

Успехи физиологических наук, 2022, T. 53, № 3, стр. 15-44
Фактор, индуцируемый гипоксией и ингибиторы пролилгидроксилазы – новая фармакологическая мишень и класс лекарственных препаратов, стимулирующих эритропоэз и не только
Д. В. Куркин a, *, Д. А. Бакулин a, Е. Е. Абросимова a, И. Н. Тюренков a
a ФГБОУ Волгоградский Государственный медицинский университет
400087 Волгоград, Россия
* E-mail: strannik986@mail.ru
Поступила в редакцию 22.03.2022
После доработки 25.03.2022
Принята к публикации 02.04.2022
- EDN: LEPGCD
- DOI: 10.31857/S0301179822030067
Аннотация
В статье приводится обзор отечественных и зарубежных публикаций, посвященных гипоксии и средствам ее коррекции. Рассмотрены гипотезы адаптации организма и его клеток к гипоксическим условиям, а также история открытия, строение, пути активации и биологические эффекты HIF – фактора, индуцируемого гипоксией. Описаны процессы, при которых указанный фактор выступает в роли цитопротектора, а также те условия, при которых HIF может являться патологическим звеном. Рассмотрены и описаны ингибиторы пролилгидроксилазы HIF – ключевого фермента, разрушающего фактора, индуцируемого гипоксией (роксадустат, вададустат, молидустат, дапродустат, десидустат, энародустат), в том числе их применение и побочные эффекты.
ВВЕДЕНИЕ
Кислород жизненно необходим большинству организмов для осуществления метаболизма и продукции энергии. При нарушении оксигенации наступает состояние гипоксии, влекущее за собой клеточную и тканевую дисфункцию с последующей их гибелью. В наиболее общем виде гипоксию можно определить, как несоответствие энергопотребности клетки энергопродукции в системе митохондриального окислительного фосфорилирования. Причинами гипоксии, как правило, являются внешние и внутренние факторы: расстройства внешнего дыхания, кровообращения в легких, кислородтранспортной функции крови, нарушения системного, регионарного кровообращения и микроциркуляции, эндотоксемия [6]. Следовательно, улучшение адаптации к недостаточности кислорода важно для терапевтического влияния на течение и исход внутренних факторов. Первые разработки в данном направлении появились в 60-е годы XX в. на кафедре фармакологии Военно-медицинской академии им. С.М. Кирова в Ленинграде и принадлежали профессору В.М. Виноградову: были созданы средства защиты от гипоксии, затем получившие название антигипоксантов. Одними из первых представителей данной группы являлись аминотиоловые производные (гутимин, амтизол) [2].
Дальнейшие исследования привели к созданию многочисленных антигипоксических средств, имеющих различные точки приложения:
Антигипоксанты прямого энергизирующего действия – корректоры нарушений энергетического обмена (иначе корректоры дисфункции дыхательной цепи митохондрий);
Антигипоксанты непрямого энергизирующего действия (корректоры нарушений метаболических путей) [4].
Классификация антигипоксантов включает [6]:
1. Ингибиторы окисления жирных кислот.
2. Сукцинатсодержащие и сукцинатобразующие средства.
3. Естественные компоненты дыхательной цепи.
4. Искусственные редокс-системы.
5. Макроэргические соединения.
Антигипоксанты как самостоятельный класс лекарственных препаратов выделен только в России, однако их противогипоксическое действие исследуется учеными и имеет перспективы для лечения различных заболеваний (например, нейродегенеративные заболевания, опосредованные митохондриальной дисфункцией [29] и окислительным стрессом [76], коррекцией и адаптацией организма к экстремальным условиям среды [48]).
В последнее время все больше внимания исследователей в области физиологии и фармакологии уделяется фактору, индуцируемому гипоксией (HIF), как белку, участвующему в системном ответе организма на изменение концентрации кислорода (изменение его активности – один из ключевых факторов адаптации к изменению условий среды). Изучение физиологии HIF может стать объяснением феномену прекондиционирования (применяющегося в кардиохирургии и гипоксических тренировках организма) – метаболической адаптации организма, заключающейся в подготовке клеток и тканей к длительной гипоксии после ряда кратковременных эпизодов нарушения доставки кислорода [5, 7]. Экспрессия гена HIF-lα также может служить маркером – отражать специфический ответ на гипоксическое воздействие [10].
Гипоксия является неотъемлемой частью нормального эмбрионального развития человека, к примеру, это состояние стимулирует стволовые клетки к развитию [74], также концентрация кислорода важна для закрытия нервной трубки плода, регуляции апоптоза и морфогенеза в период гестации [93].
РЕАКЦИЯ КЛЕТОК НА ИЗМЕНЕНИЕ КОНЦЕНТРАЦИИ О2
Молекулярный кислород (O2) необходим для окислительного фосфорилирования – основного источника энергии для клеток всех аэробных организмов. Продуктом этого процесса является углекислый газ (CO2). Выход концентрации этих газов за пределы физиологического диапазона представляет серьезную угрозу для выживания клеток, тканей и всего организма. Перечисленное выше указывает на чрезвычайную роль процессов контроля и поддержки их эндогенных концентраций (на соответствующих уровнях) для обеспечения процессов гомеостаза.
Высшие организмы, такие как млекопитающие, в процессе эволюции развили механизмы, позволяющие улавливать изменение концентраций O2 и CO2 как в кровотоке, так и в отдельных клетках, в результате чего протекают адаптивные реакции к гипоксии или гиперкапнии.
Классический пример высокоспецифичной реакции на гипоксию чувствительных к кислороду клеток – клетки синокаротидной зоны – основные артериальные хеморецепторы, расположенные в бифуркации сонной артерии [54].
Функциональная единица каротидного тельца состоит из гломусных клеток (тип I), которые являются нейрональными по своей природе, и клеток типа II, которые более похожи на глиальные стволовые клетки, которые способствуют клеточной пролиферации при хронической гипоксемии [55]. Клетки I типа в первую очередь отвечают за восприятие кислорода. Каротидные тельца не только сильно васкуляризованы, но и сильно иннервируются как афферентными, так и эфферентными волокнами.
Существует несколько гипотез, как клетки I типа улавливают изменение концентрации кислорода.
Согласно мембранной гипотезе, в детекции снижения парциального давления кислорода (pO2) играют роль кислород-чувствительные K+-каналы, расположенные на мембране гломусных клеток. Закрытие калиевых каналов служит в качестве первичного события для ответа гломусных клеток типа I на гипоксию. За этим следует деполяризация мембраны и открытие потенциал-зависимых кальциевых каналов, что приводит к притоку кальция, что, в свою очередь, способствует высвобождению нейромедиаторов, таких как дофамин, ацетилхолин и АТФ. Затем происходит стимуляция афферентных волокон и передача сигналов в дыхательный центр. Однако стоит отметить, что калиевые каналы не действуют как прямые кислородные сенсоры: существуют вышестоящие сенсоры О2, природа которых пока до конца не установлена, однако существует несколько предположений [77], которые будут перечислены ниже.
Гипотеза метаболических сенсоров предполагает, что в условиях достаточно тяжелой гипоксии окислительное фосфорилирование в митохондриях снижается из-за ограниченной доступности конечного акцептора электронов митохондриальной цепи переноса электронов (O2). Это приводит к снижению выработки АТФ и соответствующему накоплению его предшественника АМФ. Были предложены две отдельные гипотезы, касающиеся того, как снижение концентрации АТФ связано с закрытием К-каналов в гломусных клетках I типа. Первая: связанный с Twik-ассоциациированный кислотно-чувствительный K-канал (TASK) активируется АТФ, что приводит к его закрытию при истощении АТФ. Эта прямая связь между истощением АТФ и активностью К-канала проста; однако отсутствующей частью информации в этой модели является идентификация сенсора АТФ. Согласно второй гипотезе, может иметь значение АМФ-активируемая протеинкиназа (АМФК, фермент, активирующийся при истощении АТФ и повышении в клетке отношения АМФ/АТФ). Однако остается не до конца ясным, что служит пусковым сигналом: истощение АТФ или повышение концентрации АМФ в митохондриях кислород-чувствительных клеток [24].
Гипотеза редокс сенсоров предполагает, что измененная генерация активных форм кислорода (АФК) митохондриями в ответ на гипоксию является ключевым событием в детекции концентрации О2 клетками, которое связывает гипоксию с закрытием калиевого канала и последующей деполяризацией мембраны. Было высказано предположение, что гипоксия вызывает производство АФК комплексом I цепи переноса электронов в митохондриях клеток I типа. Это подтверждается исследованиями с использованием как фармакологической, так и генетической блокады комплекса I [88].
Газотрансмиттерная гипотеза. Существует гипотеза об участии газотрансмиттеров в реализации чувствительности к кислороду в клетках типа I синокаротидной зоны. К таким газотрансмиттерам может относиться CO, продуцируемый ферментом гемоксигеназой-2, которая осуществляет превращение гема в биливердин и CO с использованием кислорода. Снижение концентрации кислорода, используемого гемоксигеназой-2, ведет к снижению концентрации CO, который, предположительно, служит активатором калиевых каналов. Таким образом, уменьшение количества CO ведет к закрытию калиевых каналов [21].
Относительно недавно было сделано предположение, что закрытие калиевых каналов, опосредованное CO, осуществляется посредством регуляции продукции другим газотрансмиттером – H2S. CO приводит к активации протеинкиназы G, которая, в свою очередь, фосфорилирует и инактивирует цистатионин-гамма-лиазу, основной клеточный источник H2S. Следовательно, снижение концентрации кислорода при гипоксии вызовет снижение концентрации CO, что, в свою очередь, приведет к снижению активности протеинкиназы G, увеличению активности цистатионин-гамма-лиазы и продукции H2S [3, 84].
Естественно, чувствительность синокаротидных клеток к гипоксии не является уникальной. Помимо каротидных телец, существуют другие периферические хеморецепторы в тканях и органах, воспринимающие острую гипоксию (например, легочные артерии, артериальный проток, мозговое вещество надпочечников или нейроэпителиальные тельца в легких), которые вместе составляют “сенсорную систему”, имеющую фундаментальное биологическое и медицинское значение [24, 78]. Клетки гладкой мускулатуры этих тканей способны к внутреннему восприятию кислорода посредством окислительно-восстановительного механизма, в котором сниженное производство АФК комплексами I и III митохондриальной цепи переноса электронов приводит к регулированию чувствительных к кислороду потенциал-зависимых калиевых каналов, которые индуцируют сужение сосудов за счет усиления внутриклеточной кальций-зависимой передачи сигналов [26]. Таким образом, в разных химиочувствительных клетках организма существует множество механизмов острой чувствительности к кислороду.
Острые физиологические реакции на изменение концентрации O2 и CO2 дополняются более медленной и более устойчивой реакцией, которая возникает на клеточном уровне и дополняет респираторную адаптацию посредством регуляции факторов транскрипции и последующей экспрессии генов.
В условиях хронический гипоксии адаптационный ответ клеток и тканей опосредуется преимущественно фактором, индуцируемым гипоксией (HIF).
HIF: ИСТОРИЯ ОТКРЫТИЯ
Этот фактор был независимо обнаружен тремя научными группами, под руководством У. Келина, Г. Семензы и П. Ратклиффа, которые независимо друг от друга рассматривали молекулярный механизм восприятия кислорода с разных точек зрения: У. Келин опирался на свой опыт в онкологии и на биохимические исследования; исследования П. Ратклиффа начались с его роли нефролога, а Г. Семенза использовал свои знания в области медицинской генетики. Исследования показали, что в условиях гипоксии активация транскрипции многих регуляторных генов начинается со связывания специфического белка с регуляторным участком на промоторе. Этот белок получил название гипоксией индуцируемый фактор-1α (hypoxia inducible factor, HIF-1α) [56]. За описание молекулярного механизма, посредством которого работает путь восприятия кислорода клетками, в 2019 г. данным ученым была присуждена Нобелевская премия.
HIF: СТРОЕНИЕ
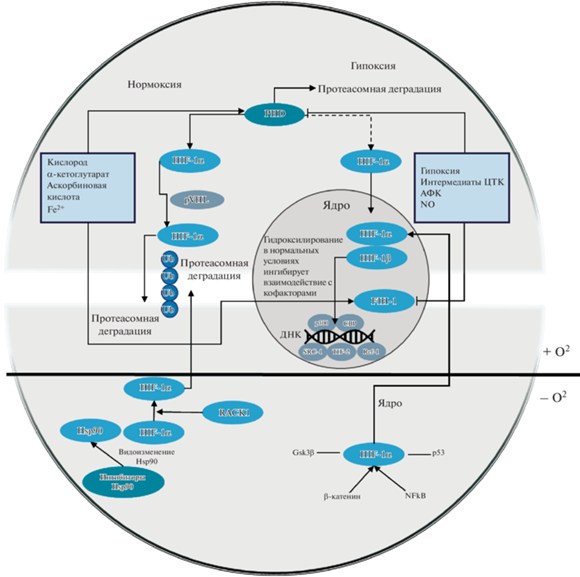
HIF-1 представляет собой гетеродимер, состоящий из постоянно экспрессируемой β-субъединицы и индуцибельной α-субъединицы (HIF-1α и HIF-1β). Обе субъединицы содержат мотив спираль–поворот–спираль, служащий для связывания ДНК и димеризации фактора. HIF-1α содержит домен кислород зависимой деградации (ODD), который является мишенью пролил-гидроксилазы-2 (PHD-2), которая запускает убиквитин-зависимую протеасомную деградацию α-субъединицы в условиях нормоксии. Также α-субъединица содержит два трансактивационных домена (TAD), регулирующих гены-мишени HIF: белок, связывающий фактор транскрипции CREB и p300, коактиваторы фактора транскрипции. Эти активаторы могут использоваться в качестве мишеней для изменения активности HIF посредством регуляции его транскрипции и трансляции. Бета-субъеденица также известна как арил-углеводородный ядерный транслокатор (ARNT), поскольку был обнаружен раньше HIF-1α и считался компонентом арил-углеводородного рецептора (AhR), обеспечивающим его перемещение в ядро [56].
Помимо HIF-1α, существуют также 2α и 3α, все они могут связываться с β-субъединицей, различаются только типами экспрессирующих их клеток: 1а экспрессируется почти всеми клетками, в то время как 2 и 3 представлены менее широко [33].
КИСЛОРОД-ЗАВИСИМЫЙ ПУТЬ АКТИВАЦИИ HIF-1α
Большинство выявленных к настоящему времени HIF-1α-взаимодействующих белков регулируют стабильность HIF-1α либо O2-зависимым, либо O2-независимым образом.
В нормоксических условиях HIF-1α имеет очень короткий период полураспада (около 5 минут) [56], поскольку подвергается быстрой деградации посредством гидроксилирования чувствительными к кислороду HIF-1α-специфическими пролилгидроксилазами (PHD-1,2,3). В основном активность HIF ограничивается активностью PHD-2 (присутствует почти во всех клетках). Помимо PHD-2 также существует еще две изоформы пролилгидрокислаз: PHD1 и PHD-3, присутствующие и в цитозоле, и в ядре, их функции не различаются. Данные белки принадлежат к суперсемейству Fe (II) и 2-оксоглутарат-зависимых оксигеназ. В условиях нормоксии, α-субъединица HIF гидроксилируется в доменах кислород-зависимой деградации, что создает сайт связывания для белка фон Хиппеля–Ландау (pVHL) – опухолевого супрессора, рекрутирующего убиквитин- лигазный белковый комплекс E3, который катализирует образование ковалентной связи убиквитина с остатками лизина в HIF-1α, что является сигналом для протеасомной деградации α-субъединицы [43, 67]. Субъединицы HIF-1α также являются субстратами для аспарагинилгидроксилазы, FIH-1 (фактор, ингибирующий HIF-1α).
FIH-1 также распознает кислород, а гидроксилирование с помощью FIH-1 нарушает критическое взаимодействие между субъединицами HIF-1α и ко-активаторами, такими как p300/CBP, нарушая транскрипционную активность HIF.
Противоположная ситуация наблюдается в условиях гипоксии: пролилгидроксилазы инактивируются (в том числе под действием интермедиатов цикла трикарбоновых кислот (ЦТК)), нарушается гидроксилирование остатков пролина, α-субъединица стабилизируется и объединяется с β-субъединицей с образованием активного транскрипционного фактора HIF-1, выполняющего свои функции (рис. 1).
Рис. 1.
Кислород-зависимый и кислород-независимый пути активации HIF-1α (https://www.abcam.com/pathways/HIF-1αlpha-pathway).
КИСЛОРОД-НЕЗАВИСИМЫЙ ПУТЬ АКТИВАЦИИ HIF-1α
Регуляция стабильности HIF-1α также может быть опосредована кислород-независимым путем. В цитоплазме HIF-1α связан с белком теплового шока 90 (Hsp90), повышающим стабильность HIF-1α. Ингибирование Hsp90 приводит к убиквитинированию и деградации HIF-1α (рис. 2).
Рис. 2.
Механизм активации HIF-1 и гены, кодируемые при его активации. Описание рисунка: (а) в условиях нормоксии индуцируемый гипоксией фактор 1 (HIF-1) подвергается протеасомной деградации; (б) в условиях гипоксии HIF-1 активирует транскрипцию генов, кодирующих следующие: транспортеры глюкозы и гликолитические ферменты, которые увеличивают поток глюкозы в пируват; PDK1 (пируватдегидрогеназная киназа), которая инактивирует PDH (пируватдегидрогеназу), митохондриальный фермент, который преобразует пируват в ацетилКоА для входа в цикл трикарбоновой кислоты (ЦТК); ЛДГ (лактатдегидрогеназа), которая преобразует пируват в лактат; (в) митохондриальные белки bnip3 и bnip3l, которые индуцируют митохондриально-селективную аутофагию. Шунтирование субстрата от митохондрий снижает продукцию АТФ, но предотвращает избыточную продукцию АФК, которая возникает из-за неэффективного транспорта электронов в гипоксических условиях.

БИОЛОГИЧЕСКИЕ ЭФФЕКТЫ HIF
На уровне организма к ответу на изменение активности HIF можно отнести эритропоэз, ангиогенез, метаболическую активность, воспалительные реакции и регуляцию иммунного ответа [32].
У HIF много генов-мишеней, часть из них ответственна за сосудистые реакции после ишемии/гипоксии: ростовые факторы и цитокины, взаимодействующие с рецепторами мембран клеток. В первую очередь это эндотелиальные клетки-предшественники, эндотелиальные клетки, другие ангиогенные клетки, мезенхимальные стволовые клетки, миоциты (рис. 3). Также среди генов-мишеней находятся те, что кодируют ферменты пируватдегидрогеназного комплекса (ПДК), транспортеры (TRPC1, 5; семейство переходных потенциальных катионных каналов рецепторов) и митохондриальные белки, снижающие утилизацию О2, регулирующие переключение с окислительного фосфорилирования на гликолиз [36].
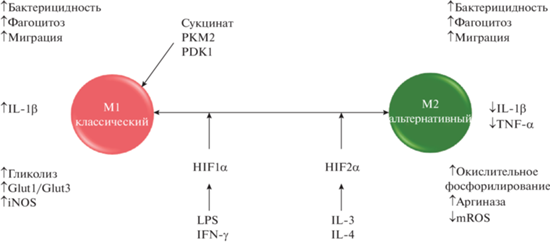
Рис. 3.
Роль HIF в реализации иммунного ответа. Описание рисунка: HIF-1α и HIF-2α играют разные роли в поляризационных макрофагах, причем HIF-1α благоприятствует M1 или классически активированным клеткам, и HIF-2α благоприятствует M2 или альтернативно активированным клеткам. Эта поляризация приводит к изменениям в продукции и метаболизме воспалительных цитокинов. Кроме того, метаболиты и метаболические ферменты, включая сукцинат, PDK1 и PKM2 (пируваткиназа типа K, изоферментом из гликолитического фермента пируваткиназы), также могут играть роль в HIF-1α-зависимой поляризации M1.
Фактор, индуцируемый гипоксией, играет непосредственную роль в эритропоэзе млекопитающих, активируя транскрипцию генов следующих белков [33]:
• транспортер двухвалентного железа 1 (DMT1) и дуоденальный цитохром B, который катализирует восстановление Fe3+ до Fe2+, необходимого для поглощения железа с пищей в двенадцатиперстной кишке млекопитающих, чтобы увеличить кишечную абсорбцию железа (Fe);
• трансферрин, который переносит Fe к рецепторам трансферрина в костном мозге;
• рецептор эритропоэтина и эндогенный эритропоэтин.
Таким образом, HIF обладает плейотропностью и является неотъемлемым компонентом клеточного и системного ответа на изменение концентрации кислорода, что нашло применение в терапии различных заболеваний. Важно понимать, что активность HIF может оказывать не только протективное действие, но и быть основным элементом патогенеза некоторых состояний. Следовательно, можно выделить две принципиальных ситуации, а именно, когда защита HIF целесообразна и когда она не желательна.
СОСТОЯНИЯ, ПРИ КОТОРЫХ HIF ВЫСТУПАЕТ КАК ЦИТОПРОТЕКТОР И ПОВЫШЕНИЕ ЕГО АКТИВНОСТИ ИЛИ НАКОПЛЕНИЕ В КЛЕТКЕ НЕОБХОДИМО
Многие заболевания, сопряженные с гипоксией, протекают на фоне меняющейся активности HIF. К таким состояниям можно отнести сердечно-сосудистые патологии: ИБС, нарушения мозгового кровообращения, эндоартериит и др., обусловленные эндотелиальной дисфункцией, воспалением, атеросклеротическим процессом и, в конечном результате, стенозом сосудов. В этих условиях HIF способствует образованию сосудистых коллатералей. Исследования роли HIF в тяжести протекания сердечно-сосудистых патологий показали, что среди пациентов с критическим стенозом коронарной артерии (просвет сужен >70%) точечная мутация HIF в 582 остатке (замена пролина на серин) у пациентов без коллатералей встречалась в 5 раз чаще, чем у пациентов с коллатералями. В другом исследовании у пациентов с первичными проявлениями коронарной недостаточности точечные мутации в локусе HIF-1α были больше ассоциированы со стенокардией напряжения, как начальным проявлением коронарной недостаточности, чем с инфарктом миокарда. Можно предположить, что HIF – один из главных модификаторов коронарной недостаточности и играет важную роль в процессе адаптации клеток к хронической гипоксии. Так, в эксперименте феномен прекондиционирования не развивался у мышей, гетерозиготных по гену HIF-1α (с одним нулевым аллелем по локусу этого гена, генотип HIF-1α+/–). Схожая закономерность справедлива для стеноза периферических артерий, особенно нижних конечностей (т.н. перемежающаяся хромота). Длительная ишемия ведет к снижению жизнеспособности тканей и их гибели, что обусловливает высокий риск ампутаций. В условиях эксперимента у животных с перевязанной бедренной артерией наблюдалось повышение уровня HIF-1α и экспрессии HIF-зависимых генов, кодирующих сосудистые ростовые факторы: фактор роста эндотелия сосудов (VEGF), сигнальный белок, вырабатываемый клетками для стимулирования васкулогенеза (образование эмбриональной сосудистой системы) и ангиогенеза (рост новых сосудов в уже существующей сосудистой системе), SDF-1 (фактор стволовых клеток, играет важную роль в эмбриональном развитии и гематопоэзе, может стимулировать пролиферацию клеток и способствовать их выживанию), ПФР (плацентарный фактор роста), Ang-1 и 2 (ангиопоэтин 1 и 2), PDGF (тромбоцитарный фактор роста В) и (SCF) фактор стволовых клеток. Дополнительно к этому, ангиогенные клетки костного мозга мигрируют в ишемический очаг и совместно с факторами ангиогенеза усиливают восстановление гемоперфузии тканей. При этом полученные результаты хуже у старых и гетерозиготных по гену HIF-1α (HIF-1a+/–) мышей [15].
Исследования показали, что лечение, нацеленное лишь на один ангиогеннный фактор (например, только VEGF), не дает значительных результатов. Было предположено, что HIF-1, как основной регулятор, ответственный за активацию многих генов, кодирующих ангиогенные факторы, может оказать лучший терапевтический эффект, чем только один сосудистый фактор роста. В исследовательских целях был сконструирован рекомбинантный аденовирус AdCa5 с искусственной, постоянно активной формой HIF-1α. Единичная внутримышечная инъекция препарата, содержащего этот вирус в ишемизированную конечность 8-месячных мышей, была достаточной для улучшения кровотока, но для 13-месячных эффективной была инъекция AdCa5, сопровождающаяся перерывом в 24 ч и последующим внутривенным введением ангиогенных клеток костного мозга (BMDACs), культивированных в течение 4 дней в присутствии сосудистых факторов роста и DMOG (диметилоксалиглицин, конкурентный антагонист α-кетоглутарата, который ингибирует гидроксилазы и индуцирует HIF-1-зависимую транскрипцию). Этапность и способ введения были обусловлены несколькими причинами. Во-первых, AdCa5 индуцирует продукцию ангиогенных факторов, что служит сигналом хоминга для BMDACs (ишемия индуцирует выработку ангиогенных цитокинов и возвращение в исходное состояние происходящих из костного мозга ангиогенных клеток, но эти адаптивные ответы ухудшаются при старении из-за сниженной экспрессии HIF-1α). Во-вторых, местное введение большого количества клеток в очаг ишемии может увеличить их гибель вследствие гипоксии, в то время как системное введение способствует отбору субпопуляций, способных к миграции в ишемизированную ткань для участия в ангиогенезе. Введение BMDACs не имело смысла без культивирования вместе с DMOG, т.к. активация HIF-1 в BMDACs имела два важных последствия: первое – стимуляция экспрессии β2-интегринов, способствующих увеличению адгезии циркулиющих BMDACs к сосудистым эндотелиальным клеткам, таким образом усиливая их закрепление в ишемическом очаге, второе – HIF-1 увеличивал их выживаемость в нем. Терапия была эффективна даже при условии, что и донор BMDAC, и реципиент с ишемией были в возрасте 17-ти месяцев. Это послужило в качестве модели аутологичной BMDAC-терапии для пожилых пациентов с ишемией конечностей [69].
В настоящее время для людей c ишемическими болезнями разрабатываются подходы генной терапии, основанные на внедрении в клетки-мишени активной формы HIF-1α с использованием вирусного вектора [23].
В России для лечения пациентов с атеросклерозом артерий нижних конечностей применяется генный препарат Неоваскулген, разработанный “Институтом стволовых клеток человека” (Москва). Механизм действия препарата основан на проникновении в клетку плазмидной конструкции, которая, не интегрируясь в геном, обеспечивает временный синтез фактора роста VEGF165, который выступает в качестве ауто- и паракринного регулятора роста сосудов in situ. Препарат безопасен для пациентов, имеет хорошую переносимость. У пациентов клинической группы установлено статистически значимое улучшение физического компонента здоровья. После применения препарата “Неоваскулген” при оценке основного (“дистанция безболевой ходьбы”), вторичных критериев эффективности (“лодыжечно-плечевой индекс”, “транскутанно определяемое напряжение кислорода”, “линейная скорость кровотока”) и данным ангиографии было установлено существенное улучшение клинической картины у пациентов с хронической ишемией нижних конечностей IIa–III ст. по А.В. Покровскому-Фонтейну [9].
Роль HIF в неоваскуляризации
Неоваскуляризация благоприятно сказывается на заживлении ран: рост капилляров происходит из сосудов, для чего в предсуществующие сосуды мигрируют ангиогенные клетки. Здесь HIF-1α-зависимый выброс цитокинов из раны вызывает мобилизацию и хоминг ангиогенных клеток костного мозга, которые могут участвовать в ангиогенезе как непосредственно, так и стимулировать его паракринно. Относительная гипоксия важна для заживления ран, поскольку обычно играет ключевую роль в регулировании всех критических процессов, связанных с восстановлением тканей. HIF является критическим фактором транскрипции, который регулирует адаптивные ответы на гипоксию. Заживление ран ухудшается с возрастом или при сопутствующих патологиях, например, при сахарном диабете: у молодых мышей линии db/db в эксцизионных ранах экспрессия HIF-1α была значительно ниже, чем у однопометных мышей без диабета [69].
Показано, что гипергликемия комплексно влияет как на стабильность, так и на активацию HIF-1α, что приводит к подавлению экспрессии генов-мишеней HIF-1, необходимых для заживления ран как in vitro, так и in vivo. Блокирование гидроксилирования HIF-1α посредством химического ингибирования, может обратить вспять этот негативный эффект гипергликемии и улучшить процесс заживления ран (то есть грануляцию, васкуляризацию, регенерацию эпидермиса и рекрутирование клеток-предшественников эндотелия). Локальный перенос двух стабильных конструкций HIF с помощью рекомбинантного аденовируса AdCa5 введенного внутримышечно, продемонстрировал, что стабилизация HIF-1α необходима и достаточна для ускорения заживления ран в условиях диабета [16].
В фибробластах db/db-мышей с гипергликемией активность HIF-1 была снижена, однако это устранялось введением ингибиторов пролилгидроксилазы диметилоксалиглицина (DMOG) или дисферриоксиамина, последний улучшал васкуляризацию и заживление ран у db/db-мышей. Внутримышечное введение AdCa5 таким мышам с лигированной бедренной артерией также улучшало восстановление перфузии и способствовало сохранению конечности. Этот подход был применим для лечения ожоговых ран: у мышей дикого типа с ожоговыми ранами наблюдалось повышение концентрации HIF-1α в ране, SDF-1 в плазме и циркулирующих BMDACs на 2-ой день, что приводило к васкуляризации и перфузии раны на 7 день. У HIF-1α+/–-мышей и старых животных данные реакции были выражены в меньшей степени. После пересадки BMDAC, которые получали от молодых доноров и пересаживали старым реципиентам не наблюдалось улучшения миграции в рану. Хоминг BMDAC был замедленным у донора, и у реципиента если они были нокаутными по HIF-1α в клеточной линии Tie2 [65].
Роль HIF в восстановлении микроциркуляции в трансплантате
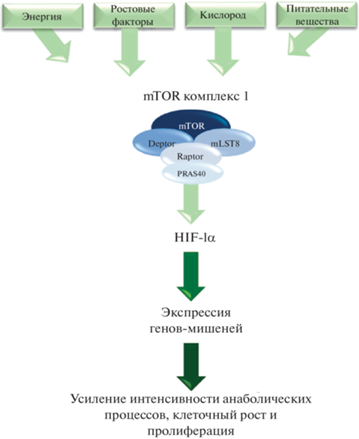
Восстановление микроциркуляции в трансплантате необходимо для улучшения его приживания. Хроническое отторжение (посттрансплантационные реакции “трансплантат против хозяина”) после пересадки легкого сопровождается констриктивным бронхиолитом (облитерирующий бронхиолит; фиброз дыхательных путей), который влияет на 5-летнюю выживаемость 50% пациентов, что является самым худшим результатом среди трансплантатов. Легкое – это сосудистый регион с интенсивным кровообращением, поддержание гемоперфузии в значениях, близких к нормальным, является критическим фактором для жизнеспособности трансплантата. Данные аутопсии свидетельствуют о том, что облитерирующему бронхиолиту предшествует нарушение кровоснабжения дыхательных путей. В ортотопической модели трансплантации трахеи, выполненных на нокаутированных по HIF-1 мышах показал, что HIF-1-зависимый набор реципиентных ангиогенных клеток Tie2+ (одна из субпопуляций циркулирующих эндотелиальных прогениторных клеток) и репарация микроциркуляторного русла дыхательных путей являются критическими детерминантами выживаемости трансплантата. Кроме того, лечение доноров трансплантатов AdCA5 (рекомбинантный аденовирус с искусственной, постоянно активной формой HIF-1α) до трансплантации, увеличило перфузию, уменьшило фиброз и повысило выживаемость трансплантата. Эти результаты объясняют процесс отторжения трансплантата не только с точки зрения иммунологического механизма, но и с позиции ведущей роли сосудистых реакций на возникающую хроническую или транзиторную ишемию. Разрушение микроциркуляторного русла также является одной из причин хронического отторжения почечных трансплантатов. В аллогенной модели трансплантации почки лечение крыс-доноров ингибитором пролилгидроксилазы перед пересадкой почки улучшило выживаемость реципиентов. Было показано, что два основных класса иммуносупрессивных препаратов, кальциневрин и ингибиторы мишени млекопитающих для рапамицина (mTOR), блокируют активность HIF-1 в культивируемых клетках (объяснение ниже в разделе “злокачественные новообразования”). Поэтому эти препараты могут оказывать непреднамеренное контртерапевтическое действие на препараты, механизм действия которых связан с регуляцией активности HIF [68].
Ингибиторы mTOR (mammalian target of rapamycin; мишень для рапамицина (Rapamune, Pfizer, USA) и его аналогов (эверолимус, темсиролимус, дактолизиб)) используются для иммуносупрессивной и противоопухолевой терапии. Одним из первых представителей является Рапамицин – метаболит, продуцируемый актиномицетами Streptomyces rapamycinicus, Streptomyces iranensis и Actinoplanes. Помимо описанных выше свойств было обнаружено, что Рапамицин обладает противогрибковой, нейропротективной/нейрогенеративной активностью, а также замедляет старение организма. В настоящее время ведутся исследования, направленные на изучение данного препарата и разработку его аналогов (т.н. рапалоги) с целью снижения побочных эффектов [83].
В клетках и тканях, подверженных гипоксии, нарушение доставки кислорода приводит к снижению выработки энергии и общему подавлению энергозатратных процессов, таких как синтез белка. При тяжелой гипоксии потребность в АТФ для синтеза белка падает примерно до 7% нормоксических клеток, что связано с резким снижением скорости трансляции белка. Зависимое от гипоксии снижение синтеза белка происходит в основном на уровнях инициации трансляции (ограничивает или замедляет скорость трансляции). Гипоксия предотвращает эукариотическую инициацию трансляции по двум различным путям, распространяемым мишенью рапамицина (рапамицина-мишени; mTOR) и стресс-чувствительной протеинкиназой R(PKR)-подобной эндоплазматической ретикулум-киназой (PERK) [40].
Участие HIF-1α в воспалительных процессах
Улучшение микроциркуляции важно при лечении воспалительных заболеваний. Патогенез хронических воспалительных заболеваний желудочно-кишечного тракта, таких как болезнь Крона и язвенный колит, включает микрососудистые нарушения и гипоксию тканей. Тяжесть колита, вызванного введением химических или бактериальных токсинов, была выше у мышей нокаутных по HIF-1α. Введение мышам дикого типа DMOG уменьшило тяжесть энтероколита, моделируемого введение химических или бактериальных колитиндуцирующих агентов.
В эпителиальных клетках, модуляция активности HIF или блокирование генов предполагала защитную роль белков HIF при экспериментальном колите мышей. Однако результаты, полученные на мышах с заблокированным HIF-1α или VHL в кишечном эпителии оказались противоречивыми. В исследовании Karhausen et al. [44], делеция HIF-1α в зрелых энтероцитах (вызванная промотором Fabp) приводила к утяжелению колита, индуцированного тринитробензолсульфатной кислотой (TNBS-индуцированного), тогда как делеция VHL приводила к защите от TNBS-индуцированного колита. Напротив, в других исследованиях сообщается, что делеция HIF-1α в предшественниках эпителиальных клеток кишечника (вызванная промотором VILIN) не оказывала влияния на течение колита (индуцированного декстрансульфат натрием – DSS), тогда как делеция VHL воспалительный ответ усиливала. Подобные противоречивые результаты могут быть объяснены особенностями химических агентов, используемых в этих двух исследованиях (TNBS и DSS), удаленной клеточной мишенью (зрелые энтероциты против клеток-предшественников) или нацеливанием субъединиц HIF-1α в организме мышей нокаутированных по VHL, разрешение этих противоречий требует дополнительных экспериментов.
Изучение участия HIF-1α в воспалительных процессах проводилось на более физиологической модели мышиного колита, в которой трансгенные мыши CEABAC10 (экспрессирующих человеческие CEACAM (гликопротеин семейства раково-эмбриональных антигенов, продукт гена человека CEACAM1)) инфицировались вирулентным штаммом AIEC LF82 (адгезивно-инвазивный штамм Escherichia coli, штамм LF82). В результате было установлено, что бактерии, ассоциированные с болезнью Крона, повышают уровень HIF-1α в эпителиальных клетках. Как у мышей, так и у человека было отмечено, что некоторые клетки, связанные с воспалением, содержат HIF-1α. Этот вывод согласуется с наблюдением, что HIF-1α, экспрессируемый макрофагами, является важным регулятором врожденного иммунитета [58].
Регуляция иммунного ответа
HIFs и PHD-2s и врожденный иммунитет. Макрофаги и нейтрофилы в ответ на гипоксию экспрессируют HIF-1α и HIF-2α [50]. Выживаемость в условиях гипоксии позволяет нейтрофилам функционировать в агрессивной среде, однако избыточная активация HIF, приводящая к персистенции нейтрофилов, может способствовать замедлению разрешения воспаления и повреждению тканей. Для выживания нейтрофилов при гипоксии важен HIF-1α, в то время как HIF-2α способствует сохранению активности и выживанию нейтрофилов во время асептического воспаления, что указывает на специфическую роль этих изоформ. Различные профили временной экспрессии с ранней активацией HIF-1α и отсроченной индукцией HIF-2α определяют функциональную дивергенцию во время различных фаз воспалительного ответа. Учитывая эту важнейшую роль HIF в выживании и функционировании нейтрофилов, неудивительно, что манипуляции с активностью PHD-2 оказывают важное влияние на выживание и функционирование нейтрофилов. Миелоидно-специфическая генетическая делеция PHD-2 или фармакологическое ингибирование ее активности вызывают усиление воспаления опосредованной нейтрофилами в ответ на инвазию Streptococcus pneumoniae и на модели острого повреждения легких, вызванной липополисахаридом (ЛПС). Это происходит из-за избыточной реакции нейтрофилов, с увеличенной миграцией, высокой выживаемостью и активацией гликолиза. Важно отметить, что эти выводы связаны с высокой стабильностью HIF-1α (обусловлена ингибированием PHD-2). В отличие от этих результатов, блокада PHD-3 в нейтрофилах была связана с уменьшением воспаления. Обнаружено, что как гипоксия, так и стимуляторы воспаления повышают уровни PHD-3 в нейтрофилах (что согласуется с данными, полученными для других типов клеток). Важно отметить, что сохраненная транскрипционная активность HIF, потеря PHD-3 приводила к потере способности к выживанию нейтрофилов в условиях гипоксии. Это важно для уменьшения степени повреждения легких, вызванного ЛПС, и уменьшением воспаления в модели колита [75].
Роль HIFs и PHD-2s в адаптивном иммунитете
HIF/PHD-2 в T-клетках. Гипоксия, воспалительная среда и метаболический фенотип участвуют в регуляции количества и функций CD4+ Т-клеток. В экспериментальных исследованиях гипоксия и HIF-1α были определены в качестве позитивных регуляторов развития Тh17 (клетки T-хэлперы 17 типа), которые представляют собой подмножество провоспалительных клеток T helper, определяемых по их продукции интерлейкина 17 (IL-17) и по множеству механизмов: усиление гликолиза, активация ключевого фактора транскрипции RORgt и деградация фактора транскрипции Treg FOXP3 (Treg – регуляторные Т-лимфоциты, которые поддерживают толерантность к собственным антигенам, Treg являются иммуносупрессивными и обычно подавляют индукцию и пролиферацию эффекторных Т-клеток; ROR отвечает за транскрипцию и развитие Th17, а FOXP3 за семейство Treg).
Как было сказано выше, VHL обладает активностью убиквитинлигазы E3 и отвечает за нацеливание HIF-1α на убиквитин-опосредованную деградацию после гидроксилирования. Потеря VHL (и накопление HIF-1α) у Treg привели к нарушению функции Treg и потере FOXP3 в результате HIF-1α-опосредованной экспрессии IFN-g и перехода Treg к более воспалительному фенотипу TH1. В отличие от этих результатов, Clambey с соавт. [22] обнаружили, что гипоксия и HIF-1α способствуют транскрипции FOXP3. В целом, эти данные предполагают, что активация HIF-1α способствует развитию TH17 по сравнению с дифференцировкой Treg, но HIF-1α также необходим для оптимального развития и функционирования Treg. Недавнее исследование, изучающее роль мРНК в Treg, также идентифицировало HIF-2α в качестве потенциального негативного регулятора развития Treg. Таким образом и HIF-1α (является основной), и HIF-2α являются важными изоформами этого белка (рис. 4) [75].
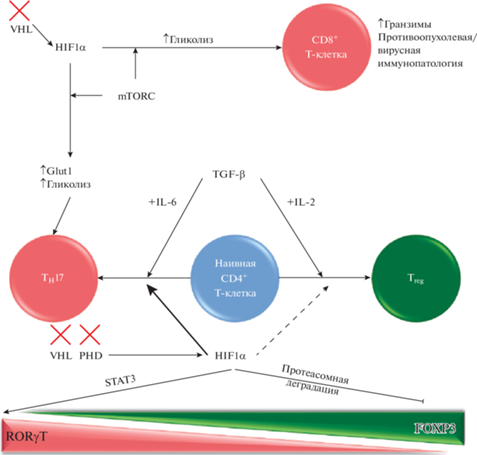
Рис. 4.
Влияние HIF на СD4 + и CD8. Описание рисунка: СD4+ и CD8+ T-клетки также поляризуются и активируются изоформами HIF. TGF-b необходим для разработки как TH17, так и Treg. IL-2 способствует развитию Treg, тогда как IL-6 способствует развитию TH17. Хотя некоторые данные указывают на то, что HIF-1α необходим для функции Treg (пунктирная стрелка), общая активация HIF-1α способствует воспалительной поляризации TH17 (толстая стрелка) за счет активизации RORgT посредством активности STAT3 и подавления (посредством протеасомной деградации) FOXP3. HIF-1α также активирует CD8 + цитотоксические Т-клетки, что приводит не только к увеличению производства гранзима и усилению противовирусной и противоопухолевой активности, но также к усилению иммунопатологии. Опять же, метаболизм играет ключевую роль в поляризации, а активность HIF-1α усиливает гликолиз и поглощение глюкозы, способствуя провоспалительным реакциям. Сокращения: mАФК, митохондриальные активные формы кислорода; STAT3, преобразователь сигнала и активатор транскрипции 3; TH17, T helper 17 cell; Treg, регуляторная Т-клетка.
HIF-1 является критическим фактором фенотипа сепсиса, благодаря участию в продуцировании провоспалительных цитокинов, приводящих к тахикардии, гипотензии и гипотермии. Инокуляция живых или неактивных грамположительных бактерий в макрофаги индуцировала HIF-1α, в то время как мыши с дефицитом миелоидного HIF-1 были устойчивы к грамположительным бактериям в условиях эндотоксемии. Блокирование PHD-3, фермента, ответственного за деградацию HIF, значительно сократила выживаемость мышей с моделью абдоминального сепсиса из-за сниженного врожденного иммунного ответа. Повышенная провоспалительная активность у этих мышей коррелирует с усилением стабилизации белка HIF-1α в макрофагах. В совокупности эти данные показывают, что HIF-1α вносит вклад в негативную роль макрофагов в патологии сепсиса. Фармакологическое ингибирование HIF-1α с использованием 2-метоксиэстрадиола защищало мышей от сепсиса, вызванного ЛПС и CLP. Подавленная продукция iNOS/NO и цитокинов, индуцированная HIF-1α, была обнаружена в перитонеальных макрофагах, что свидетельствует о важной роли миелоидного HIF-1α в выживаемости клеток и организма при сепсисе. Можно сделать вывод, что HIF влияет на исход сепсиса, определяя его тяжесть и летальность, чем его больше, тем они выше [30].
Динамические изменения в экспрессии HIF происходят во время сепсиса, который оказывает существенное влияние на выработку цитокинов, метаболизм, адаптацию клеток и клиническую симптоматику. Поэтому HIF рассматривается в качестве потенциального биомаркера при сепсисе, хотя его роль остается спорной.
Функционально активные генетические варианты в HIF-1α и PHD-2 могут влиять на экспрессию мРНК HIF-1α. Однако они не являются независимыми факторами риска 30-дневной смертности при тяжелом сепсисе. В настоящее время завершено клиническое исследование потенциала HIF-1α в качестве нового биомаркера тяжести септического шока, и данные по первичным исходам еще не опубликованы. Использование HIF в качестве биомаркера может быть осложнено, учитывая его дифференцированную роль в зависимости от типа клеток, но этот фактор предложен в качестве потенциальной терапевтической мишени при сепсисе [URL: https://clinicaltrials.gov/ct2/show/NCT02163473]. Однако до настоящего времени исследования были сосредоточены на косвенном таргетировании HIF. Эдаравон – индуцирующий HIF-1α препарат, который подавляет окислительный стресс и защищает сердце от септической травмы и дисфункции миокарда. Дальнейшие исследования с использованием специфических ингибиторов HIF потребуются для полного выяснения потенциала этого сигнального пути в качестве терапевтической мишени при сепсисе [30, 61].
Во многих фармацевтических и биотехнологических компаниях были инициированы программы поиска лекарственных средств для разработки ингибиторов пролилгидроксилазы (PHI), которые, как описано выше для DMOG, индуцируют активность HIF для лечения расстройств, при которых HIF опосредует защитные физиологические реакции.
СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОВЫШЕНИЕ АКТИВНОСТИ HIF И ЕГО НАКОПЛЕНИЕ НЕЖЕЛАТЕЛЬНО
Эритроцитозы состояния, сопровождающиеся повышенным количеством эритроцитов, что обусловлено чрезмерной активацией гена эритропоэтина. Были идентифицированы лица с избыточной продукцией эритроцитов из-за мутаций зародышевой линии в генах, кодирующих VHL, PHD-2 и HIF-2α, демонстрируя существенную роль этого пути в регуляции эритропоэза. Эти мутации нарушают гидроксилирование и убиквитинирование, тем самым повышая уровни HIF-1α и HIF-2α при любом заданном парциальном давлении O2 (PO2). У людей появлялись глобальные физиологические изменения, которые включали измененные вентиляционные и легочные сосудистые реакции на гипоксию, а также измененные метаболические реакции на физические нагрузки [69].
Злокачественные новообразования
Злокачественные новообразования (при которых, васкуляризация и адаптация к гипоксии ишемизированных очагов опухоли нежелательна) включают гипоксические области, формирующиеся из-за высоких темпов пролиферации клеток в сочетании с образованием сосудистой сети, которая является структурно и функционально ненормальной и не способна обеспечить адекватную гемоперфузию, в результате чего вначале формируется область ишемии, переходящая в некроз. Повышенные уровни HIF-1α или HIF-2α в диагностических биопсиях опухоли связаны с высоким риском смерти при онкологии мочевого пузыря, головного мозга, молочной железы, толстой кишки, шейки матки, эндометрия, головы/шеи, легких, яичников, поджелудочной железы, простаты, прямой кишки или желудка. Экспериментальные методы, повышающие экспрессию HIF-1α, приводили к увеличению роста опухоли, тогда как потеря активности HIF приводит ее ограничению. HIF могут активироваться генетическими изменениями при онкологии у человека, сопровождающимися потерей функции VHL. Учитывая обширную валидацию HIF-1 в качестве потенциальной терапевтической мишени, были идентифицированы препараты, ингибирующие HIF-1, и показано, что на моделях ксенотрансплантата они оказывают противоопухолевое действие (рис. 5).
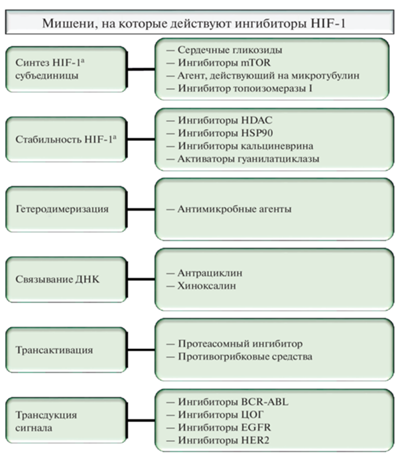
Рис. 5.
Группы препаратов, которые действуют на HIF-1α или его эффекты. Объяснение рисунка. Терапевтический подход, предполагающий ингибирование HIF на одном или нескольких уровнях регуляции, представляется целесообразным, поскольку HIF активируют транскрипцию генов, которые играют ключевую роль в критических аспектах онкобиологии, включая: поддержание стволовых клеток, выживание клеток, эпителиально-мезенхимальный переход, генетическая нестабильность, васкуляризация, метаболизм глюкозы, регулирование рН, иммунное уклонение, инвазия и метастазирование, а также радиорезистентность.
В США более 100 женщин каждый день умирают от рака молочной железы. Среднее значение pO2 при раке молочной железы составляет 10 мм рт. ст. по сравнению с >60 мм рт. ст. в нормальной ткани молочной железы. Сниженное рО2 связано с повышенным риском метастазирования и летальности. Высокие уровни белка HIF-1α, выявленные при биопсии опухолей иммуногистохимическими методами, ассоциированы с повышенным риском метастазирования (метастазирование предполагает >90% смертности от рака молочной железы) и летального исхода у пациенток с раком молочной железы. Роль HIF-1α при метастазировании рака молочной железы была изучена на модельных организмах (у трансгенных мышей, склонных к спонтанно возникающим опухолям) и при проведении ортотопических трансплантаций (инъекции клеток рака молочной железы человека в жировую ткань молочной железы) у иммунодефицитных мышей. Первичные опухоли производят рекрутинг клеток костного мозга в легкие и другие места метастазирования. При опухоли молочной железы гипоксия индуцирует экспрессию лизилоксидазы, секретируемого белка, который ремоделирует коллаген в местах образования метастатической ниши [28]. В дополнение к LOX, онкология молочной железы запускает секрецию LOX-подобных протеинов 2 и 4, которые вместе с LOX кодируются генами, экспрессия которых регулируется в том числе и HIF-1. Ингибирование HIF-1 блокирует образование метастатической ниши, независимо от того, какой белок LOX/LOXL экспрессируется, в то время, как применение доступных ингибиторов LOX оказалось неэффективным из-за их высокой селективности только к LOX, в то время как для достижения эффекта необходима блокада всех белков LOXL [79]. Эти результаты иллюстрируют роль HIF-1 как главного регулятора, который контролирует экспрессию нескольких генов, участвующих в одном (патофизиологическом) процессе [70]. Таким образом, в раковых клетках под действием HIF-1 экспрессируется LOX и ряд LOX-подобных протеинов, ремоделирующих коллаген и создающих т.н. преметастатическую нишу (когда туда попадают раковые клетки, ниша становится метастатической).
Несмотря на то, что роль HIF в патогенезе злокачественных новообразований очевидна, есть данные, что она не первостепенна: большую значимость отводят белку mTOR, как главному регулятору клеточного роста и пролиферации, который также регулирует и активность HIF независимо от гипоксии (рис. 6). Блокирование mTOR также снижает и продукцию HIF, даже в условиях гипоксии (in vitro). При переносе данных результатов на модели in vivo (подкожный ксенотрансплантат nude-мышам) было обнаружено, что ингибирование mTOR вызывает значительное снижение роста опухолей различного происхождения в основном за счет торможения клеточной пролиферации. Более того, данный эффект может быть получен независимо от уровня HIF в опухолевой клетке. Концентрация HIF может не зависеть от активности mTOR, вероятно, из-за клеточного микроокружения: стойкая гипоксия ведет к прочной стабилизации HIF (альфа-субъединица не подвергается деградации; чем она стабильнее, тем больше накапливается и объединяется с бета-субъединицей). Таким образом, эффективное лечение злокачественных новообразований может не затрагивать его активность, т.е. активность HIF может не влиять на эффективность противоопухолевой терапии [46].
Легочная гипертензия
Легочная гипертензия (избыточная васкуляризация вследствие длительной гипоксии и гипертрофии правого желудочка) также является состоянием, при котором нежелательно повышение экспрессии (усиление сигналинга) HIF-1α [62]. Длительное воздействие альвеолярной гипоксии, возникающей у пациентов с хроническими заболеваниями легких, приводит к ремоделированию легочной сосудистой системы и повышению легочного артериального давления, а также гипертрофии правого желудочка. В исследовании были идентифицированы множественные целевые гены HIF-1α, которые играют ключевую роль в ответе гладкомышечных клеток легочной артерии на гипоксию. Так HIF-1α+/– и HIF-2α+/– мыши защищены от гипоксической легочной гипертензии, что указывает на то, что HIF-1α и HIF-2α играют ключевую патогенную роль. HIF также вовлечен в химически индуцированные и генетические формы легочной артериальной гипертензии, при которых гипоксия не являлась этиологическим фактором.
Большое количество доказательств показывает, что устойчивая гипоксия окружающей среды может вызывать легочную гипертензию через HIF-опосредованный путь. Действительно, первые доказательства роли HIF в легочной гипертензии in vivo получены на животных, дефицитных по HIF. У мышей с гетерозиготной делецией зародышевой линии HIF-1α или HIF-2α с подверженной хронической гипоксией, наблюдалось нарушение развития легочной гипертензии, частично из-за ограниченных возможностей ремоделирования сосудов легких. Роль HIF в развитии легочной гипертензии доказаны результатами исследований патологических наследственных мутаций, приводящих к гиперактивации HIF. Гомозиготность по аллелю VHL, содержащему мутацию R200W, которая разлагает субъединицы HIF-1α менее эффективно, чем нормально функционирующий VHL, связана с HIF-2α-зависимой полицитемией и легочной гипертензией. Кроме того, мутация, приводящая к усилению функции HIF-2α (G537W у человека или G536W у мышей), стала причиной развития более тяжелой легочной гипертензии с сопутствующим обострением эритроцитоза. Эти первоначальные исследования выявили вклад HIF в развитие гипертонической болезни. Более поздние исследования in vivo позволили по-новому взглянуть на вклад изоформ HIF-1α и HIF-2α в развитие легочной гипертензии, а также определить последующие HIF-зависимые события, относящиеся к этому патологическому сценарию [73].
Обструктивное апноэ во сне
Обструктивное апноэ во сне (чередующиеся состояния гиперкапнии и гипоксии ведут к накоплению АФК, а увеличение катехоламинов и симпатическая стимуляция ведут к гипертензии) обусловленное западением мягких тканей глотки и закупориванием дыхательных путей, что ведет к снижению парциального давления кислорода в крови. Эта гипоксемия ощущается хеморецепторами синокаротидной зоны, что приводит к возбуждению, очищению дыхательных путей и реоксигенации. Цикл гипоксии и реоксигенации за ночь повторяется десятки раз и приводит к повышению уровня АФК в синокаротидной зоне и головном мозге. Симпатическая активация и повышение концентрации катехоламинов в плазме приводят к системной гипертензии (рис. 7а). Обструктивное апноэ во сне связано как с гипоксией, так и с гиперкапнией, но воздействие на грызунов хронической прерывистой/интермиттирующей гипоксии (ХИГ) достаточно, чтобы вызвать гипертонию. Лечение мышей скавенджером супероксида Mn TMPyP (миметик супероксиддисмутазы) преграждает опосредованные хронической интермиттирующей гипоксией увеличение HIF-1α, катехоламинов и артериального давления (АД), показывая, что АФК участвует в нисходящей регуляции синтеза HIF. Однако, у HIF-1α+/– мышей отсутствуют ХИГ-опосредованные увеличения АФК, катехоламинов и АД, предполагая, что HIF-1 участвует в восходящей регуляции АФК, таким образом демонстрируются спорные данные по поводу того, что первично: накопление АФК активирует HIF или наоборот. Подтверждена прямая связь индукции HIF-1α через образование АФК, что приводит к транскрипции Nox2-гена, кодирующего НАДФ-оксидазу, вызывающую генерацию супероксидных радикалов. HIF-1α в низких концентрациях экспрессируется в каротидных тельцах в нормоксических условиях (нормальном PO2) и индуцируется хронической интермиттирующей гипоксией. Напротив, экспрессия HIF-2α высока в каротидных тельцах в нормоксических условиях и снижается из-за кальпаин-зависимой деградации в ответ на ХИГ. Снижение уровня HIF-2α связано со снижением экспрессии гена Sod2, который кодирует митохондриальную супероксиддисмутазу, преобразующую супероксидный анион-радикал в пероксид водорода. Введение подверженным ХИГ крыс ингибитора кальпаина блокирует деградацию HIF-2α, восстанавливает активность супероксиддисмутазы (SOD2), предотвращает окислительный стресс и гипертензию. Таким образом, нарушение баланса между уровнями HIF-1α и HIF-2α в каротидных тельцах играет ключевую роль в патогенезе гипертензии, индуцированной ХИГ. Постоянная гипоксия индуцирует HIF-1α и HIF-2α, что приводит к легочной гипертензии, тогда как ХИГ индуцирует HIF-1α, но ингибирует HIF-2α, что приводит к системной (артериальной) гипертензии. Когда изолированные каротидные тельца от мышей дикого типа перфузируются гипоксической газовой смесью, повышается деполяризация O2-чувствительных гломусных клеток и активность каротидного синусового нерва, но эти ответы отсутствуют в каротидных тельцах, полученных от HIF-1α+/– мышей. Напротив, каротидные тельца от HIF-2α+/– мышей имеют более выраженный ответ к острой гипоксии, что сопровождается увеличением концентрации катехоламинов и АД в нормоксических условиях. MnTMPyP лечение HIF-2α+/– мышей нормализует артериальное давление и реакцию клетки синокаротидной зоны. Таким образом, даже в нормоксических условиях баланс между HIF-1α и HIF-2α контролирует функцию клеток синокаротидной зоны и сердечно-сосудистый гомеостаз.

Функциональный антагонизм. HIF 1 изоформа отвечает за негативные проявления, а 2 изоформа их блокирует. В норме между ними и есть функциональный антагонизм, который нарушается при апноэ. Хроническая прерывистая гипоксия приводит к системной артериальной гипертонии из-за активации HIF-1a и деградации HIF-2α. Повышенная HIF-1a-зависимая экспрессия NADPH-оксидазы 2 (NOX2), которая генерирует супероксид-анион, и пониженная HIF-2α-зависимая экспрессия супероксид-дисмутазы 2 (SOD2), которая потребляет супероксид, приводят к повышению уровня АФК в теле сонной артерии и активации симпатической нервной системы и системной гипертонии (рис. 7б). Эти результаты дают новое представление о логике, лежащей в основе приобретения паралога HIF-1α во время эволюции позвоночных [63, 68].
Вышесказанное становится особенно актуальным, если учесть, что синдром обструктивного апноэ является распространенным и может быть не только независимым фактором риска формирования заболеваний сердечно-сосудистой системы, но и отягощать другие патологические процессы [8].
Диабет
Нарушение реакции крупных сосудов на ишемию – болезнь коронарных и периферических артерий, мелких – на повреждение кожи в той или иной степени являются следствием снижения активности HIF. Однако диабет также связан с чрезмерной пролиферацией мелких сосудов (т.е. глазной неоваскуляризацией) и HIF-1, по-видимому, играет ключевую роль в патогенезе этого осложнения.
Сахарный диабет (СД) – это группа метаболических (обменных) заболеваний, характеризующихся хронической гипергликемией, которая является результатом нарушения секреции инсулина, действия инсулина или обоих этих факторов. Хроническая гипергликемия при СД сопровождается повреждением, дисфункцией и недостаточностью различных органов, особенно глаз, почек, нервов, сердца и кровеносных сосудов [1]. Гипергликемия часто связана с явлением, называемым псевдогипоксия, которое характеризуется повышенным соотношением NADH/NAD+ из-за увеличенного потока глюкозы через полиольный путь. В условиях гипоксии дисбаланс возникает в результате нарушения окисления НАДН [18].
При снижении транспорта глюкозы в клетки, в них возникает состояние псевдогипоксии, при этом изменений в количестве NADH и NAD + не происходит, что было обнаружено при сравнении изолированных сетчатки полученной от крыс с диабетом и без него. Исследования с применением пимонидазола (связывается с тиолсодержащими белками, особенно в гипоксических клетках, применяется для качественной и количественной оценки гипоксии), маркера гипоксической ткани, показывают повышенные уровни гипоксии в ткани животных с диабетом, а экспрессия мРНК HIF-1α в их сердце была выше. На уровне белка HIF-1α стабилизируется не только гипоксией, но и продуктами аэробного гликолиза, главным образом пируватом. Гипергликемия влияет на трансактивацию HIF-1 посредством модификации его коактиватора p300 и, таким образом, уменьшает транскрипционную активность HIF-1 (р300 – коактиватор, помогает HIF “сесть” на нужный участок; гипергликемия влияет на р300, не влияя на α1-субъединицу), не влияя на стабильность белка HIF-1α. Гипергликемия активирует HIF-1-опосредованную сигнальную трансдукцию через белок, реагирующий на глюкозу, отвечающий за углеводный элемент (ChREBP), что было показано на модели диабетической гломерулопатии [39].
ChREBP играет функциональную роль в гликолитической и липогенной регуляции генов. Эксперименты с клетками, культивируемыми в среде, сочетающей гипергликемию и гипоксию, показывают повышенную деградацию белка HIF-1α. Таким образом, диабет не только вызывает гипоксию, но также нарушает передачу сигналов HIF-1. Неспособность клеток/тканей адекватно реагировать на гипоксию увеличивает риск осложнений у пациентов с диабетом. Например, у крыс с диабетом, индуцированным стрептозотоцином, экспрессия мРНК HIF-1α ниже, а размер инфаркта миокарда выше, чем у животных с нормогликемией [37].
Гипергликемия вызывает клубочковую гиперфильтрацию и увеличивает канальцевую реабсорбцию натрия и глюкозы через SGLT, которые усиливают натрий-калий-АТФазную активность, что приводит к увеличению потребности в кислороде. Таким образом, проксимальные канальцевые клетки в диабетической почке подвержены хронической гипоксии и высокой экспрессии HIF-1α. Стабильная экспрессия HIF-1α в трубчатых эпителиальных клетках приводит к тубулоинтерстициальному фиброзу. Кроме того, ингибитор активатора плазминогена-1 (PAI-1), основной ген-мишень HIF-1, также является важным фактором для прогрессирования фиброза почек, и в специализированных исследованиях показано, что нокаутирование по гену, кодирующему PAI-1, облегчает течение диабетической нефропатии у мышей [14].
В другом исследовании, проведенном T. Cai с соавт. [17], на образцах почки человека с СД или мыши с воспроизведенной моделью СД авторы обнаружили, что развитие диабетической почечной недостаточности и прогрессирование почечного фиброза повлекло за собой глубокие изменения в метаболизме проксимальных канальцев, характеризующиеся переключением с утилизации жирных кислот на гликолиз и накопление липидов, что связано с повышенной экспрессией HIF-1α. Добавление дапаглифлозина к клеточной культуре (2 мкмоль/л) устраняло повышенный уровень HIF-1α в проксимальных канальцах почек. Также дапаглифлозин защищал от индуцированного глюкозой метаболического сдвига в PTC (Primary tubular epithelial cells) посредством ингибирования HIF-1α, что может быть одним из механизмов нефропротекторного действия ингибиторов SGLT2 при сахарном диабете.
Профибротический эффект HIF-1 оказывает за счет активизации лизилоксидаз. Тубулоинтерстициальная гипоксия обусловлена гломерулосклерозом и разрежением капилляров, которое обычно встречается в почках пациентов с хронической болезнью почек (ХБП). Вследствие гипоксии в эпителиальных клетках почек HIF-1α стабилизируется, что приводит к повышенной экспрессии генов лизилоксидазы и других профиброгенных факторов, способствуя тем самым EMT (epithelial-mesenchymal transition – эпителиально-мезенхимальный переход) и накоплению ECM (extracellular matrix – внеклеточный матрикс).
Почечный эпителиальный HIF-1α является фиброгенным на модели хронического повреждения почек и, таким образом, оказывает стимулирующее действие на болезнь. Это открытие противоречит его предполагаемой цитопротекторной роли при острых и хронических ишемических повреждениях и предполагает, что HIF-1 может играть специфическую для контекста и/или типа клеток биологическую роль в отношении исхода заболевания почек [35].
HIF–VEGF
HRM (реагирующие на гипоксию РНК) представляют собой специфическую группу микроРНК, которые регулируются гипоксией. Недавние исследования показали, что несколько HRM, включая микроРНК семейства let-7, были высоко индуцированы в ответ на активацию HIF при гипоксии, и они участвовали в ангиогенезе путем взаимодействия с AGO1 (argonaute1) и положительной регуляцией VEGF. Клетки, находящиеся в гипоксической среде, сигнализируют о необходимости стимулировать рост новых кровеносных сосудов, чтобы получить больше кислорода и питательных веществ. HIF-1 при гипоксии ускоряет высвобождение проангиогенных цитокинов, таких как VEGF, основной индуктор ангиогенеза. Промежуточные сигнальные события, связывающие HIF-1 и VEGF, строго контролируются микроРНК (miR), которые являются эндогенными некодирующими молекулами РНК в пути HIF–miR–VEGF [87, 91].
МикроРНК (miRs) представляют собой эндогенные небольшие некодирующие молекулы РНК (~22 нуклеотида), которые обеспечивают экспрессию генов на посттранскрипционном уровне. РНК-полимеразы II и III участвуют в транскрипции генов микроРНК с образованием первичных транскриптов miR (pri-miRs), которые обычно имеют длину в несколько сотен нуклеотидов и содержат консервативные шпильки. Эти pri-miRs процессируются ферментом RNase III Drosha в промежуточные шпильки (~60–70 нуклеотидов), которые называются предшественниками miRs (pre-miRs), и pre-miRs активно транспортируются из ядра через нуклеоцитоплазматический челнок Экспортин-5 (XPO-5) с помощью GTP-связывающего ядерного белка Ran. Пре-miRs в цитоплазме расщепляется другим ферментом RNase III Dicer (рибонуклеаза) для превращения в miR-дуплексы, которые затем включаются в РНК-индуцируемый комплекс выключения (сайленсинга) гена (miRISC). Внутри miRISC белки семейства аргонавтов (AGO) необходимые для функции miR у человека, поскольку они способствуют активации miRISC, катализируя диссоциацию ведущей цепи miR (зрелого miR) из сопутствующей цепи (расщепляемой позднее); только AGO1 и AGO2 из восьми белков AGO у человека могут опосредовать такую диссоциацию цепи во время созревания miR. Идентифицировано, что AGO1 связано с miR-опосредованной трансляционной репрессией; однако только AGO2-содержащий RISC способен катализировать расщепление мРНК-мишеней. Хотя в предыдущих исследованиях подробно изучалась роль AGO в координации деятельности miRISC, существует недостаточно информации об экспрессии AGO в ответ на клеточный стресс и его физиологическом значении для ремоделирования сосудов [49].
Было подтверждено, что многие miRs связаны с патофизиологией различных сердечно-сосудистых заболеваний. Поскольку эндотелиоциты контролируют образование новых кровеносных сосудов (ангиогенез), что является критическим для сосудистого гомеостаза, дисфункция эндотелия в ответ на неблагоприятные гемодинамические изменения и патологические стимулы, такие как воспаление или хроническая гипоксия, может привести к неадекватному или аномальному ангиогенезу. Это предрасполагает к развитию многих сосудистых заболеваний, включая PAD (заболевание периферических артерий) и CAD (заболевание коронарной артерии). Семейство микроРНК let-7 (lethal-7) является одним из наиболее многообещающих кандидатов miR в качестве новых регуляторов ангиогенеза, учитывая его высокую экспрессию в эндотелиоцитах, и оно напрямую нацелено на некоторые факторы, связанные с ангиогенезом, такие как TSP-1 (тромбоспондин 1), TIMP-1 (тканевой ингибитор металлопротеиназ 1) и TGFBR1 (трансформирующий фактор роста β рецептор-1). В своей работе Chen Z. с соавт. показали, что сигнальный путь HIF-1–let-7–AGO1–VEGF важен для контроля ангиогенеза эндотелиальных клеток при гипоксии. Члены семейства let-7 miR идентифицированы как HRM (микроРНК, реагирующие на гипоксию), уровни которых сильно повышены с помощью фактора транскрипции HIF-1 при гипоксии. Зрелый let-7 нацеливается на мРНК AGO1 и снижает уровень miRISC, образуемый AGO1 и другими miRs, которые нацелены на VEGF, следовательно, освобождая VEGF от трансляционной репрессии, чтобы способствовать ангиогенезу. Подтвержденные экспериментами in vitro и in vivo, эти данные подтверждают аргумент важной ангиогенной оси, соединяющей HIF, miRs и AGO1 в EC, которые могут потенциально служить ценной мишенью для про- и антиангиогенной терапии. При нормоксии из-за взаимодействия с miRISC мРНК VEGF недоступна для трансляции. Активированный при гипоксии HIF-1, индуцировал биогенез let-7, что приводило к подавлению образования мРНК AGO1, белка и miRISC, что активировало трансляцию мРНК VEGF (рис. 8) [20].
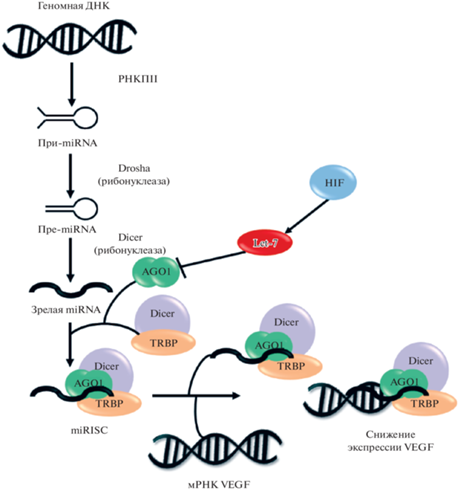
С помощью сконструированной компьютерной модели были определены различные характеристики в клеточных механизмах определения кислорода и в регуляторной сети miR, которая контролирует канонический путь HIF–VEGF. Модель предсказывает, что стабилизация HIF-1α подчиняется гипотетическому переключающему режиму и отрицательно регулируется дестабилизатором мРНК при гипоксии. Чтобы продемонстрировать, на чем сфокусировано исследование, было показано, что let-7 и AGO1 являются инициаторами и координаторами высвобождения VEGF, в то время как они оказывают отрицательное влияние на контроль обратной связи друг на друга и способны минимизировать влияние возможных внешних возмущений; также иллюстрирована роль miR-15a в качестве конечной эффекторной молекулы, которая находится под контролем AGO1, поскольку обилие miR-15a напрямую определяет, сколько мРНК VEGF доступно для трансляции. Вместе эти результаты показывают интегрированное изображение множества miR, каждый с разными мишенями, которые работают совместно с miR-процессирующими белками (например, Dicer, AGO1), чтобы противодействовать неблагоприятным физиологическим стрессам путем стимулирования синтеза VEGF и ангиогенеза [86].
В настоящее время модель рассматривает let-7 и miR-15a в качестве ключевых регуляторов miR процесса подавления VEGF, вызванного гипоксией. Продукция miRs в модели следовала хорошо установленному пути биогенеза miR, который подвергается транскрипции, ядерно-цитоплазматическому транспорту, эндонуклеолитическому процессингу и загрузке miRISC. Модель объединяет процессинг Drosha и транспорт XPO-5 в одностадийную реакцию, и образование miRISC вместе с диссоциацией дуплекса miR упрощается как один обратимый процесс ассоциации между белком AGO1 и miR. Комплекс, образованный белком AGO1 и let-7, может возвращаться в ядро и стимулировать процессинг pri-let-7, который составляет положительную ауторегуляторную петлю. Let-7 подавляет трансляцию двух целей, AGO1 и Dicer, и этот сайленсинг негативно влияет на созревание и стабилизацию let-7. МРНК AGO1 и Dicer обрабатываются с помощью let-7 miRISC и направляются в цитоплазматические домены, называемые p-телами (содержат белки, участвующие в различных посттранскрипционных процессах, таких как деградация мРНК, нонсенс-опосредованный распад мРНК (NMD), репрессия трансляции и РНК-опосредованное замалчивание генов). Поскольку обнаружено, что р-тела участвуют в общем обороте мРНК, было предположено, что, как только мРНК попадут в р-тела, они будут сохранены, недоступными для трансляции со значительно более медленной скоростью деградации по сравнению с таковой цитоплазматических мРНК, в то время как очень малая часть из них все еще может выйти из p-тел и снова войти в трансляционный механизм. Поскольку ось let-7/AGO1 будет влиять на экспрессию группы miRs, приводящую к измененной динамике многих генов-мишеней, мы выбрали VEGF, поскольку он играет решающую роль в ангиогенезе и обширной поддержке литературных данных, в качестве гена для механистической демонстрации детали того, как этот каскад контролирует специфическую экспрессию генов во время проангиогенного ответа. Для текущих целей модели miR-15a выбран для представления группы VEGF-нацеливающих miRs, поскольку miR-15a была экспериментально подтверждена для прямой репрессии синтеза VEGF и заметно влияет на ангиогенез в EC [86, 87]. Кроме того, показано, что гипоксия ослабляет ассоциацию AGO1 со многими направленными на VEGF мишенями, включая miR-15a, и вызывает значительное подавление этих miR. Эти данные дополнительно связывают динамику miR-15a с координацией по оси let-7/AGO1. МРНК VEGF, на которые нацеливается miR-15a, также проходят серию этапов, включающих хранение p-тела, сходное с механизмом let-7-обеспечиваемого молчания мРНК [88].
ПРЕДСТАВИТЕЛИ HIF-ПРОТЕКТОРОВ
Во многих фармацевтических и биотехнологических компаниях инициированы программы обнаружения лекарственных средств для разработки ингибиторов пролилгидроксилазы, которые индуцируют активность HIF. Локальная и кратковременная индукция активности HIF с помощью ингибиторов пролилгидроксилазы, генной терапии или других средств, вероятно, будет перспективной для лечения многих из обозначенных выше заболеваний. В случае управления сердечно-сосудистыми заболеваниями, местная терапия необходима для обеспечения передачи сигналов для рекрутирования ангиогенных клеток костного мозга. К длительному системному применению ингибиторов пролилгидроксилаз следует подходить с большой осторожностью, поскольку люди, у которых конститутивно активируется HIF, подвержены большему риску развития сердечно-сосудистых заболеваний и увеличению смертности. Предполагается, что активация HIF и HIF-зависимых генов может выступать прокальцифицирующим фактором при минеральном дисбалансе и вызывать кальцификацию сосудов. Данные события приводят к развитию сердечно-сосудистых осложнений [59]. С другой стороны, глубокое ингибирование активности HIF и сосудистых реакций на ишемию, которые связаны со старением, позволяют предположить, что системная заместительная терапия может рассматриваться в качестве профилактической меры для пациентов, у которых могут быть документированы нарушенные реакции HIF на гипоксию. Потеря функции VHL в С. elegans, увеличивает продолжительность жизни HIF-1-зависимом образом, что может указывать на геропротекторное действие HIF-1.
Исследование сердечного ритма с нормальным гематокритом было первым крупномасштабным рандомизированным клиническим исследованием по оценке эффекта агентов стимулирующих эритропоэз у пациентов, находящихся на диализе. Определяя смерть и нефатальный инфаркт миокарда как первичную конечную точку, было проведено сравнение между группой с высоким гематокритом (42%) и группой с низким (30%). Данное исследование не было завершено из-за тенденции к неожиданному увеличению числа смертей и чрезмерному количеству добавок железа [13]. Это исследование вызывает опасения, что нормализация гематокрита с помощью агентов, стимулирующих эритропоэз не всегда может быть необходимой для лучших результатов, и, таким образом, установка целевого уровня гематокрита строго на уровне нормы в целом не рекомендуется. В исследованиях CREATE, CHOIR и TREAT обсуждение нормализации анемии с помощью агентов, стимулирующих эритропоэз было позже распространено на пациентов на стадии до диализа. Исследование CREATE показало, что терапия агентами, стимулирующими эритропоэз, направленная на достижение нормального уровня гемоглобина, не оказывает положительного влияния на профилактику сердечно-сосудистых заболеваний [53]. Во-вторых, исследование CHOIR показало, что возрастает уровень смертности, застойной сердечной недостаточности, нефатального инфаркта миокарда и церебральной апоплексии [38]. В-третьих, исследование TREAT показало, что частота церебральной апоплексии значительно выросла в группе с высоким уровнем гемоглобина [62]. После этих испытаний исследование RED-HF, направленное на достижение целевого уровня гемоглобина выше 130 г/л у пациентов с хронической сердечной недостаточностью, привело к увеличению частоты церебрального тромбоза [57]. Результаты этих клинических исследований показали, что целевой уровень гемоглобина выше 130 г/л или близкий к норме может иметь выраженный сердечно-сосудистый риск, который может привести к плохому прогнозу пациентов с ХБП. Примечательно, что апостериорный анализ исследований CREATE, CHOIR и TREAT показал, что истинной причиной неожиданных нежелательных явлений является не лечение агентами, стимулирующими эритропоэз, нацеленное на высокий уровень гемоглобина как таковое, а гипореактивность ESA, при которой либо высокая доза агентов, стимулирующих эритропоэз (в результате ослабленной реакции на ЭСС) или добавление больших доз железа [47]. Основной аномалией, объясняющей гипореактивность ЭСС, является хроническое воспаление и сопутствующая ему дисрегуляция железа у пациентов с ХБП.
Основываясь на вышеупомянутых исследованиях, руководство KDIGO довольно осторожно относится к использованию агентов, стимулирующих эритропоэз. В руководстве рекомендуется, чтобы терапия препаратами железа была в первую очередь, а агенты, стимулирующие эритропоэз не использовалась, даже если гемоглобин пациента >100 г/л. Более того, он рекомендует, чтобы целевой уровень гемоглобина был менее 115 г/л [45]. Однако, в отличие от рекомендаций KDIGO, в рекомендациях JSDT особое внимание уделялось установке индивидуального целевого значения гемоглобина для конкретных стадий, особенно для различения пациентов, находящихся на диализе, от пациентов, находящихся на стадиях до диализа. Исходя из этого, агенты, стимулирующие эритропоэз, следует начинать, когда повторно измеряемое значение гемоглобина составляет менее 110 г/л, если пациенты находятся на додиализной стадии или на перитонеальном диализе, и ниже 100 г/л, если пациенты на гемодиализе. Кроме того, целевое значение гемоглобина должно быть установлено на уровне 100–120 г/л, если пациенты находились на гемодиализе, и 110–130 г/л, если они находились на стадии перед диализом или на перитонеальном диализе [80].
Как упоминалось выше, лечение анемии у пациентов с ХБП высокими дозами ЭСС может быть связано с повышенной смертностью у этих пациентов. Что касается концентрации эритропоэтина, его уровни после введения агентов, стимулирующих эритропоэз, очевидно, увеличиваются по сравнению с уровнями со стабилизаторами HIF-1α. Стабилизаторы HIF-1α могут контролировать почечную анемию, сохраняя при этом эритропоэтины в почти нормальном физиологическом диапазоне. Это может быть преимуществом у пациентов с ХБП с анемией, которым для лечения потребуются высокие дозы агентов, стимулирующих эритропоэз [66].
Анемия является распространенным осложнением хронической болезни почек (ХБП). Из-за ограничений у препаратов, стимулирующих эритропоэз, применяющихся в соответствии с современными стандартами лечения, существует необходимость в разработке новых методов лечения. Ингибиторы пролилгидроксилазы, индуцируемой гипоксией, могут быть новым перспективным вариантом лечения. В настоящее время проводятся клинические испытания 5 ингибиторов пролилгидроксилаз для лечения анемии, основные результаты которых описаны ниже.
РОКСАДУСТАТ (FG-4592/ASP1517)
Роксадустат (рис. 9) (2-(4-гидрокси-1-метил-7-феноксиизохинолин-3-карбоксамидо) уксусная кислота) является ингибитором HIF-PH второго поколения от FibroGen, Astellas и AstraZeneca, который в ряде стран одобрен для лечения анемии у пациентов с хронической почечной недостаточностью (ХПН): Китай (c 2018), Японии (c 2019) и Европейском союзе (c 2021). В США (в 2021) FDA не одобрил Роксадустат для использования у пациентов, находящихся и не находящихся на диализе, рекомендовав провести дополнительные клинические исследования сердечно-сосудистой безопасности препарата.
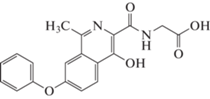
Роксадустат является пероральным ингибитором PHD-2 (t1/2 составляет 12–13 ч для здоровых людей) стабилизирует HIF-1α, увеличивает биодоступность железа (уменьшая потребность в парентеральном введении препаратов железа) и уровень эндогенного Эритропоэтина (Эритропоэтин) в пределах физиологического диапазона, что стимулирует эритропоэз.
На ClinicalTrials.gov отмечено 35 завершенных КИ Роксадустата (FG-4592), связанные с оценкой фармакокинетики, безопасности и эффективности препарата у здоровых людей, при анемии в условиях хронической болезни почек (различной стадии), при вызванной химиотерапией анемии. Продолжающиеся КИ (II и III фазы) (более 10) преимущественно нацелены на оценку эффективности при сопутствующей патологии сердца и у детей.
По результатам проведенных исследований у пациентов с ХПН с диализом, введение Роксадустата в течение 7 недель приводило к увеличению среднего уровня Hb на ≥20 г/л. В другом исследовании в течение первых 16 нед. лечения уровень Hb увеличивались в среднем на 18.3 ± 0.9 г/л (P < 0.001). В настоящее время проводится ряд исследований фазы 3 у пациентов с терминальной стадией почечной недостаточности и ХБП с НБ, продолжительностью от 24 нед. до 3 лет [71]. В сравнительных исследованиях эффективности пероральный Роксадустат не уступал парентеральному α-Эритропоэтинэтину в качестве терапии анемии у пациентов, проходящих диализ [19]. При этом Роксадустат дозозависимо и значительно снижал уровни общего холестерина у пациентов с НБЗ ХБП, что было подтверждено в исследовании, которое проводилось на пациентах, находящихся на диализе. В исследованиях Zhang с соавторами также показано положительное влияние Роксадустата в отношении снижения триглицеридов и протективное действие в отношении атеросклероза, что является важным, учитывая распространенность нарушений липидного обмена [89].
Появляется все больше свидетельств того, что почечная гипоксия встречается при остром почечном повреждении (ОПП) различной этиологии и играет важную роль как при ишемической, так и при токсической острой почечной недостаточности. Во многих исследованиях сообщалось, что предварительная активация HIF защищает от ишемии-реперфузии почки и повреждения, вызванного цисплатином. Использование малых молекул для стабилизации HIF-1α является новой стратегией лечения ОПП. Например, было показано, что предварительное введение роксадустата ослабляет ишемическое ОПП.
Введении нефротоксина цисплатина приводит к значительному увеличению содержания провоспалительных факторов в плазме (TNF-α, IL-6, IL-1β и моноцитарного хемоаттрактантного белка-1 (MCP-1)) и ткани почек (TNF-α, IL-6, IL-1β, MCP-1, сосудистый белок клеточной адгезии 1 (VCAM-1) и циклооксигеназы-2), является валидной моделью ОПП. Применение Роксадустата предупреждает развитие воспаления в почках и повышение концентрации указанных медиаторов, после введения цисплатина. Роксадустата и последующее за его введением повышение концентрации белка HIF-1α вызывает увеличение экспрессии генов-мишеней HO-1 и эритропоэтина, что имеет выраженный терапевтический потенциал при ОПП и ХБП [82].
Эксперименты in vivo показали, что Роксадустат эффективно ускоряет заживление ран, сокращает время заживления и вызывает гиперплазию эпидермиса, имитируя положительное влияние острой гипоксии на активацию эпителиальных стволовых клеток и запуская ускоренную реэпителизацию во время заживления ран. Это исследование продемонстрировало, что как гипоксия, так и Роксадустат улучшают пролиферацию и подвижность эпителиальных стволовых клеток путем стабилизации HIF-1α, что может применяться для ускорения заживление ран [72]. В исследованиях введение Роксадустата ускоряло заживление кожных ран и способствовало ангиогенезу в раневых участках крыс со стрептозотоциновой моделью сахарного диабета [92]. Это особенно важно, учитывая фактическое отсутствие лекарственных препаратов, которые были бы эффективны при терапии диабетической стопы – ведущей причины ампутаций при СД.
Применение ингибиторов пролилгидроксилазы, индуцируемых гипоксией (HIF), в качестве терапевтического средства лечения пациентов, страдающих анемией, было продемонстрировано для различных клинических условий. Однако, помимо этого предполагаемого применения, стабилизаторы HIF могут стать предметом неправильного использования в любительских и элитных видах спорта из-за их эритропоэтических свойств, что недавно было подтверждено несколькими случаями неблагоприятных аналитических результатов в тестах на допинг-контроль. [34].
Применение Роксадустата повышает устойчивость к ишемически/реперфузионному повреждению миокарда ишемии у мышей [25].
Роксадустат был эффективен при почечной анемии у пациентов с НБЗ и ХБП [90], улучшая обмен гемоглобина и железа, не увеличивая частоту возникновения нежелательных и тяжелых нежелательных явлений [42].
Роксадустат не только корректирует депрессивное поведение, но также улучшает вызванное хроническим непредсказуемым легким стрессом ухудшение памяти, что может объясняться значимой ролью HIF-1α в нейрогенезе, в том числе гиппокампа и синаптической пластичности. На молекулярном уровне было обнаружено, что введение Роксадустата активирует пути передачи сигналов HIF-1α и цАМФ-чувствительного элемента, связывающего белок/нейротрофический фактор мозга, происходящие in vivo, а также способствует экспрессии белков постсинаптической плотности (PSD), PSD95 и Homer1. Исследование первичных нейронов гиппокампа показало, что Роксадустат способствует росту дендритов. Роксадустат является многообещающей терапевтической стратегией для лечения болезни Паркинсона через улучшение митохондриальной функции в условиях окислительного стресса. Результаты, полученные Ли Г. с соавт. [51], не только дают экспериментальное обоснование для клинического применения Роксадустата при депрессии, но также подтверждают аргумент, что сигнальный путь HIF-1α является многообещающей мишенью для лечения заболеваний ЦНС. Это утверждение также подтверждается исследованиями Li X. [52], в которых Роксадустат защищает от индуцированной МРТР гибели TH-позитивных нейронов черной субстанции и ослабляет нарушения поведения мышей.
ВАДАДУСТАТ (AKB-6548)
Вададустат (Akebia, рис. 10) – ингибитор HIF-PH, в настоящее время находится в фазе 3 стадии клинических исследований в качестве препарата для лечения вторичной анемии при ХБП (t1/2 составляет 4.5 ч при пероральном приеме у здоровых участников).

На ClinicalTrials.gov отмечено 25 завершенных КИ Вададустата (AKB-6548), связанные с оценкой фармакокинетики, лекарственного взаимодействия, кардиобезопасности, эффективности при нарушенной функцией печени, при анемии в условиях хронической болезни почек (различной стадии). Продолжающиеся 4 КИ (II и III фазы) нацелены на оценку эффективности при анемии у детей и взрослых и для профилактики и лечения острого респираторного дистресс-синдрома у госпитализированных пациентов с COVID-19.
Среди побочных эффектов Вададустата наиболее часто отмечали следующие: диарея (4.3%) и тошнота (4.3%). Прекратили исследование из-за побочных эффектов десять (7.2%) пациентов, получавших Вададустат и 3 (4.2%) получавших плацебо. Артериальная гипертензия чаще отмечалась как побочное действие в группе Вададустат, чем в группе плацебо, хотя у всех пациентов, получавших Вададустат, у которых сообщалось о гипертонии, в анамнезе было повышенное артериальное давление, этот показатель на фоне терапии не изменялся. Терапия Валдадустатом не влияла на уровень холестерина в крови.
В 2020 г. завершилась 3 фаза КИ для Вададустата с участием 2600 пациентов на диализе с анемией, связанной с хроническим заболеванием почек – INNO2VATE, в котором вададустат не уступал дарбэпоэтину альфа в отношении сердечно-сосудистой безопасности, а также коррекции и поддержания концентраций гемоглобина [27].
В сентябре 2020-го года Akebia Therapeutics представила основные данные клинических испытаний фазы III PRO2TECT вададустата как средства лечения анемии, связанной с хроническим заболеванием почек (ХБП), у взрослых, не находящихся на диализе. PRO2TECT – это вторая глобальная программа компании по оценке сердечно-сосудистых заболеваний Фазы III.
В двух испытаниях PRO2TECT оценивалась эффективность и безопасность препарата по сравнению с дарбэпоэтином альфа. Оба исследования соответствовали первичной и ключевой вторичной конечной точке эффективности с не меньшей эффективностью по сравнению с дарбэпоэтином альфа. Неменьшую эффективность определяли, как среднее изменение гемоглобина (Hb) между исходным уровнем и периодом первичной оценки с 24 по 36 нед. и периодом вторичной оценки с 40 по 52 нед. Однако препарат не соответствовал первичной конечной точке безопасности программы PRO2TECT, которая не уступает по времени первому возникновению серьезных неблагоприятных сердечно-сосудистых событий (MACE) [21].
МОЛИДУСТАТ (BAY 85-3934)
Фармацевтическая компания Bayer Healthcare проводит оценку ингибитора HIF-PH, молидустата (BAY 85-3934, рис. 11). В доклинических исследованиях было показано, что молидустат эффективен при почечной и воспалительной анемии и, в отличие от терапии Эритропоэтином он снижает артериальное давление на модели ХБП. Уровни эндогенного Эритропоэтина, индуцированные во время лечения, были близки к нормальному физиологическому диапазону. На здоровых мужчинах введение однократных доз (37.5 и 50 мг) дозозависимо увеличивало уровень Эритропоэтина и количество ретикулоцитов.
На ClinicalTrials.gov отмечено 15 завершенных и 1 продолжающееся КИ (I, II и III фазы) Молидустата, связанные с оценкой безопасности, фармакокинетики и эффективности при анемии в условиях хронической болезни почек (различной стадии) у пациентов на диализе и без диализа.
В рандомизированном, двойном слепом, плацебо-контролируемом исследовании фазы 2b длительностью 16-недель (121 участник) Молидустат применялся у пациентов с анемией, не страдающих ЭКА с НБЗ в фиксированной дозе один и два раза в день. До конца исследования дошли 40% пациентов, получавших Молидустат, и 90% пациентов, получавших плацебо. Прекращение лечения молидустатом происходило главным образом из-за уровней Hb > 130 г/л или повышения >10 г/л через 2 нед. (44 из 61; ни один из них из-за Hb < 80 г/л); более высокие дозы молидустата привели к более высокой частоте прекращения приема из-за достижения критериев Hb.
Влияние молидустата на метаболизм железа оценивалось в трех 16-недельных рандомизированных контролируемых исследованиях фазы 2, оценивающих безопасность и эффективность молидустата при лечении анемии, связанной с ХБП, в различных популяциях: пациентам, не получавшим лечение, и пациентам, ранее получавшим агенты стимулирующие эритропоэз, не находящимся на диализе, и ранее леченные Эритропоэтином пациенты на гемодиализе [33]. У пациентов, не получавших ранее лечение и/или диализ, коэффициент насыщения трансферрина, концентрации гепсидина, ферритина и железа снижались на фоне применения молидустата, тогда как общая железосвязывающая способность сыворотки увеличивалась. Аналогичные результаты наблюдались у ранее леченных Эритропоэтином пациентов, не находящихся на диализе, хотя изменения в этих параметрах были более значительными у пациентов, не получавших лечение, чем у ранее леченных Эритропоэтин пациентов. У ранее получавших стимуляторы эритропоэза пациентов, получавших гемодиализ, концентрация гепсидина и общая железосвязывающая способность сыворотки оставались стабильными с молидустатом, тогда как коэффициент насыщения трансферрина, концентрации ферритина и железа увеличивались. В целом, аналогичные тенденции наблюдались во вторичных анализах подгрупп пациентов, не получавших добавки железа [11].
MIYABI Non-Dializ-Correction (MolIdustat once dailY improves renal Anaemia By Inducing erythropoietin; ND-C) и MIYABI Non-Dialysis-Maintenance (ND-M) – рандомизированные, открытые, многоцентровые исследования в параллельных группах, целью которых является оценка эффективности лечения молидустатом по сравнению с дарбепоэтином альфа у пациентов с анемией и ХБП без диализа. Вторичными задачами являются оценка безопасности, фармакокинетики и фармакодинамики лечения молидустатом. Средние значения (стандартное отклонение) исходных уровней гемоглобина составляли 98.4 (6.4) г/л для Молидустата и 100 (6.1) г/л для Дарбэпоэтина. Среднее (95% доверительный интервал [ДИ]) средних уровней гемоглобина в течение периода оценки для молидустата (112.8 [110.7; 115] г/л) и дарбэпоэтина (117 [115; 119 ] г/л) находилось в пределах целевого диапазона. Таким образом, Молидустат не уступал Дарбэпоэтину в изменении среднего уровня гемоглобина в течение периода оценки. Доля пациентов, сообщивших по крайней мере об одном нежелательном явлении, возникшем во время лечения (TEAE), составила 93.9% для Молидустата и 93.7% для Дарбэпоэтина [81].
ДАПРОДУСТАТ (GSK-1278863)
Фармацевтическая компания GlaxoSmithKline разрабатывает ингибитор HIF-PH, дапродустат (GSK-1278863, рис. 12).
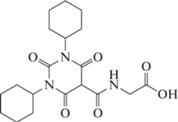
На ClinicalTrials.gov отмечено 35 завершенных и 1 продолжающееся КИ (I, II и III фазы) Дапродустата, связанные с оценкой фармакокинетики, лекарственного взаимодействия, кардиобезопасности, эффективности при анемии в условиях хронической болезни почек (различной стадии). Также завершены КИ противоишемического действия Дапродустата в отношении почек и сердца в процессе хирургического лечения аневризмы грудной аорты, а также его влияние на функциональное восстановление мышц после интенсивных физических упражнений.
В фазе I дапродустат хорошо переносился и дозозависимо увеличивал уровни Эритропоэтин (t1/2 составляет 4 ч при пероральном приеме у здоровых участников). В фазе 2а исследования пациенты с NDD-CKD (Диализ-независимые пациенты с ХБП (non-dialysis-dependent)) и терминальной стадии почечной недостаточности в течение 4 недель принимали ежедневно дапродустат (в дозе 0.5, 2 и 5 мг). Среднее повышение уровня Hb на 10 г/л было достигнуто в группе, получавшей 5 мг в популяции NDD-CKD не получавших Эритропоэтин. В гемодиализной популяции уровни Hb оставались стабильными после перехода от Эритропоэтина в группе лечения 5 мг, но не при более низких (0.5 и 2 мг) дозах дапродустата. Исследование, в котором изучались скорость повышения уровня гемоглобина, безопасность и переносимость, показало, что суточные дозы 10 и 25 мг обеспечивают эффективный эритропоэз при умеренной суточной выработке эндогенного Эритропоэтин. Применение этих доз у некоторых пациентов приводило к повышению Hb до высокого уровня (>130 г/л), что привело к раннему прекращению исследования. Подобное высокое повышение уровня Hb также имело место после ежедневного применения дапродустата в дозах 50 и 100 мг для групп с ХЗП 3–5 стадий и наряду с другими нежелательными явлениями, не связанными с переносимостью уровня Hb, приводило к раннему прекращению исследования.
Как и другие агенты в классе, наиболее распространенным побочными эффектами, наблюдаемым в исследованиях фазы 2, была тошнота [33].
Дапродустат, как и все представители этого класса, позиционируется в качестве эффективной альтернативы для лечения анемии у пациентов с ХБП. Дапродустат вызывал дозозависимые эффекты связанные и не связанные с эритропоэзом. Не было клинически значимого увеличения VEGF по сравнению с группами плацебо, что означает, что ингибирование HIF-PH не влияет на регуляцию VEGF посредством эритропоэтина. Значительное число пациентов вышло из исследования из-за быстрого увеличения уровней Hb на ≥10 г/л или уровня Hb > 135 г/л в течение 2-недельного периода. Эти устойчивые фармакодинамические ответы в течение таких коротких периодов времени могут указывать на потенциальную возможность развития сердечно-сосудистых или канцерогенных побочных эффектов от применения дапродустата при обследовании в течение более длительного периода времени. Общие побочные эффекты, такие как тошнота и диспепсия, хорошо переносились и были в основном доброкачественными. Основываясь на предварительном клиническом исследовании, дапродустат неэффективен для лечения заболевания периферических артерий. Эти данные в основном ограничены в зависимости от продолжительности исследований и небольшой численности пациентов. Наконец, существуют другие ингибиторы HIF-PH, которые в настоящее время находятся в фазе III разработки. Хотя эти агенты имеют одинаковый механизм действия, они различаются в зависимости от дозировки и администрации. Если в III фазе испытаний дапродустата будут обнаружены положительные результаты, он станет одним из четырех ингибиторов HIF-PH, поданных для одобрения FDA для лечения анемии у пациентов с ХБП в ближайшие годы [12].
ДЕСИДУСТАТ (ZYAN1)
Десидустат (ZYAN1, рис. 13) Zydus Cadila является пероральным HIF-PHI, который стимулирует эритропоэз.
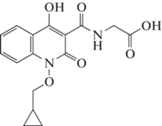
На ClinicalTrials.gov отмечено 3 завершенных и 1 продолжающееся КИ (I, II и III фазы) Десидустата, связанные с оценкой фармакокинетики, эффективности при анемии в условиях хронической болезни почек (различной стадии). Также в КИ оценивается эффективность Десидустата для лечения анемии у пациентов, получающих химиотерапию, а также для оценки его эффективности и безопасности для лечения пациентов с COVID-19 легкой, средней и тяжелой степени тяжести, где Дезидустат вводили в течение 14 дней вместе с рекомендуемой стандартной терапией.
У пациентов, получавших Десидустат (6 нед.; 100, 150 и 200 мг соответственно) Hb в среднем увеличился на 15.7; 22.2 и 29.2 г/л. Частота ответа (увеличение ≥10 г/л) составляла 66% для 100 мг, 75% для 150 мг и 83% в группах лечения 200 мг в популяции MITT. Фармакокинетические характеристики и безопасность Десидустата также оценивали в исследованиях, проведенных Parmar, 2019 с соавт. [Регистрационный номер клинического испытания CTRI/2017/05/008534 (зарегистрирован 11 мая 2017 г.) [60].
Хронические воспалительные заболевания часто связаны с анемией. В таких условиях анемию обычно лечат с помощью агентов, стимулирующих эритропоэз, которые связаны с потенциально опасными побочными эффектами и плохими результатами. Считается, что субоптимальный эритропоэз при хроническом воспалении вызван повышенным уровнем гепсидина, который вызывает блокаду железа в тканевых запасах. В работе с использованием моделей воспаления у грызунов Десидустат был оценен как эффективное лечение анемии воспаления. У мышей BALB/c однократное введение Десидустата оказывало противовоспалительное действие, вызванное липополисахаридом (ЛПС) или скипидаром, повышало уровень Эритропоэтина в сыворотке, количество железа и ретикулоцитов и снижало уровни гепсидина в сыворотке. При анемии, вызванной скипидарным маслом, у мышей BALB/c повторная терапия Десидустатом дозозависимо увеличивала содержание гемоглобина, эритроцитов и гематокрита. У самок крыс Льюиса лечение Десидустатом заметно снижало вызванную PGPS анемию и увеличивало количество гемоглобина, эритроцитов и лейкоцитов (WBC), гематокрит, сывороточное железо и селезенку. Эти эффекты Десидустата были связаны со снижением экспрессии гепсидина (HAMP), а также со снижением сывороточного гепсидина и увеличением экспрессии эритропоэтина в печени и почках. Лечение Десидустатом вызвало значительное увеличение экспрессии дуоденального цитохрома B (DcytB), ферропортина (FPN1) и транспортера 1 двухвалентного металла (DMT1) в двенадцатиперстной кишке и FPN1 и белка-1 хемоаттрактанта моноцитов (MCP-1) в печени, что свидетельствует об общем влиянии на метаболизм железа. Таким образом, фармакологическое ингибирование ферментов пролилгидроксилазы может быть полезным при лечении анемии воспаления [41]. Вопрос об универсальности этого эффекта для всех представителей HIF–PHI остается открытым, но для Десидустата эффективность (при анемии и при COVID-19) в некоторых КИ связывают со снижением уровня воспалительных цитокинов (таких как IL-6) и окислительного стресса.
ЭНАРОДУСТАТ (JTZ-951)
Энародустат (JTZ-951, рис. 14), разрабатываемый Japan Tobacco и Torii Pharmaceutical, пероральный ингибитор HIF-PH был одобрен в сентябре 2020 г. в Японии для лечения анемии, связанной с ХБП.
Клинические исследования фазы 3 подтвердили эффективность и безопасность Энародустата у пациентов с анемией и ХБП, не находящихся на диализе, на перитонеальном диализе и на гемодиализе [31]. Во время исследования уровень Hb оставался в пределах целевого диапазона, Энародустат снижал уровни гепсидина и ферритина, повышал общую железосвязывающую способность и в целом хорошо переносился.
ЗАКЛЮЧЕНИЕ
Соотношение кислорода и углекислого газа в организме подвержено строгому контролю со стороны различных систем. Изменение концентраций любого из этих газов быстро улавливается, что влечет за собой ответ организма и адаптацию к меняющимся условиям как внешней, так и внутренней среды. Сигнальная роль кислорода и углекислого газа, несомненно, играет ключевую роль в развитии адаптационного ответа, однако длительное нарушение гомеостаза этих газов служит звеном патогенеза многих заболеваний. Состояние гипоксии эволюционно важно для развития организма, но также сопровождает большое количество патологических процессов, поэтому необходимо понимать механизмы ее формирования с целью поиска способов фармакологической коррекции. Перспективным направлением является стабилизация α-субъединицы фактора, индуцируемого гипоксией (HIF-протекторы). Разработка лекарственных препаратов путем подавления метаболизма эндогенных биологически активных веществ, не одно десятилетие является распространенной стратегией многих фармацевтических компаний. Многие блокбастеры были созданы именно так (ингибиторы ДПП-4, МАО, иАПФ). В качестве протекторов HIF предложены вещества ингибирующие пролилгидроксилазу, препятствующие его деградации, что делает возможным осуществление его эффектов, а именно запуск экспрессии нижестоящих факторов ответа на гипоксию. Препараты, защищающие HIF, по мнению некоторых экспертов имеют высокие шансы пополнить список лекарств-блокбастеров. Однако стоит понимать, что ввиду своего плейотропного действия, стабилизация и, как следствие, избыточное накопление HIF, может быть нежелательными при ряде заболеваний, также необходимы дальнейшие исследования плейотропных эффектов данных препаратов, которые могут не зависеть от HIF.
Список литературы
Дедов И.И., Шестакова М.В., Майоров А.Ю. и др. Сахарный диабет 2 типа у взрослых // Сахарный диабет. 2020. Т. 23. № 2S. С. 4. https://doi.org/10.14341/
Зарубина И.В., Шабанов П.Д. Молекулярная фармакология антигипоксантов. СПб.: Н-Л, 2004. 368 с.
Зинчук В.В. Кислородтранспортная функция крови и газотрансмиттер сероводород // Успехи физиологических наук. 2021. Т. 52. № 3. С. 41–55. https://doi.org/10.31857/S0301179821030085
Левченкова О.С., Новиков В.Е. Антигипоксанты: возможные механизмы действия и клиническое применение // Вестник Смоленской государственной медицинской академии. 2011. № 4. С. 43–57.
Левченкова О.С., Новиков В.Е. Возможности фармакологического прекондиционирования // Вестник РАМН. 2016. № 1. https://doi.org/10.15690/vramn626
Оковитый С.В., Шуленин С.Н., Смирнов А.В. Клиническая фармакология антигипоксантов и антиоксидантов. СПб.: ФАРМиндекс, 2005. 72 с.
Рыбникова Е.А., Самойлов М.О. Современные представления о церебральных механизмах гипоксического пре и посткондиционирования // Успехи физиологических наук. 2016. Т. 47. № 4. С. 3–17.
Сысоев Ю.И., Крошкина К.А., Пьянкова В.А., Карев В.Е., Оковитый С.В. Изменение амплитудных и спектральных параметров электрокортикограмм крыс, перенесших черепно-мозговую травму // Биомедицина. 2019. Т. 15. № 4. С. 107. https://doi.org/10.33647/2074-5982-15-4-107-120
Швальб П.Г., Гавриленко А.В., Калинин Р.Е. и др. Эффективность и безопасность применения препарата “Неоваскулген” в комплексной терапии пациентов с хронической ишемией нижних конечностей (IIb-III фаза клинических испытаний) // Гены и клетки. 2011. № 3. С. 76–83.
Шустов Е.Б., Каркищенко Н.Н., Дуля М.С. и др. Экспрессия гипоксия-индуцибельного фактора HIF1α как критерий развития гипоксии тканей // Биомедицина. 2015. Т. 1. № 4. С. 4.
Akizawa T., Macdougall I.C., Berns J.S. et al. Iron regulation by molidustat, a daily oral hypoxia-inducible factor prolyl hydroxylase inhibitor, in patients with chronic kidney disease // Nephron. 2019. V. 143. № 4. P. 243. https://doi.org/10.1159/000502012
Becker K.A., Jones J.J. An emerging treatment alternative for anemia in chronic kidney disease patients: a review of daprodustat // Adv. Ther. 2018. V. 35. № 1. P. 5. https://doi.org/10.1007/s12325-017-0655-z
Besarab A., Chernyavskaya E., Motylev I. et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients // J. Am. Soc. Nephrol. 2016. V. 27. № 4. P. 1225. https://doi.org/10.1681/ASN.2015030241
Bessho R., Takiyama Y., Takiyama T. et al. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy // Scientific reports. 2019. V. 9. № 1. P. 1. https://doi.org/10.1038/s41598-019-51343-1
Bosch-Marce M., Okuyama H., Wesley J.B. et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia // Circ. Res. 2007. V. 101. № 12. P. 1310. https://doi.org/10.1161/CIRCRESAHA.107.153346
Botusan I.R., Sunkari V.G., Savu O. et al. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice // Proc. Natl. Acad. Sci. USA. 2008. V. 105. № 49. P. 19426 https://doi.org/10.1073/pnas.0805230105
Cai T., Ke Q., Fang Y. et al. Sodium–glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice // Cell Death Dis. 2020. V. 11. № 5. P. 390. https://doi.org/10.1038/s41419-020-2544-7
Cerychova R., Pavlinkova G. HIF-1, metabolism, and diabetes in the embryonic and adult heart // Front. Endocrinol. 2018. № 9. P. 460. https://doi.org/10.3389/fendo.2018.00460
Chen N., Hao C., Peng X. et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis // N. Engl. J. Med. 2019. V. 381. № 11. P. 1001. https://doi.org/10.1056/NEJMoa1813599
Chen Z., Lai T.C., Jan Y.H. et al. Hypoxia-responsive miRNAs target argonaute 1 to promote angiogenesis // J. Clin. Invest. 2013. V. 123. № 3. P. 1057. https://doi.org/10.1172/JCI65344
Chertow G.M., Pergola P.E., Farag Y.M.K. et al. Vadadustat in patients with anemia and non-dialysis-dependent CKD // N. Engl. J. Med. 2021. V. 384. № 17. P. 1589. https://doi.org/10.1056/NEJMoa2035938
Clambey E.T., McNamee E.N., Westrich J.A. et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa // Proc. Natl. Acad. Sci. USA. 2012. V. 109. № 41. P. E2784. https://doi.org/10.1073/pnas.1202366109
Creager M.A., Olin J.W., Belch J.J. et al. Effect of hypoxia-inducible factor-1α gene therapy on walking performance in patients with intermittent claudication // Circulation. 2011. V. 124. № 16. P. 1765. https://doi.org/10.1161/CIRCULATIONAHA.110.009407
Cummins E.P., Strowitzki M.J., Taylor C.T. Mechanisms and consequences of oxygen and carbon dioxide sensing in mammals // Physiol. Rev. 2020. V. 100. № 1. P. 463. https://doi.org/10.1152/physrev.00003.2019
Deguchi H., Ikeda M., Ide T. et al. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice // Circ. J. 2020. V. 84. № 6. P. 1028. https://doi.org/10.1253/circj.CJ-19-1039
Dunham-Snary K.J., Hong Z.G., Xiong P.Y. et al. A mitochondrial redox oxygen sensor in the pulmonary vasculature and ductus arteriosus // Pflügers Arch. 2016. V. 468. № 1. P. 43. https://doi.org/10.1007/s00424-015-1736-y
Eckardt K.U., Agarwal R., Aswad A. et al. Safety and efficacy of vadadustat for anemia in patients undergoing dialysis // N. Engl. J. Med. 2021. V. 384. № 17. P. 1601. https://doi.org/10.1056/NEJMoa2025956
Erler J.T., Weaver V.M. Three-dimensional context regulation of metastasis // Clin. Exp. Metastasis. 2009. V. 26. № 1. P. 35. https://doi.org/10.1007/s10585-008-9209-8
Ferro J.M., Bousser M.G., Canhão P. et al. European Stroke Organization guideline for the diagnosis and treatment of cerebral venous thrombosis – Endorsed by the European Academy of Neurology // Eur. J. Neurol. 2017. V. 2. № 3. P. 195. https://doi.org/10.1177/2396987317719364
Fitzpatrick S.F. Immunometabolism and sepsis: a role for HIF? // Front. Mol. Biosci. 2019. V. 6. P. 85. https://doi.org/10.3389/fmolb.2019.00085
Fukui K., Tanaka T., Nangaku M. Enarodustat to treat anemia in chronic kidney disease // Drugs Today (Barc). 2021. V. 57. № 8. P. 491–497. https://doi.org/10.1358/dot.2021.57.8.3304877
Grampp S., Schmid V., Salama R. et al. Multiple renal cancer susceptibility polymorphisms modulate the HIF pathway // PLoS Genet. 2017. V. 13. № 7. e1006872. https://doi.org/10.1371/journal.pgen.1006872
Gupta N., Wish J.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD // Am. J. Kidney Dis. 2017. V. 69. № 6. P. 815. https://doi.org/10.1053/j.ajkd.2016.12.011
Hansson A., Thevis M., Cox H. et al. Investigation of the metabolites of the HIF stabilizer FG-4592 (roxadustat) in five different in vitro models and in a human doping control sample using high resolution mass spectrometry // J. Pharm. Biomed. Anal. 2017. V. 134. P. 228. https://doi.org/10.1016/j.jpba.2016.11.041
Higgins D.F., Kimura K., Bernhardt W.M. et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition // J. Clin. Invest. 2007. V. 117. № 12. P. 3810. https://doi.org/10.1172/JCI30487
Huang D., Li T., Li X. et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression // Cell reports. 2014. V. 8. № 6. P. 1930. https://doi.org/10.1016/j.celrep.2014.08.028
Iizuka K., Bruick R.K., Liang G. et al. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis // Proc. Natl. Acad. Sci. USA. 2004. V. 101. № 19. P. 7281. https://doi.org/10.1073/pnas.0401516101
Inrig J.K., Barnhart H.X., Reddan D. et al. Effect of hemoglobin target on progression of kidney disease: a secondary analysis of the CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trial // Am. J. Kidney Dis. 2012. V. 60. № 3. P. 390. https://doi.org/10.1053/j.ajkd.2012.03.009
Isoe T., Makino Y., Mizumoto K. et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein // Kidney Int. 2010. V. 78. № 1. P. 48. https://doi.org/10.1038/ki.2010.99
Ivanova I.G., Park C.V., Kenneth N.S. Translating the hypoxic response — the role of HIF protein translation in the cellular response to low oxygen // Cells. 2019. V. 8. № 2. P. 114. https://doi.org/10.3390/cells8020114
Jain M., Joharapurkar A., Patel V. et al. Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation // Eur. J. Pharmacol. 2019. V. 843. P. 113. https://doi.org/10.1016/j.ejphar.2018.11.023
Jia L., Dong X., Yang J., Jia R., Zhang H. Effectiveness of hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat on renal anemia in non-dialysis-dependent chronic kidney disease: a systematic review and meta-analysis // Ann. Transl. Med. 2019. V. 7. № 23. P. 720. https://doi.org/10.21037/atm.2019.12.18
Kaelin W.G. Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer // Nat. Rev. Cancer. 2008. V. 8. № 11. P. 865. https://doi.org/10.1038/nrc2502
Karhausen J., Furuta G.T., Tomaszewski J.E. et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis // J. Clin. Invest. 2004. V. 114. № 8. P. 1098. https://doi.org/10.1172/JCI21086
Khwaja A. KDIGO clinical practice guideline for acute kidney injury // Nephron Clin. Pract. 2012. V. 2. № 1. P. 1. https://doi.org/10.1159/000339789
Knaup K.X., Guenther R., Stoeckert J. et al. HIF is not essential for suppression of experimental tumor growth by mTOR inhibition // J. Cancer. 2017. V. 8. № 10. P. 1809. https://doi.org/10.7150/jca.16486
Kuragano T., Kitamura K., Matsumura O. et al. ESA hyporesponsiveness is associated with adverse events in maintenance hemodialysis (MHD) patients, but not with iron storage // PLoS One. 2016. V. 11. № 3. e0147328. https://doi.org/10.1371/journal.pone.0147328
Laukka T., Mariani C.J., Ihantola T. et al. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes // J. Biol Chem. 2016. V. 291. № 8. P. 4256. https://doi.org/10.1074/jbc.M115.688762
Lee Y., Kim M., Han J. et al. MicroRNA genes are transcribed by RNA polymerase II // EMBO J. 2004. V. 23. № 20. P. 4051. https://doi.org/10.1038/sj.emboj.7600385
Lei Z., Li B.O., Yang Z. et al. Regulation of HIF-1α and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration // PloS One. 2009. V. 4. № 10. e7629. https://doi.org/10.1371/journal.pone.0007629
Li G., Zhao M., Cheng X. et al. FG-4592 improves depressive-like behaviors through HIF-1-mediated neurogenesis and synapse plasticity in rats // Neurotherapeutics. 2020. V. 17. № 2. P. 664. https://doi.org/10.1007/s13311-019-00807-3
Li X., Zou Y., Xing J. et al. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3β/Nrf2 pathway // Oxid. Med. Cell. Longev. 2020. P. 6286984. https://doi.org/10.1155/2020/6286984
Locatelli F., Eckardt K.U., Macdougall I.C. et al. Value of N-terminal brain natriuretic peptide as a prognostic marker in patients with CKD: results from the CREATE study // Curr. Med. Res. Opin. 2010. V. 26. № 11. P. 2543. https://doi.org/10.1185/03007995.2010.516237
López-Barneo J., Pardal R., Ortega-Sáenz P. Cellular mechanism of oxygen sensing // Annu. Rev. Physiol. 2001. V. 63. № 1. P. 259. https://doi.org/10.1146/annurev.physiol.63.1.259
López-Barneo J., González-Rodríguez P., Gao L. et al. Oxygen sensing by the carotid body: mechanisms and role in adaptation to hypoxia // Am. J. Physiol. Cell Physiol. 2016. V. 310. № 8. P. 629. https://doi.org/10.1152/ajpcell.00265.2015
Masoud G.N., Wei L. HIF-1α pathway: role, regulation and intervention for cancer therapy // Acta Pharm. Sin. B. 2015. № 5. P. 378. https://doi.org/10.1016/j.apsb.2015.05.007
McMurray J.J., Anand I.S., Diaz R. et al. Design of the Reduction of Events with Darbepoetin alfa in Heart Failure (RED-HF): a Phase III, anaemia correction, morbidity–mortality trial // Eur. J. Heart Fail. 2009. V. 11. № 8. P. 795. https://doi.org/10.1093/eurjhf/hfp098
Mimouna S., Gonçalvès D., Barnich N. et al. Crohn disease-associated Escherichia coli promote gastrointestinal inflammatory disorders by activation of HIF-dependent responses // Gut Microbes. 2011. V. 2. № 6. P. 335–346. https://doi.org/10.4161/gmic.18771
Mokas S., Larivière R., Lamalice L. et al. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification // Kidney International. 2016. V. 90. № 3. P. 598. https://doi.org/10.1016/j.kint.2016.05.020
Parmar D., Kansagra K. MON-318 A phase II trial to assess safety, tolerability and efficacy of PHD-2 inhibitor (desidustat-zyan1) in the treatment of anemia in pre-dialysis chronic kidney disease patients // Kidney Int. Rep. 2019. V. 4. № 7. S430. https://doi.org/10.1016/j.ekir.2019.05.1131
Petrov D., Mansfield C., Moussy A., Hermine O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? // Front. Aging Neurosci. 2017. V. 9. P. 68. https://doi.org/10.3389/fnagi.2017.00068
Pfeffer M.A., Burdmann E.A., Chen C.Y. et al. Baseline characteristics in the trial to reduce cardiovascular events with aranesp therapy (TREAT) // Am. J. Kidney Dis. 2009. V. 54. № 1. P. 59. https://doi.org/10.1053/j.ajkd.2009.04.008
Prabhakar N.R., Semenza G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2 // Physiol. Rev. 2012. V. 92. № 3. P. 967. https://doi.org/10.1152/physrev.00030.2011
Prabhakar N.R., Peng Y.J., Nanduri J. Hypoxia-inducible factors and obstructive sleep apnea // J. Clin. Invest. 2020. V. 130. № 10. P. 5042. https://doi.org/10.1172/JCI137560
Sarkar K., Rey S., Zhang X. et al. Tie2-dependent knockout of HIF-1 impairs burn wound vascularization and homing of bone marrow-derived angiogenic cells // Cardiovasc. Res. 2012. V. 93. № 1. P. 162. https://doi.org/10.1093/cvr/cvr282
Satoru K., Maruyama Y., Honda H. A new insight into the treatment of renal anemia with HIF stabilizer // Renal Replacement Therapy. 2020. № 6. P. 63. https://doi.org/10.1186/s41100-020-00311-x
Semenza G.L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy // Trends Pharmacol. Sci. 2012. V. 33. № 4. P. 207. https://doi.org/10.1016/j.tips.2012.01.005
Semenza G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease // Annu. Rev. Physiol. 2014. V. 76. P. 39. https://doi.org/10.1146/annurev-physiol-021113-170322
Semenza G.L. Hypoxia-Inducible Factors in Physiology and Medicine // Cell. 2012. V. 148. № 3. P. 399. https://doi.org/10.1016/j.cell.2012.01.021
Semenza G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells // Trends Mol. Med. 2012. V. 18. № 9. P. 534. https://doi.org/10.1016/j.molmed.2012.08.001
Soleymani Abyaneh H., Gupta N., Radziwon-Balicka A. et al. STAT3 but not HIF-1α is important in mediating Hypoxia-Induced chemoresistance in MDA-MB-231, a triple negative breast cancer cell line // Cancers. 2017. V. 9. № 10. P. 137. https://doi.org/10.3390/cancers9100137
Tang M., Zhu C., Yan T. et al. Safe and Effective Treatment for Anemic Patients With Chronic Kidney Disease: An Updated Systematic Review and Meta-Analysis on Roxadustat // Front. Pharmacol. 2021. № 12. P. 658079. https://doi.org/10.3389/fphar.2021.658079
Urrutia A.A., Aragonés J. HIF oxygen sensing pathways in lung biology // Biomedicines. 2018. V. 6. № 2. P. 68. https://doi.org/10.3390/biomedicines6020068
Wang Y., Zhang S., Li F. et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics // Nucleic Acids Res. 2020. V. 48. № D1. P. D1031. https://doi.org/10.1093/nar/gkz981
Watts E.R., Sarah R.W. Inflammation and hypoxia: HIF and PHD isoform selectivity // Trends Mol. Med. 2019. V. 25. № 1. P. 33. https://doi.org/10.1016/j.molmed.2018.10.006
Wei J.F., Huang F.Y., Xiong T.Y. et al. Acute myocardial injury is common in patients with COVID-19 and impairs their prognosis // Heart. 2020. V. 106. № 15. P. 1154. https://doi.org/10.1136/heartjnl-2020-317007
Weir E.K., López-Barneo J., Buckler K.J., Archer S.L. Acute oxygen sensing mechanisms // N. Engl. J. Med. 2005. V. 353. № 19. P. 2042. https://doi.org/10.1056/NEJMra050002
Weir E.K., Stephen L.A. The role of redox changes in oxygen sensing // Respir. Physiol. Neurobiol. 2010. V. 174. № 3. P. 182. https://doi.org/10.1016/j.resp.2010.08.015
Wong C.C., Gilkes D.M., Zhang H. et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation // Proc. Natl. Acad. Sci. USA. 2011. V. 108. № 39. P. 16369. https://doi.org/10.1073/pnas.1113483108
Yamamoto H., Nishi S., Tomo T. et al. 2015 Japanese Society for Dialysis Therapy: guidelines for renal anemia in chronic kidney disease // Renal Replacement Therapy. 2017. № 3. P. 36. https://doi.org/10.1186/s41100-017-0114-y
Yamamoto H., Nobori K., Matsuda Y. et al. Efficacy and safety of molidustat for anemia in ESA-naive nondialysis patients: a randomized, phase 3 trial // Am. J. Nephrol. 2021. V. 52. № 10–11. P. 871–883. https://doi.org/10.1159/000518071
Yang Y., Yu X., Zhang Y. et al. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury // Clin. Sci. 2018. V. 132. № 7. P. 825. https://doi.org/10.1042/CS20171625
Yoo Y.J., Kim H., Park S.R., Yoon Y.J. An overview of rapamycin: from discovery to future perspectives // J. Ind. Microbiol. Biotechnol. 2017. V. 44. № 4–5. P. 537–553. https://doi.org/10.1007/s10295-016-1834-7
Yuan G., Vasavda C., Peng Y.J. et al. Protein kinase G-regulated production of H2S governs oxygen sensing // Sci. Signal. 2015. V. 8. № 373. P. ra37. https://doi.org/10.1126/scisignal.2005846
Yin K.J., Olsen K., Hamblin M. e. al. Vascular endothelial cell-specific microRNA-15a inhibits angiogenesis in hindlimb ischemia // J. Biol. Chem. 2012. V. 287. № 32. P. 27055 https://doi.org/10.1074/jbc.M112.364414
Zhao C., Popel A.S. Computational Model of MicroRNA Control of HIF-VEGF Pathway: Insights into the Pathophysiology of Ischemic Vascular Disease and Cancer // PLoS Comput Biol. 2015. V. 11. № 11. P. e1004612. https://doi.org/10.1371/journal.pcbi.1004612
Zhao Q., Li Y., Tan B.B. et al. HIF-1α induces multidrug resistance in gastric cancer cells by inducing MiR-27a // PloS One. 2015. V. 10. № 8. e0132746. https://doi.org/10.1371/journal.pone.0132746
Zhao Z.Y., Gao Y.Y., Gao L. et al. Protective effects of bellidifolin in hypoxia-induced in pheochromocytoma cells (PC12) and underlying mechanisms // J. Toxicol. Environ. Health A. 2017. V. 80. № 22. P. 1187. https://doi.org/10.1080/15287394.2017.1367114
Zhang P., Du J., Zhao H. et al. Radioprotective effects of roxadustat (FG-4592) in haematopoietic system // J. Cell. Mol. Med. 2019. V. 23. № 1. P. 349. https://doi.org/10.1111/jcmm.13937
Zhang Y., Ren S., Xue H. et al. Roxadustat in treating anemia in dialysis patients (ROAD): protocol and rationale of a multicenter prospective observational cohort study // BMC Nephrology. 2021. V. 22. № 1. P. 28. https://doi.org/10.1186/s12882-021-02229-w
Zhu H., Wang X., Han Y. et al. Icariin promotes the migration of bone marrow stromal cells via the SDF-1α/HIF-1α/CXCR4 pathway // Drug Des. Devel. Ther. 2018. № 12. P. 4023. https://doi.org/10.2147/DDDT.S179989
Zhu Y., Wang Y., Jia Y., Xu J., Chai Y. Roxadustat promotes angiogenesis through HIF-1α/VEGF/VEGFR2 signaling and accelerates cutaneous wound healing in diabetic rats // Wound Repair Regen. 2019. V. 27. № 4. P. 324. https://doi.org/10.1111/wrr.12708
Ziello J.E., Jovin I.S., Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia // Yale J. Biol. Med. 2007. V. 80. № 2. P. 51.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук