболевании COVID-19 чрезмерная иммунная реакция на короновирус, приводящая к цитокиновому шторму и, как следствие, к неблагоприятному исходу болезни, наблюдается чаще всего у пациентов пожилого возраста с ослабленным иммунитетом [78]. Цитокиновый шторм может быть вызван причинами не только инфекционного (вирусы, бактерии), но и неинфекционного характера (ожоги, панкреатит, онкология и лечение химиопрепаратами, проведение хирургических операций) [19, 84]. Следствием развития цитокинового шторма является поликлональная активация клеток иммунной системы, т.е. потеря специфичности иммунитета, когда уже не только чужеродные, но и собственные клетки организма хозяина связываются антигенами и уничтожаются в ходе дальнейшей реакции иммунного ответа.
Развитие цитокинового шторма происходит на фоне чрезмерной активации системы врожденного иммунитета. Клетками врожденного иммунитета, участвующими в патогенезе цитокинового шторма, являются нейтрофилы, макрофаги, Т-клетки и естественные киллеры. При усиленной активации эти клетки секретируют чрезмерное количество цитокинов, инициируя цитокиновый шторм, который сопровождается выбросом большого количества биологически активных веществ. В развитии цитокинового шторма принимают участие как провоспалительные, так и противовоспалительные цитокины различных семейств: интерлейкины (ИЛ), интерфероны (ИФН), хемокины, колонии стимулирующие факторы, факторы некроза опухоли (ФНО) [52, 83]. Однако ведущая роль в патогенезе цитокинового шторма принадлежит ИЛ-1β, ФНО-α, ИФН-γ, ИЛ-6. При этом в индуцировании цитокинового шторма центральная, критическая роль отводится ИЛ-1, который рассматривается в качестве эффекторной молекулы [42]. С помощью метода полимеразной цепной реакции было, например, показано, что при гиперцитокинемии, характерной для реакции “трансплантат против хозяина”, уровень транскриптов мРНК ИЛ-1 увеличивается в несколько сотен раз, тогда как уровень транскриптов ФНО-α возрастает только в 4–6 раз [13]. Установлено, что при повреждении легких ИЛ-1β является ключевым цитокином, управляющим провоспалительной активностью [31, 75]. ИЛ-1β, экспрессируемый в ответ на проникновение в организм патогенов, антигенное раздражение или повреждение тканей, вызывает экспрессию генов и синтез ФНО-α, ИЛ-6 и других провоспалительных цитокинов в макрофагах и тучных клетках. Этот эффект, сопровождающийся усилением синтеза оксида азота и провоспалительных продуктов метаболизма арахидоновой кислоты, таких как простагландины и тромбоксаны, способствует развитию цитокинового шторма.
ОСТРЫЙ РЕСПИРАТОРНЫЙ ДИСТРЕСС-СИНДРОМ (ОРДС) В ПАТОГЕНЕЗЕ ЦИТОКИНОВОГО ШТОРМА
Цитокиновый шторм имеет сложный патогенез, существенный вклад в который вносит гиперактивация системы комплемента, являющаяся важным компонентом как врожденного, так и приобретенного иммунитета. Система комплемента представляет собой группу защитных белков, которые постоянно присутствуют в крови и являются протеолитическими ферментами, способными нарушать целостность клеточной мембраны, вызывая этим гибель клетки [10]. Провоспалительные цитокины (ИЛ-1, ИЛ-6 и ФНО-α) могут увеличивать продукцию белков комплемента, вызывая слишком мощную активацию этой системы. В результате она начинает воздействовать не только на клетку-патоген, но и на собственные клетки организма: протеолитические ферменты повреждают ткани, разрушают эритроциты и тромбоциты.
Цитокиновый шторм имеет широкий спектр неблагоприятных последствий [82, 86, 28, 80 ]. Однако чаще всего и в наибольшей степени от неконтролируемого выброса цитокинов страдает дыхательная система. Иммунопатологические изменения в легких, вызванные цитокиновым штормом приводят к развитию острого респираторного дистресс синдрома [26, 51, 54, 60]. Синонимами ОРДС являются “шоковое”, “влажное”, “травматическое” легкое. ОРДС приводит к острой дыхательной недостаточности – патологическому состоянию, при котором не обеспечивается поддержание нормального газового состава крови либо оно достигается за счет более интенсивной работы аппарата внешнего дыхания, что приводит к снижению функциональных возможностей организма. Именно ОРДС является основной причиной смерти пациентов от коронавирусной инфекции SARS-CoV, MERS-CoV, SARS-CoV-2 [36, 83].
Несмотря на многообразие факторов, приводящих к ОРДС, в его основе лежат повреждения легочных структур [12]. Появление провоспалительных цитокинов в кровеносном русле (ИЛ-8, ИЛ-1β, ФНО-α), активирует нейтрофилы, которые мигрируют в просвет альвеол. В альвеолах активированные нейтрофилы секретируют множество деструктивных факторов (таких как лейкотриены, оксиданты, протеазы, фактор активации тромбоцитов), повреждающих альвеолярный эпителий. Повреждение эпителия альвеол вызывает выход фибрина и других белков в просвет альвеол, что способствует формированию в альвеолах гиалиновых мембран, состоящих из протеинов плазмы, остатков цитоплазмы и ядер слущенных клеток эпителия. Происходит утолщение аэрогематического барьера, затрудняется диффузия кислорода. Установлено, что экссудативное и диффузное повреждение альвеол является основной причиной тяжелой гипоксемии при COVID-19.
К основным морфологическим изменениям в легких при ОРДС относится также повреждение эндотелия легочных капилляров, их базальных мембран, увеличение проницаемости капилляров, что ведет к накоплению внесосудистой жидкости и экссудации белков с формированием некардиогенного отека легких [7]. В воздушное пространство альвеол поступает отечная жидкость, возникает дефицит сурфактанта, вызванный повреждением пневмоцитов II типа. Нарушение сурфактантного слоя, выстилающего бронхиолы и альвеолы, вызывает ателектаз, т.е. спадение альвеол. В результате часть легочного кровотока проходит по невентилируемым участкам легких, шунтируется. Венозная кровь, притекающая к легким и попадающая в шунты, не изменяет свой газовый состав. На выходе из легких она встречается с кровью, оттекающей от нормально работающих альвеол. В результате смешивания этих двух потоков образуется артериальная кровь, напряжение кислорода в которой снижено из-за примеси неоксигенированной крови.
Повышение проницаемости стенок легочных капилляров при цитокиновом шторме вызывает усиленный транспорт жидкости, богатой альбумином в интерстициальную ткань легкого и развитие интерстициального отека легких. Выход фибрина в интерстиций способствует фиброзированию легочной ткани, снижению ее эластичности. Нормальная легочная ткань разрушается, а затем заменяется соединительной тканью. В результате в эластичной ткани образуются нерастяжимые участки, рубцы. Легкие становятся менее эластичными и менее растяжимыми, уменьшается функциональная емкость легких [22, 37]. Кроме того, заполнения альвеол жидкостью снижает воздушность легочной ткани. Все это увеличивает сопротивление дыханию и создает дополнительную нагрузку на дыхательные мышцы. В результате паталогических изменений в легких, экссудативного и диффузного повреждения альвеол, инициируемых цитокиновым штормом, вызванным иммунной дисрегуляцией, теряется самое главное – диффузионная способность легких, ухудшается оксигенация крови, развивается гипоксемия и дыхательная недостаточность.
Гиперреакция иммунной системы приводит к истощению работы иммунитета. В результате возникает вторичная реакция – иммунная недостаточность. Иммуносупрессивное состояние способствует развитию оппортунистических бактериальных и микотических инфекций респираторного тракта, которые утяжеляют состояние больных короновирусной инфекцией перенесших цитокиновый шторм [27].
ВЛИЯНИЕ ПРОВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ НА РЕФЛЕКТОРНУЮ РЕГУЛЯЦИЮ ДЫХАНИЯ
К настоящему времени накоплен целый ряд экспериментальных фактов, указывающих на то, что влияние цитокинового шторма на систему внешнего дыхания не ограничивается морфологическими повреждениями легочных структур. Показано, что повышение системного уровня провоспалительных цитокинов влияет на рефлекторные механизмы регуляции дыхания [1, 2, 14, 15, 43, 55], снижает устойчивость организма к гипоксии [3], ухудшает способность к спонтанному восстановлению дыхания [48, 49].
Влияние провоспалительных цитокинов на артериальные и медуллярные хеморецепторы
Важнейшими элементами компенсаторных и адаптивных реакций системы внешнего дыхания являются гипоксический и гиперкапнический дыхательные хеморефлексы. Эти рефлексы участвуют в поддержании газового гомеостаза артериальной крови. Хеморефлексы осуществляются при участии хеморецепторов каротидных телец, расположенных в бифуркации сонной артерии, которые возбуждаются при снижении напряжения кислорода, повышении напряжения углекислого газа и уменьшении pH артериальной крови. При гипоксии гломусные клетки каротидных телец деполяризуются в ответ на недостаток кислорода и выделяют нейромедиаторы, которые активируют сенсорные нервные волокна, передающие афферентную информацию в дыхательный центр ствола мозга.
В настоящее время имеются данные об участии воспалительных цитокинов в физиологии и пластичности каротидного тела. Обнаружено, что циркулирующие цитокины могут влиять на артериальные хеморецепторы [74]. Как оказалось, системное воспаление, для которого характерна гиперцитокинемия, вызывает морфологические изменения в каротидных тельцах, которые снижают их чувствительность к гипоксии [43, 55]. Установлено, что гломусные клетки каротидных телец постоянно экспрессируют рецепторы воспалительных цитокинов, включая рецепторы ФНО-α (ФНО-R1 и ФНО-R2), ИЛ-1β (IL-1R1) и ИЛ-6 [43, 55, 87, 88]. Показано, что циркулирующие цитокины, и в частности ФНО-α, могут распознаваться этими мембранными рецепторами и провоцировать высвобождение гломусными клетками тормозного медиатора дофамина [40, 93]. В экспериментах, проведенных in vitro установлено, что в каротидном теле ФНО-α может уменьшать хемосенсорные разряды, вызванные гипоксией [40].
В совокупности, эти данные позволяют предполагать, что цитокиновый шторм может ослаблять функцию каротидных телец, снижая тем самым компенсаторную реакцию дыхательной системы на уменьшение напряжения кислорода в артериальной крови. Данные, полученные в нашей лаборатории, подтверждают это предположение, показывая, что повышение системного уровня ключевых провоспалительных цитокинов ИЛ-1β и ФНО-α, увеличивая вентиляцию легких при спокойном дыхании воздухом, в то же время снижает чувствительность респираторной системы к гипоксии ослабляя вентиляционный гипоксический ответ [16, 17].
Провоспалительные цитокины могут влиять не только на периферические, артериальные, но и на центральные, медуллярные хеморецепторы, роль которых выполняют хемочувствительные нейроны, расположенные на вентральной поверхности продолговатого мозга. Возбуждение медуллярных хеморецепторов, усиливающее инспираторную активность дыхательного центра, происходит при повышении концентрации ионов водорода во внеклеточной жидкости мозга, которое происходит при гиперкапнических изменениях в газовом составе артериальной крови. Сдвиг внутритканевого рН является наиболее выраженным фактором в механизме действия углекислоты на центральные хеморецепторы [6, 61]. При усилении эндогенной продукции ФНО-α было обнаружено снижение вентиляционной чувствительности к гиперкапнии [46]. Ослабление вентиляционного ответа на гиперкапнию наблюдается и после экзогенного повышения церебрального уровня ИЛ-1β [14, 15]. Эти данные свидетельствуют о влиянии провоспалительных цитокинов на центральные механизмы регуляции дыхания. Возможность таких влияний определяется тем, что цитокины и их рецепторы экспрессируются в большинстве областей мозга и участвуют в нейроиммунных взаимодействиях, оказывая прямое или опосредованное действие на клетки центральной нервной системы [8]. Иммуногистохимические исследования показали наличие экспрессии цитокинов и их рецепторов в ядре солитарного тракта и в вентролатеральном отделе продолговатого мозга, т.е. в респираторно зависимых районах ствола мозга [29, 33, 45, 72].
Экспериментальные факты, свидетельствующие о снижении вентиляционного ответа на изменение газового состава крови при повышении системного и церебрального уровня провоспалительных цитокинов, дают основание полагать, что цитокиновый шторм ослабляет компенсаторные возможности системы внешнего дыхания. Нарушение рефлекторных механизмов регуляции дыхания усугубляет ухудшение дыхательной функции, вызванное морфологическими повреждениями дыхательных путей, так как ослабление вентиляционного ответа на гипоксию и гиперкапнию препятствует восстановлению нормального газового состава артериальной крови.
Влияние провоспалительных цитокинов на бронхопульмональные сенсорные волокна
Рассматривая патофизиологические механизмы цитокинового шторма, следует также отметить способность цитокинов модулировать активность не только артериальных и медуллярных хеморецепторов, но и хемочувствительных рецепторов и ноцицепторов дыхательных путей, таких как быстроадаптирующиеся (ирритантные) рецепторы и рецепторы C-волокон (J-рецепторы). Быстро адаптирующиеся высокопороговые рецепторы представляют собой Aδ-миелинизированные афферентные нервные волокна, идущие от немиелинизированных терминалей, локализованных на всем протяжении трахеобронхиального дерева. Легочные C-волокона являются капсаицин-чувствительными афферентными волокнами, имеющими функциональные связи с тучными клетками. Афферентная активность, возникающая в бронхопульмональных сенсорных терминалях проводится главным образом блуждающими нервными волокнами, которые проецируются на уровень продолговатого мозга в ядро одиночного тракта.
Высокопороговые рецепторы Aδ имеют много общих характеристик с рецепторами C-волокон, включая хемочувствительность: и те, и другие стимулируются перекисью водорода, арахидоновой кислотой, провоспалительными цитокинами ФНО-α и ИЛ-1β [59, 62, 92]. С другой стороны, нейропептиды, выделяющиеся из окончаний C-волокон, могут воздействовать на иммунные клетки [11]. Недавние исследования показали, что чувствительность бронхолегочных С-волокон значительно повышена при воспалительных заболеваниях дыхательных путей [56].
Эти данные демонстрируют, что ноцицепторы дыхательных путей могут быть активированы провоспалительными цитокинами и подтверждают гипотезу о том, что бронхопульмональные сенсорные волокна передают иммунные сигналы из легких в головной мозг и осуществляют нейроиммунное взаимодействие между легкими и мозгом [58]. Известно, что стимуляция С-волокон при низком уровне интенсивности вызывает учащенное поверхностное дыхание, а при высокой интенсивности – апноэ, т.е. остановку дыхания. Наши данные показывают, что повышение системного уровня ИЛ-1β или ФНО-α снижает возможность спонтанного восстановления дыхания после апноэ, вызванного гипоксическим воздействием, увеличивая летальность при тяжелой степени острой гипоксии [3]. Возможно, что в основе этих явлений наряду с другими механизмами лежит и усиленная активация легочных ноцицепторов провоспалительными цитокинами. Логично предположить, что этот механизм будет активирован и при цитокиновом шторме, что усугубит неблагоприятное действие гипоксии на организм и увеличит возможность неблагоприятного исхода.
Роль циклооксигеназных и NO-синтазных путей в механизмах влияния цитокинов на респираторную функцию
Влияние цитокинов на физиологические функции может быть опосредовано множественными путями, через высвобождение простаноидов, норэпинефрина, кортикотропинрилизинг фактора, оксида азота (NO) [47, 48, 69, 71, 89]. Вывод о том, что провоспалительные цитокины могут действовать на механизмы регуляции дыхания не прямо, а опосредовано подтверждается экспериментальными данными. Так, например, было показано, что системное введение ИЛ-1β индуцирует экспрессию средне-раннего гена c-fos в ряде структур головного мозга, в том числе в ядре одиночного тракта, в латеральных парабрахиальных ядрах, в вентролатеральном отделе продолговатого мозга, т.е. в тех областях, где расположены дыхательные нейроны [38]. При этом анализ распределения мРНК, кодирующей белок рецептора ИЛ-1 первого типа (IL-1R1) проведенный этими же исследователями не обнаружил мРНК IL-1R1 среди тех нейронов, которые отвечали на внутривенное введение ИЛ-1β индукцией транскрипционного фактора fos [39]. Следовательно, нейроны, отвечающие на цитокиновый сигнал, не имели соответствующих рецепторов. Более того, в одной из работ было показано, что прямое действие ИЛ-1β на структуры мозгового ствола in vitro не изменяет респираторно-зависимую нейрональную активность этого отдела мозга [71]. Эти данные доказывают, что действие ИЛ-1β на центральные механизмы регуляции дыхания не может реализовываться через прямое влияние ИЛ-1β на респираторные нейроны. Необходимы посредники, участвующие в передаче цитокинового сигнала.
Результаты экспериментальных исследований указывают на важную роль циклооксигеназных и NO-синтазных механизмов в путях проведения влияния провоспалительных цитокинов на рефлекторный контроль дыхания. Так, эксперименты с церебровентрикулярным и внутривенным введением ИЛ-1β на фоне действия диклофенака показали, что данный препарат устраняет угнетающее влияние ИЛ-1β и ФНО-α на гипоксический и гиперкапнический вентиляционные ответы [15–17]. В экспериментах на новорожденных крысятах было показано, что ослабление дыхания после интраперитониального введения ИЛ-1β опосредовано простагландин-зависимыми путями [71]. Как известно, диклофенак является препаратом, угнетающим активность циклооксигеназы (СОХ), фермента необходимого для синтеза простагландинов из арахидоновой кислоты. ИЛ-1β, взаимодействуя с рецептором интерлейкина-1 (ИЛ-1R1), индуцирует активность циклооксигеназы-2 (СОХ-2) и микросомальной синтазы-1 простагландина Е (mPGES-1). СОХ-2 катализирует образование простагландина Н2 (РGН2) из арахидоновой кислоты, а mPGES-1 катализирует синтез простагландина Е2 (PGE2) из РGН2. Отсутствие влияния ИЛ-1β на центральный и периферический хеморефлексы на фоне действия диклофенака позволяет сделать вывод о том, что одним из основных механизмов реализации обнаруженных респираторных эффектов ИЛ-1β является синтез РGЕ2. В соответствии с современными данными, простагландины рассматриваются как один из тормозных модуляторов, вносящих вклад в респираторную депрессию [18]. Установлено, что при повышении системного уровня цитокинов простагландины в больших количествах экспрессируются клетками церебрального эндотелия [68, 91]. Будучи небольшими растворимыми молекулами, PG легко проникают через клеточные мембраны и гематоэнцефалический барьер. Посредством этих молекул цитокины могут влиять на функции даже тех нейронов, которые не имеют рецепторов цитокинов и модулировать центральные механизмы регуляции дыхания, так как высокий уровень экспрессии рецепторов простагландинов обнаруживается в области ядра одиночного тракта, амбигуального ядра, прабрахиальных ядер, т.е. в респираторно-зависимых областях мозгового ствола [66, 70].
Простагландины могут опосредовать действие воспалительных цитокинов и на периферическую хеморецепцию, участвуя в модуляции активности каротидного тела. Установлено, что гломусные клетки каротидных тел экспрессируют рецепторы PGE2 и цитокинов воспаления (ИЛ-1β, ИЛ-6, ФНО-α). Поэтому при системном введении не исключается возможность торможения простагландинами гломусных клеток каротидного тела, т.к. показано, что PGE2 тормозит гипоксически индуцированное высвобождение катехоламина из клеток 1 типа в каротидных телах, которое является маркером деполяризации [44].
Цепочку событий, происходящих после повышения церебрального и системного уровня ИЛ-1β можно описать следующим образом. Повышение церебрального уровня ИЛ-1β вызывает индукцию СОХ-2 клеточными элементами мозга, имеющими рецепторы к ИЛ-1. Это могут быть и глиальные, и нервные клетки. Повышение уровня ИЛ-1β в кровеносной системе способствует индукции СОХ-2 эндотелием церебральных сосудов и клетками каротидного тела, имеющего большое количество рецепторов ИЛ-1. В результате и в первом, и во втором случае усиливается синтез простагландинов Е2, которые высвобождаются в межклеточное пространство и оказывают тормозное действие на нервные клетки имеющие рецепторы к РGЕ2.
Результаты исследований указывают также на важную роль оксида азота в путях проведения влияния воспаления и гиперцитокинемии на рефлекторный контроль дыхания [1, 17, 50, 57, 67]. В организме NO синтезируется в результате окислительной реакции, катализируемой ферментом NO-синтазой (NOS) из L-аргинина. В экспериментах на наркотизированных крысах с повышенным системным уровнем ИЛ-1β и ФНО-альфа было установлено, что действие неспецифического ингибитора NO-синтаз L-нитро-аргинин-метил-эфира (L-NAME) значительно ослабляет модулирующее влияние провоспалительных цитокинов на паттерн дыхания и респираторные хеморефлексы [5, 17]. Эти факты указывает на участие оксида азота в реализации респираторных влияний провоспалительных цитокинов.
Усиление синтеза NO при повышении системного уровня провоспалительных цитокинов происходит при взаимодействии цитокинов с их рецепторами, расположенными в эндотелии кровеносных сосудов. Обладая высокой проникающей способностью, они легко диффундируют через мембрану близлежащих клеток и влияют на внутриклеточные процессы, не взаимодействуя с мембранными рецепторами [21]. Диффундируя в соседние клетки, оксид азота активизирует в них образование циклического гуанозинмонофосфата (цГМФ), способного влиять на проводимость ионных каналов и, таким образом, изменять электрогенез нейронов. Установлено, что NO способен модулировать возбудимость гломусных клеток и сенсорных нейронов каротидных тел. Внедрение животным аденовируса, экспрессирующего нейрональную NO-синтазу, уменьшает базовые разряды хеморецепторов каротидных тел и ослабляет ответ на гипоксию [57]. В других исследованиях было показано двойственное дозозависимое влияние оксида азота на артериальную хеморецепцию: при нормоксии NO усиливает каротидные хемосенсорные разряды, а при гипоксии – ослабляет [50, 65]. Этот факт объясняет обнаруженный нами двойственный респираторный эффект ИЛ-1β и ФНО-α: экзогенное повышение системного уровня этих цитокинов увеличивает базовую вентиляцию при нормоксии, но уменьшает вентиляционный ответ на гипоксию.
Анализ данных литературы указывают на возможность участия провоспалительных цитокинов в модуляции хеморефлекторного контроля дыхания не только в патологических, но и в нормальных физиологических условиях, посредством активации конститутивных форм NOS, нейрональной и эндотелиальной, в гломусных клетках каротидных тел [85]. Однако в условиях цитокинового шторма роль оксида азота в модуляции дыхательных хеморефлексов может резко возрастать, так как провоспалительные цитокины способствуют усилению макрофагального синтеза индуцибельной NO-синтазы (iNOS). При этом синтез оксида азота может в сотни раз превышать синтез, осуществляемый конститутивными изоформами фермента. В этом случае действие фермента iNOS перестает быть физиологическим, так как высокие дозы NO токсичны для клеток.
Известные на сегодняшний день данные указывают на то, что активация и взаимодействие циклооксигеназных и NO-синтазных путей может быть одним из основных специфических механизмов, посредством которого провоспалительные цитокины способны изменять функциональное состояние дыхательной системы. Впервые о возможности взаимодействия между циклооксигеназными и NO-синтазными путями сообщалось в работе Salvemini в 1993 [76]. Это исследование показало, что NO активирует циклооксигеназу. Впоследствии, многие исследования подтвердили, что активность циклооксигеназы, а соответственно и синтеза простагландинов, может регулироваться оксидом азота [32, 63–65]. На моделях воспаления было доказано, что молекулы NO и простагландинов могут продуцироваться одновременно в одних и тех же тканях [32, 34]. Дальнейшие исследования подтвердили, что нитрооксиданты (NO, супероксид, пероксинитрит) модулируют биосинтез простагландинов через циклооксигеназные пути [53, 77]. Однако в полной мере конкретные молекулярные механизмы, с помощью которых NO регулирует выработку простагландинов пока остаются не выясненными. Известные в настоящее время данные позволяют предположить, что в основе негативного влияния провоспалительных цитокинов на гипоксический и гиперкапнический хеморефлексы может лежать усиление синтеза простагландинов, вызванное активацией циклооксигеназных и NO-синтазных путей при цитокин-рецепторном взаимодействии на гломусных клетках каротидного тела и эндотелии церебральных сосудов. На рис. 1 представлена возможная схема такого взаимодействия.
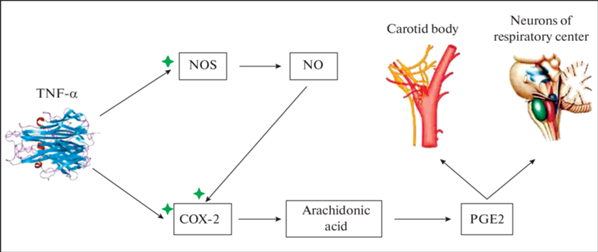
ЗАКЛЮЧЕНИЕ
Анализ литературных данных убеждает в актуальности проведения исследований механизмов респираторных эффектов провоспалительных цитокинов с целью выяснения путей влияния цитокинового шторма на функцию внешнего дыхания и его роли в развитии дыхательной недостаточности. Патогенез цитокинового шторма имеет множественные причины, характеризуясь как морфологическими повреждениями дыхательных путей, так и нарушением рефлекторных механизмов регуляции дыхания. Снижение вентиляционного ответа на гипоксию и гиперкапнию при повышении системного и церебрального уровня провоспалительных цитокинов свидетельствует о том, что цитокиновый шторм ослабляет компенсаторные возможности системы внешнего дыхания, препятствуя восстановлению нормального газового состава артериальной крови. Это усугубляет ухудшение вентиляционной функции легких, вызванное диффузным и экссудативным поражением альвеол, способствующим развитию острого респираторного дистресс-синдрома. Исследование влияния провоспалительных цитокинов на нейрогенные, рефлекторные механизмы регуляции висцеральных функций расширяют представления о патогенезе цитокинового шторма и могут иметь существенное значение для профилактики и лечения его последствий.
Список литературы
Александров В.Г., Александрова Н.П., Туманова Т.С. и др. Участие NO-ергических механизмов в реализации респираторных эффектов провоспалительного цитокина интерлейкина 1-бета // Рос. физиол. журн. им. И.М. Сеченова. 2015. Т. 101 № 10. С. 1190.
Александрова Н.П., Меркурьев В.А., Туманова Т.С., Александров В.Г. Механизмы модуляции рефлекторного контроля дыхания при повышении системного уровня провоспалительного цитокина интерлейкина 1-β // Рос. физиол. журн. им. И.М. Сеченова. 2015. Т. 101. № 10. С. 1158.
Донина Ж.А., Баранова Е.В., Александрова Н.П. Влияние провоспалительного цитокина интерлейкина 1-β на резистентность организма к острой гипоксии. Российский физиол. журн. им. И.М. Сеченова. 2016. Т. 102. № 11. С. 1333.
Кетлинский С.А., Симбирцев А.С. Цитокины. Спб.: Фолиант, 2008. С. 552.
Клинникова А.А., Данилова Г.А., Александрова Н.П. Роль NO-синтазных путей в реализации влияния провоспалительных цитокинов на паттерн дыхания и вентиляционный ответ на гипоксию // Рос. физиол. журн. им. И.М. Сеченова. 2021. Т. 107. № 11. С. 1. https://doi.org/10.31857/S0869813921110042
Менакер С. Гуморальная и нервная регуляция дыхания // Патофизиология легких. Москва: Бином, 2008. С. 220.
Мороз В.В., Голубев А.М., Марченков Ю.В. и др. Морфологические признаки острого повреждения легких различной этиологии (экспериментальное исследование) // Общая реаниматология. 2010. Т. 6. № 3. С. 29. https://doi.org/10.15360/1813-9779-2010-3-29
Мюльберг А.А., Гришина Е.В. Цитокины как медиаторы нейроиммунных взаимодействий // Успехи физиологических наук. 2006. Т. 37. № 1. С. 18.
Ройт А., Бростофф Д., Мейл Д. Иммунология. Москва: Мир, 2000.
Тарасова И.В. Система комплемента // Аллергология и иммунология в педиатрии. 2010. № 2(21). С. 45.
Филипова Л.В., Ноздрачев А.Д. Бронхолегочный нервнорецепторный аппарат // Вестник СПб университета. 2010. Сер. 3. Вып. 3. С. 54.
Чучалин А.Г. Тяжелый острый респираторный синдром // Архив патологии. 2004. № 3. С. 5–11.
Abhyankar S., Gilliland D.G., Ferrara J.L. Interleukin-1 is a critical effector molecule during cytokine dysregulation in graft versus host disease to minor histocompatibility antigens // Transplantation. 1993. V. 56(6). P. 1518. https://doi.org/10.1097/00007890-199312000-00045
Aleksandrova N.P., Danilova G.A. Effect of intracerebroventricular injection of interleukin-1-beta on the ventilatory response to hyperoxic hypercapnia // Eur. J. Med. Res. 2010. V. 15(II). P 3.
Aleksandrova N.P., Danilova G.A., Aleksandrov V.G. Cyclooxygenase pathway in modulation of the ventilatory response to hypercapnia by interleukin-1β in rats // Respir. Physiol. Neurobiol. 2015. V. 209. P. 85. https://doi.org/10.1016/j.resp.2014.12.006
Aleksandrova N.P., Danilova G.A., Aleksandrov V.G. Interleukin-1beta suppresses the ventilatory hypoxic response in rats via prostaglandin dependent pathways // Canad. J. Physiol. Pharmacol. 2017. V. 95(6). P. 681. https://doi.org/10.1139/cjpp-2016-0419
Aleksandrova N.P., Klinnikova A.A., Danilova G.A. Cyclooxygenase and nitric oxide synthase pathways mediate the respiratory effects of TNF-α in rats // Respir. Physiol. Neurobiol. 2021. V. 284: 103567. https://doi.org/10.1016/j.resp.2020.103567
Ballanyi K., Onimaru H., Homma I. Respiratory network function in the isolated brainstem-spinal cord of newborn rats // Prog. Neurobiol. 1999. V. 59. P. 583–634.
Behrens E.M., Koretzky G.A. Cytokine storm syndrome. Looking toward the precision medicine era // Arthritis Rheumatol. 2017. V. 69(6). P. 1135. https://doi.org/10.1002/art.40071
Blackwell N.S., Christman J.W. Sepsis and cytokines: current status // Br. J. Anaesth. 1996. V. 77. P.110. https://doi.org/10.1093/bja/77.1.110
Brenman J.E., Bredt D.S. Nitric oxide signaling in the nervous system // Methods Enzymol. 1996. V. 269. P. 119. https://doi.org/10.1016/s0076-6879(96)69014-4
Bulanov A. Transfusion-associated lung injury (TRALI): obvious and incomprehensible // Anest. Reanimatol. 2009. V. 5. P. 48.
Burger D., Daer J.-M. Inhibitory cytokines and cytokine inhibitors // Neurology. 1995. V. 45(6). P. S39. https://doi.org/10.1212/wnl.45.6_suppl_6.s39
Casey L.C., Balk R.A., Bone R.C. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome // Ann. Intern. Med. 1993. V. 119(8). P. 771. https://doi.org/10.7326/0003-4819-119-8-199310150-00001
Channappanavar R., Fehr A.R., Vijay R. et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice // Cell host & Microbe. 2016. V. 19(2). P. 181. https://doi.org/10.1016/j.chom.2016.01.007
Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology // Semin. Immunopathol. 2017: V. 39. P. 529.https://doi.org/10.1007/s00281-017-0629-x
Chen N., Zhou M., Dong X. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study // Lancet. 2020. V. 395. № 10223. P. 507. https://doi.org/10.1016/S0140-6736(20)30211-7
Chousterman B.G., Swirski F.K., Weber G.F. Cytokine storm and sepsis disease pathogenesis // Seminars in Immunopathology. 2017. V. 39(5). P. 517.
Churchill L., Taishi P., Wang M. et al. Brain distribution of cytokine m RNA induced by systemic administration of interleukin-1beta or tumor necrosis factor alpha // Bran Res. 2006. V. 1120. № 1. P. 64.
Cohen J. The immunopathogenesis of sepsis // Nature. 2002. V. 420. P. 885. https://doi.org/10.1038/nature01326
Conti H., Caraffa A., Gallenga C. et al. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: a promising inhibitory strategy // J. Biol. Regul. Homeost. Agents. 2020. P. 34(6). P. 1971. https://doi.org/10.23812/20-1-E
Cuzzocrea S., Salvemini D., Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes // Kidney. 2007. Int. 71 (4). P. 290. https://doi.org/10.1038/sj.ki.5002058
Dantzer R., Konsman J.P., Bluthe R.M. et al. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? // Auton. Neurosci. 2000. V. 85(1–3). P. 60.
Dantzer R., Konsman L., Silva B.R. et al. Endothelial nitric oxide synthase and cyclooxygenase are activated by hydrogen peroxide in renal hypertensive rat aorta // Eur. J. Pharmacol. 2017. V. 814. P. 87. https://doi.org/10.1016/j.ejphar.2017.07.047
Davidson S., Maini M.K., Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections // J. interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2015 V. 35(4). P. 252. https://doi.org/10.23812/20-1-E
Drosten C., Seilmaier M., Corman V.M. et al. Clinical features and virological analysis of a case of Middle East respiratory syndrome coronavirus infection // The Lancet Infectious Diseases. 2013. V. 13(9). P. 745.
Dushianthan A., Grocott M.P., Postle A.D., Cusack R. Acute respiratory distress syndrome and acute lung injury // Postgrad. Med. J. 2011. V. 87(1031). P. 612.
Ericsson A., Kovacs K.J., Sawchenko P.E. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons // J. Neurosci. 1994. V. 14. № 2. P. 897.
Ericsson A., Liu C., Hart R.P., Sawchenko P.E. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation // J. Comp. Neurol. 1995. V. 361. P.681.
Fernández R., González S., Rey S. et al. Lipopolysaccharide-induced carotid body inflammation in cats: functional manifestations, histopathology and involvement of tumour necrosis factor-alpha // Exp. Physiol. 2008. V. 93(7). P. 892. https://doi.org/10.1113/expphysiol.2008.041152
Ferrara J.L. Cytokine dysregulation as a mechanism of graft versus host disease // Curr. Opin. Immunol. 1993. V. 5(5). P. 794. https://doi.org/10.1016/0952-7915(93)90139-j
Ferrara J.L., Abhyankar S., Gilliland D.G. Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1 // Transplant. Proc. 1993. V. 25 (Pt 2). P. 1216.
Gauda E.B., Shirahata M., Masona A. et al. Inflammation in the carotid body during development and its contribution to apnea of prematurity // Respir. Physiol. Neurobiol. 2013. V. 185(1). P. 120. https://doi.org/10.1016/j.resp.2012.08.005
Gomez-Nino A., Lopez-Lopez J.R., Almaraz L., Gonzalez C. Inhibition of [3H] catecholamine release and Ca2+ currents by prostaglandin E2 in rabbit carotid body chemoreceptor cells // J. Physiology (London). 1994. V. 476. P. 269–277.
Gordon F.J. Effect of nucleus tractus solitarius lesions on fever produced by interleukin-1beta // Auton. Neurosci. 2000. V. 85. P. 102. https://doi.org/10.1016/s1566-0702(00)00228-9
Gosselin L.E., Barkley J.E., Spencer M.J. et al. Ventilatory dysfunction in mdx mice: impact of tumour necrosis factor-alpha deletion // Muscle Nerve. 2003. V. 28. P. 336. https://doi.org/10.1002/mus.10431
Graff G.R., Gozal D. Cardiorespiratory responses to interleukin-1beta in adult rats: role of nitric oxide, eicosanoids and glucocorticoids // Arch. Physiol. Biochem. 1999. V. 107(2). P. 97.
Herlenius E. An inflammatory pathway to apnea and autonomic dysregulation // Respir. Physiol. Neurobiol. 2011. V. 178. P. 449. https://doi.org/10.1016/j.resp.2011.06.026
Hofstetter A.O., Herlenius E. Interleukin-1beta depresses hypoxic gasping and autoresuscitation in neonatel DBA/1lacJ mice // Respir. Physiol. Neurobiol. 2005. V. 146. № 2–3. P.135. https://doi.org/10.1016/j.resp.2004.11.002
Iturriaga R. Nitric oxide and carotid body chemoreception // Biol. Res. 2001. V. 34(2). P. 135. https://doi.org/10.4067/s0716-97602001000200019
Jiang Y., Xu J., Zhou C. et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome // Amer. J. Respir. Crit. Care Med. 2005. V. 171(8). P. 850. https://doi.org/10.1164/rccm.200407-857OC
Kempuraj D., Selvakumar G.P., Ahmed M.E. et al. COVID-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation // Neuroscientist. 2020. V. 26(5–6). P. 402. https://doi.org/10.1177/1073858420941476
Kim S.F. The role of nitric oxide in prostaglandin biology; update // Nitric Oxide. V. 25(3). 2011. P. 255. https://doi.org/10.1016/j.niox.2011.07.002
Komorowski M., Aberegg S.K. Using applied lung physiology to understand COVID-19 patterns // British J. Anaesthesia. 2020. V. 125. № 3. P. 250. https://doi.org/10.1016/j.bja.2020.05.019
Lam S.Y., Tipoe G.L., Liong E.C., Fung M.L. Chronic hypoxia upregulates the expression and function of proinflammatory cytokines in the rat carotid body // Histochem. Cell Biol. 2008. V. 130(3). P. 549. https://doi.org/10.1007/s00418-008-0437-4
Lee L.Y. Respiratory sensations evoked by activation of bronchopulmonary C-fibers // Respir. Physiol. Neurobiol. 2009. V. 167(1). P. 26. https://doi.org/. resp.2008.05.006.https://doi.org/10.1016/j
Li Y-L., Li Y-F., Liu D. et al. Gene transfer of neuronal nitric oxide synthase to carotid body reverses enhanced chemoreceptor function in heart failure rabbits // Circ Res. 2005. V. 97(3). P. 260. https://doi.org/10.1161/01.RES.0000175722.21555.55
Li H.F., Yu J. Airway chemosensitive receptors in vagus nerve perform neuro-immune interaction for lung-brain communication // Adv. Exp. Med. Biol. 2009. V. 648. P. 421. https://doi.org/10.1007/978-90-481-2259-2_48
Lin S., Li H., Xu L. et al. Arachidonic acid products in airway nociceptor activation during acute lung injury // Exp. Physiol. 2011. V. 96. P. 966 https://doi.org/10.1113/expphysiol.2011.058263
Lin S.-H., Zhao Y.-S., Zhou D.-X. et al. Coronavirus disease 2019 (COVID-19): cytokine storms, hyper-inflammatory phenotypes, and acute respiratory distress syndrome // Genes Dis. 2020. V. 7(4). P. 520. https://doi.org/10.1016/j.gendis.2020.06.009
Loeschcke H.H. Respiratory chemosensitivity in the medulla oblongata // Acta Neurobiol. Exp. 1973. V. 33. P. 97.
Maier S.F., Goehler L.E., Fleshner M., Watkins L.R. The role of the vagus nerve in cytokine-to-brain communication // Ann. New York: Acad. Sci. 1998. V. 840. P. 289.
Molina-Holgado F., Lled’o A., Guaza C. Evidence for cyclooxygenase activation by nitric oxide in astrocytes // Glia. 1995. V. 15 (2). P. 167.
Mollace V., Muscol C., Masin E. et al. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors // Pharmacol. Rev. 2005. V. 57(2). P. 217.
Moya E.A., Alcayaga J., Iturriaga R. NO modulation of carotid body chemoreception in health and disease // Respir. Physiol. Neurobiol. 2012. V. 184(2). P. 158. https://doi.org/10.1016/j.resp.2012.03.019
M. Ek, Alcayaga E., Arias C. et al. Distribution of the EP3 prostaglandin E (2) receptor subtype in the rat brain: relationship to sites of interleukin-1-induced cellular responsiveness // J. Comp. Neurol. 2000. 428(1). 5–20. https://doi.org/10.1002/1096-9861(20001204)428: 1<5::aid-cne2>3.0.co;2-m
Murakami Y., Yokotani K., Okuma Y., Osumi Y. Nitric oxide mediates central activation of sympathetic outflow induced by interleukin-1 beta in rats // Eur. J. Pharmacol. 1996. V. 317(1). P. 61.
Nadeau S., Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier // Neuroscience. 1999. V. 93 (4). P. 1449.
Nakamori T., Morimoto A., Murakami N. Effect of a central CRF antagonist on cardiovascular and thermoregulatory responses induced by stress or IL-1β // Am. J. Physiol. 1993. V. 265(4). P. 834. https://doi.org/10.1152/ajpregu.1993.265.4.R834
Nakamura K., Kaneko T., Yamashita Y. et al. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system // J. Comp. Neurol. 2000. V. 421(4). P. 543.
Olsson A., Kayhan G., Lagercrantz H., Herlenius E. IL-1 beta depresses respiration and anoxic survival via a prostaglandin-dependent pathway in neonatal rats // Pediatr Res. 2003. V. 54. P. 326–331. https://doi.org/10.1203/01.PDR.0000076665.62641.A2
Oppenheim J.J. Cytokines, their receptors and signals // In: The Autoimmune Diseases. London: Elsevier. V. 2020. P. 275. https://doi.org/10.1016/B978-0-12-812102-3.00015-4
Parsons P.E., Eisner M.D., Thompson B.T. et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury // Critical Care Medicine. 2005. V. 33(1). P. 1–232. https://doi.org/10.1097/01.ccm.0000149854.61192.dc
Porzionato A., Macch, V., De Caro R., Di Giulio C. Inflammatory and immunomodulatory mechanisms in the carotid body // Respir. Physiol. Neurobiol. 2013. V. 187(1). P. 31. https://doi.org/10.1016/j.resp.2013.02.017
Pugin J., Ricou B., Steinberg K.P. et al. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1 // Am. J. Respir. Crit. Care Med. 1996. V. 153. P. 1850.
Salvemini D., Misko T.P., Masferrer J.L. et al. Nitric oxide activates cyclooxygenase enzymes // Proc. Natl. Acad. Sci. U. S. A. 1993. 90(15). 7240.
Salvemini D., Kim S.F., Mollace V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: relevance and clinical implications. Am. J. Physiol // Regul. Integr. Comp. Physiol. 2013. V. 304(7). P. 473. https://doi.org/10.1152/ajpregu.00355.2012
Shaw A.S., Goldstein D.R., Montgomery R.R. Age-dependent dysregulation of innate immunity // Nature reviews Immunology. 2013. V. 13(12). P. 875.
Shimabukuro-Vornhagen A., Gödel P., Subklewe M. et al. Cytokine release syndrome // J. Immuno Therapy of Cancer. 2018. V. 6(1). P. 56.
Song P., Li W., Xie J., Hou Y., You C. Cytokine storm induced by SARS-CoV-2 // Clin. Chim. Acta. 2020. V. 509. P. 280–7. https://doi.org/10.1016/j.cca.2020.06.017
Stockman L.J., Bellamy R., Garner P. SARS: systematic review of treatment effects // PLoS Medicine. 2006. V. 3(9). P. 343-e. https://doi.org/10.1371/journal.pmed.0030343
Tay M.Z., Poh C.M., Renia L. et al. The trinity of COVID-19: immunity, inflammation and intervention // Nat. Rev. Immunol. 2020. V. 20. P. 363. https://doi.org/10.1038/s41577-020-0311-8
TisoncikKorth M.J., Simmons C.P. et al. Into the eye of the cytokine storm // Microbiol. Mol. Biol. Rev. 2012. V. 76(1). P. 16. https://doi.org/10.1128/MMBR.05015-11
Tonini G., Sanini D., Vincenzi B. et al. Oxaliplatin may induce cytokine release syndrome in colorectal cancer patients // J. Biol. Regul. Homeost. Agents. 2002. P. 16.
Valdes V., Mosqueira M., Rey S. Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms // Am. J. Physiol. Lung. Cell. Mol. Physiol. 2003. V. 284(1). P. 57. https://doi.org/10.1007/978-1-4419-9280-2_45
Wang H., Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome // The Amer. J. Emerg. Med. 2008. V. 26(6). P. 711. https://doi.org/10.1016/j.ajem.2007.10.031
Wang X., Wang B.R., Duan X. L. Strong expression of interleukin-1 receptor types I in the rat carotid body // J. Histochem. Cytochem. 2002. V. 50(12). P. 1677. https://doi.org/10.1177/002215540205001213
Wang X., Zhang X.J., Xu Z. Morphological evidence for existence of IL-6 receptor alpha in the glomus cells of rat carotid body // Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006. V. 288(3). P. 292. https://doi.org/10.1002/ar.a.20310
Watanabe T., Tan N., Saiki Y. et al. Possible involvement of glucocorticoids in the modulation of interleukin-1-induced cardiovascular responses in rats // J. Physiol. 1996. V. 491(1). P. 231. https://doi.org/10.1113/jphysiol.1996.sp021211
Wong J.P., Viswanathan S., Wan M. et al. Current and future developments in the treatment of virus-induced hypercytokinemia // Future Medicinal Chemistry, 2017. V. 9. № 2. P. 169. https://doi.org/10.4155/fmc-2016-0181
Wong M.L., Bongiorno P.B., Gold P.W., Licinio J. Localization of interleukin-1beta converting enzyme mRNA in rat brain vasculature: evidence that the genes encoding the interleukin-1 system are constitutively expressed in brain blood vessels // Pathophys. Implicat. Neuroimmun. Modulat. 1995. V. 2(3). P. 141. https://doi.org/10.1159/000096884
Yu J., Lin S., Zhang J. et al. Airway nociceptors activated by pro-inflammatory cytokines // Respir. Physiol. Neurobiol. 2007. V. 156. P. 116.https://doi.org/10.1016/j.resp.2006.11.005
Zapata P., Larrain C., Reyes P., Fernández R. Immunosensory signaling by carotid body chemoreceptors // Respir. Physiol. Neurobiol. 2011. V. 178(3). P. 370. https://doi.org/doi:10.1016/j. resp.2011.03.025
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук