

Вестник РАН, 2022, T. 92, № 10, стр. 950-960
ГЕМАТОЭНЦЕФАЛИЧЕСКИЙ БАРЬЕР В НЕЙРОИММУННЫХ ВЗАИМОДЕЙСТВИЯХ И РАЗВИТИИ ПАТОЛОГИЧЕСКИХ ПРОЦЕССОВ
А. С. Дятлова a, *, Н. С. Новикова a, **, Б. Г. Юшков b, ***, Е. А. Корнева a, ****, В. А. Черешнев b, *****
a Институт экспериментальной медицины
Санкт-Петербург, Россия
b Институт иммунологии и физиологии Уральского отделения Российской академии наук
Екатеринбург, Россия
* E-mail: anst.diatlova@gmail.com
** E-mail: novikiem@gmail.com
*** E-mail: b.yushkov@iip.uran.ru
**** E-mail: korneva_helen@mail.ru
***** E-mail: v.chereshnev@mail.ru
Поступила в редакцию 17.01.2022
После доработки 20.03.2022
Принята к публикации 01.07.2022
- EDN: JNUYRW
- DOI: 10.31857/S0869587322100048
Аннотация
Гематоэнцефалический барьер (ГЭБ) представляет собой своеобразный фильтр, обладающий высокой избирательностью по отношению к веществам различных типов. ГЭБ обеспечивает иммунный статус мозга, а также является важным регулятором нейроиммунных взаимодействий. В данном обзоре анализируются некоторые молекулярные и клеточные особенности ГЭБ, а также пять основных путей нейроиммунной коммуникации, опосредуемых ГЭБ. Обсуждаются функции ГЭБ в нейроиммунных взаимодействиях при различных заболеваниях: рассеянном склерозе, болезнях Альцгеймера и Паркинсона. Рассмотрены последние данные о нарушении функции ГЭБ при коронавирусной инфекции COVID-19, вызываемой вирусом SARS-CoV-2.
Взаимодействие нервной и иммунной систем обеспечивает возможность адаптивного реагирования последней на поступление в организм чужеродных агентов – бактерий, вирусов, грибков и т.д. Изучение участия гематоэнцефалического барьера (ГЭБ) в этом процессе представляет особый интерес. ГЭБ обеспечивает поддержание гомеостаза центральной нервной системы (ЦНС) и выполняет защитную функцию [1]. Это своего рода фильтр, пропускающий в ЦНС кислород, ионы и другие жизненно важные молекулы, но ограничивающий прохождение многих других веществ. Высокая избирательность ГЭБ по отношению к веществам различных типов объясняется его гистологическим строением.
Говоря о гематоэнцефалическом барьере, обычно подразумевают его эндотелиальную составляющую, однако в литературе выделяют эпителиальные клетки сосудистых сплетений как образующие барьер, обеспечивающий сохранение постоянства состава спинномозговой жидкости. В 2012 г. сформулирована концепция глимфатической системы – единой очистительной системы мозга, включающей в себя некоторые компоненты ГЭБ [2]. В данном обзоре представлены данные, касающиеся сосудистой части ГЭБ, но не особенностей барьера спинномозговой жидкости и глимфатической системы.
Эндотелиальные капилляры ГЭБ представляют собой микроваскулярную сеть, основу которой, как и в прочих сосудах организма, составляют эндотелиальные клетки. Капилляры ЦНС – непрерывные нефенестрированные [3], то есть их базальная мембрана непрерывна, без межклеточных щелей и пор (фенестраций) в плазматической мембране. Таким образом, эндотелиальная выстилка капилляров мозга – сплошная.
Помимо базальной мембраны и эндотелиальных клеток, важным компонентом ГЭБ являются глиальные и муральные клетки, а также нейроны, иммунные клетки ЦНС и периферической крови, суммарно формирующие нейрососудистую единицу. Функции ГЭБ в основном обеспечиваются эндотелиальными клетками, взаимодействующими с другими компонентами нейрососудистой единицы [4].
КОМПОНЕНТЫ ГЭБ И ЕГО ФУНКЦИИ
Молекулярный и клеточный состав ГЭБ определяет его функции. ГЭБ состоит не только из эндотелиальных клеток капилляров ЦНС, базальной мембраны капилляров, но и нескольких других типов клеток – муральных, включающих в себя гладкомышечные клетки и клетки-перициты, а также нейронов, астроцитов, периваскулярных макрофагов и микроглии, в некоторых случаях иммунных клеток периферической крови.
Эндотелиальные клетки капилляров ЦНС обладают уникальными свойствами, отличающими их от эндотелиальных клеток капилляров других органов. Отсутствие пор в плазматической мембране эндотелиоцитов обусловливает низкую скорость трансцитоза. Эти клетки соединены друг с другом непрерывными комплексами белков – плотными контактами, что также резко снижает вероятность параклеточного транспорта и позволяет регулировать движение ионов, молекул и клеток между кровью и мозгом. Молекулярную основу плотных контактов образует ряд белков, среди которых кадгерины, катенины, окклюдин, клаудины и каркасные белки ZO-1, ZO-2 и ZO-3 [5]. Клаудины необходимы для формирования параклеточного барьера, а каркасные белки ZO-1, -2 и -3 стабилизируют структуру и связывают плотные контакты с цитоскелетом. Кроме того, плотные контакты связаны с базальными адгерентными контактами, соединяющими все эндотелиальные клетки, что усиливает прочность конструкции. Плотные контакты являются своеобразными фильтрами с пропускной способностью менее 4 нм: они, как физический барьер, позволяют свободно проникать в мозг и из него только небольшим газообразным и липофильным молекулам, тогда как перенос больших молекул осуществляется посредством специальных транспортных систем [6].
К люминальной поверхности эндотелиальной клетки поляризованы efflux-транспортёры, или транспортёры оттока. Они представляют наибольший интерес для изучения, так как препятствуют проникновению фармакологических препаратов в ЦНС. Efflux-транспортёры выводят из мозга потенциально вредные соединения, такие как глутамат, даже против градиента концентрации, что требует АТФ в качестве источника энергии. К ним относят суперсемейство ABC-транспортёров – белков с общей доменной организацией (наличие трансмембранных и АТФ-связывающих доменов, которые также называют ABC-доменами). Наиболее изученным представителем этого суперсемейства в ГЭБ является P-гликопротеин (Pgp), обладающий широкой субстратной специфичностью и осуществляющий активный транспорт разнообразных веществ. Считается, что основная физиологическая роль Pgp – выведение ксенобиотиков. В эндотелиоцитах ГЭБ и астроцитах представлено большее количество Pgp, чем в других клетках организма [7].
Второй тип транспортёров – высокоспецифичные переносчики питательных веществ, которые облегчают транспортировку определённых молекул через ГЭБ в ЦНС (influx-транспортёры, или транспортёры притока). Транспортёры притока облегчают доставку лекарств в ЦНС [8]. Большое количество митохондрий в эндотелиоцитах капилляров ЦНС, вероятно, имеет решающее значение для регуляции активного транспорта.
Как правило, большие гидрофильные молекулы не могут переноситься через ГЭБ, за исключением случаев специфического рецепторного или адсорбционно-опосредованного трансцитоза. Именно таким образом во внеклеточное пространство мозга переносятся трансферрин, липопротеины низкой плотности, инсулин и другие пептидные гормоны [9].
Сочетание вышеперечисленных свойств эндотелиальных клеток позволяет ГЭБ жёстко регулировать транспорт веществ из кровеносного русла в ЦНС и в обратном направлении, поддерживая гомеостаз в ткани мозга.
Муральные клетки – гладкомышечные клетки, окружающие крупные сосуды, и клетки-перициты, покрывающие эндотелиальные стенки более мелких сосудов. Перициты располагаются на аблюминальной поверхности и погружены в базальную мембрану эндотелиальных клеток. Особенностью перицитов является отсутствие специфических маркеров, что затрудняет их идентификацию среди других типов клеток. Среди молекул, которые теоретически могут быть маркерами перицитов, выделяют PDGFRβ, NG2, CD13 и CD146, но NG2 и CD13 также экспрессируются гладкомышечными клетками. Перициты содержат белки-констрикторы, что обусловливает возможность их сокращения и позволяет контролировать диаметр капилляра [10]. Перициты не прилегают к эндотелиальным клеткам, но формируют с ними адгезионные контакты по типу “вилка–розетка”, а также другие типы контактов: адгезионные бляшки, плотные и щелевые контакты [11]. К основным отличиям перицитов ЦНС от перицитов периферии относят их происхождение (нервный гребень, а не мезодерма), а также соотношение количества эндотелиоцитов и перицитов: в ЦНС этот показатель достигает 1 : 1, а на периферии – 100 : 1 [12]. Функции перицитов многообразны, в их числе регуляция ангиогенеза, синтез компонентов внеклеточного матрикса (ВКМ), участие в ранозаживлении, а также регуляция степени инфильтрации ЦНС иммунными клетками [13].
Базальная мембрана (БМ), выстилающая капилляры, подразделяется на два типа: внутреннюю васкулярную и внешнюю паренхиматозную. Васкулярная БМ представляет собой ВКМ, синтезируемый эндотелиоцитами и перицитами, а компоненты паренхиматозной БМ синтезируются астроцитами [14]. Молекулярный состав сосудистой и паренхиматозной базальных мембран несколько различается, хотя преимущественно они состоят из коллагена IV типа, ламинина, нидогена, протеогликанов и гликопротеинов. Основная функция БМ – барьерная, и разрушающее действие на неё матриксных металлопротеаз (ММРs) является одним из звеньев патогенеза различных неврологических расстройств [15].
Астроциты – клетки макроглии, отростки которых подходят к кровеносным сосудам и формируют дистрогликан-дистрофиновый комплекс, необходимый для правильного встраивания в мембрану аквапорина-4. Астроциты регулируют поступление воды в ЦНС, а также обеспечивают клеточную взаимосвязь между нейронными цепями и кровеносными сосудами. Эти нейрососудистые соединения позволяют астроцитам регулировать кровоток в ответ на сигналы, приходящие от нервных клеток, например, регулировать сокращение и расслабление гладкомышечных клеток и перицитов, окружающих кровеносные сосуды. Предполагается, что в процессе эмбрионального развития астроциты не являются необходимым условием для формирования ГЭБ, однако нормальное развитие и поддержание функций ГЭБ невозможны в отсутствие астроцитов [16].
Клетки иммунной системы мозга подразделяются на два главных типа – микроглиальные клетки и периваскулярные макрофаги. Микроглиальные клетки мигрируют в ЦНС ещё в процессе эмбриогенеза, они образуются из гематопоэтических предшественников желточного мешка, а периваскулярные макрофаги имеют моноцитарное происхождение и проходят через ГЭБ, оседая в периваскулярном пространстве Вирхова–Робина [17]. Периваскулярные макрофаги обеспечивают первую линию защиты врождённого иммунитета. Функции микроглиальных клеток не ограничены участием в иммунной защите: они способствуют ранозаживлению, регулируют нейрональное развитие в эмбриогенезе, выполняют роль антиген-презентирующих клеток в адаптивном иммунитете [18].
Другие виды клеток – нейтрофилы, макрофаги, Т-клетки – при различных формах патологии участвуют в регуляции функции ГЭБ. Активные формы кислорода, синтезируемые этими клетками, увеличивают проницаемость сосудов, что может лежать в основе нарушения ГЭБ при различных неврологических заболеваниях [19].
Таким образом, нейроваскулярную единицу ГЭБ составляют эндотелиоциты капилляров головного мозга, перициты, астроциты, микроглиальные клетки, а также нейроны, ВКМ и гликокаликс (рис. 1).
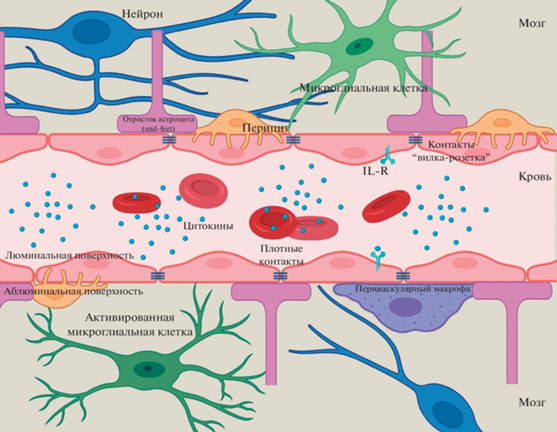
В ГЭБ есть особые участки, отграничивающие так называемые циркумвентрикулярные органы, прилегающие к стенкам III и IV желудочков мозга. К ним относят субфорникальный орган, заднее поле (area postrema) в продолговатом мозге, эпифиз и медиальную эминенцию нейрогипофиза. Гематоэнцефалический барьер здесь наиболее проницаем, что обусловлено другим типом капилляров – непрерывных фенестрированных, плотной сетью пронизывающих циркумвентрикулярные органы. Из-за повышенного кровоснабжения и проницаемости ГЭБ, циркумвентрикулярные органы оказываются важным компонентом системы нейроэндокринной регуляции, контролируя уровень цитокинов и температуру тела, водно-солевой баланс, артериальное давление, регулируя деятельность ЖКТ [20].
УЧАСТИЕ ГЭБ В НЕЙРОИММУННЫХ ВЗАИМОДЕЙСТВИЯХ
ГЭБ и клетки нейрососудистой единицы служат интерфейсом для взаимосвязи между ЦНС и периферией и являются важными регуляторами нейроиммунной коммуникации. Выделяют пять нейроиммунных осей, опосредуемых ГЭБ: модуляция проницаемости ГЭБ; модуляция транспортёров ГЭБ; захват и транспорт иммуноактивных веществ клетками ГЭБ; перенос иммунных клеток через ГЭБ в ткань мозга; секреция иммуноактивных веществ клетками ГЭБ и нейрососудистой единицы [21]. Эти оси могут функционировать независимо друг от друга, но чаще активируются совместно, обеспечивая различные варианты передачи сигналов от иммунной системы в мозг.
Нарушения проницаемости ГЭБ могут быть вызваны рядом факторов, включая активность цитокинов и хемокинов, бактерий и их компонентов, белков комплемента, белков острой фазы и т.д.
Строго говоря, термин “разрушение” обычно относят к патологическим процессам, однако даже в физиологических условиях защитная функция ГЭБ может незначительно варьироваться. Степень его проницаемости зависит от локализации: небольшие артериолы головного мозга имеют множество кавеолярных пузырьков, которые практически отсутствуют в капиллярах головного мозга. Показано, что эти кавеолы в артериолах опосредуют нервно-сосудистое соединение и физиологические функции при большей проницаемости ГЭБ [22].
Нарушения проницаемости могут быть следствием повреждающих и неповреждающих изменений, отражающих соответственно наличие или отсутствие физических повреждений. Первые подразумевают изменения на гистологическом уровне: повреждение эндотелиальных клеток, плотных контактов, и т.д. Неповреждающие изменения затрагивают молекулярный уровень реализации функций ГЭБ.
Повышение проницаемости ГЭБ может происходить посредством усиления пара- и трансцеллюлярного транспорта из-за снижения функций белков плотных и межклеточных контактов и усиления везикулярных механизмов. Разрушение плотных контактов между эндотелиоцитами ГЭБ происходит, когда синтез белков плотных контактов снижен, если они неправильно локализованы или посттрансляционно модифицированы [23]. Показано, что медиаторы воспаления модулируют синтез и активность белков плотных контактов. Например, инъекция IL-1β в паренхиму мозга приводит к потере экспрессии окклюдина и ZO-1 в эндотелиальных клетках. В эксперименте это приводило к усилению параклеточного транспорта фосфотирозина, используемого в качестве маркера, и рекрутированию нейтрофилов в сосуды, где плотные контакты отсутствовали. TGF-β1 подавляет экспрессию клаудина-5 [1]. К медиаторам воспаления, повышающим пара-клеточный транспорт через ГЭБ, относят брадикинин, гистамин, серотонин, арахидоновую кислоту и АТФ [24].
В качестве одного из механизмов, лежащих в основе модуляции белков плотных контактов медиаторами воспаления, предполагают действие кальпаина – внутриклеточной кальций-зависимой протеазы, регулирующей адгезию клеток. Показано, что ингибирование кальпаина предотвращает индуцированную IL-1β потерю белка ZO-1 в плотных контактах эндотелиоцитов и изменения в сборке цитоскелета F-актина [25].
К протективным факторам, способствующим поддержанию синтеза белков плотных контактов эндотелиоцитов, относят IL-25, нетрин-1, аннексин A1 (синтезируются эндотелиоцитами) и белки семейства SHH (синтезируются астроцитами). IL-1β снижает экспрессию SHH [26].
Ферментативная деградация белков плотных контактов также может происходить при развитии нейровоспаления. Протеазы нейтрофилов, такие как MMP9 и эластаза, способствуют разрушению ВКМ и эндотелия при ишемии и реперфузии. Интрацеребральная инъекция эластазы нейтрофилов вызывает отёк эндотелия и очаговый некроз кровеносных сосудов у крыс [27]. Ингибирование или нокаут матриксных металлопротеаз предотвращает деградацию плотных контактов и нарушение ГЭБ в острой фазе после ишемии и реперфузии мозга [28].
Другой возможный механизм нарушения проницаемости ГЭБ – физическое разрушение эндотелиальных клеток, в результате чего в них образуются везикулярные каналы, сквозь которые проходят крупные молекулы. Данный механизм продемонстрирован при отёке головного мозга, черепно-мозговой травме, сепсисе. Нарушения синтеза белка пузырьков плазмалеммы PLVAP в эндотелиоцитах ассоциированы с болезнью Альцгеймера и рассеянным склерозом [29]. Таким образом, патологическая фенестрация ГЭБ может быть важным фактором нарушения его проницаемости.
Модуляция функций ГЭБ иммунноактивными веществами. Как упоминалось выше, эндотелиоциты ГЭБ на своей поверхности содержат большое количество транспортёрных систем, среди которых есть как системы активного транспорта, так и работающие по принципу облегчённой диффузии. Функции некоторых из них изменяются при нейровоспалении и модулируются сигнальными молекулами, например, белком Pgp. Его лиганды включают в себя ингибиторы протеаз, опиаты, противоэпилептические средства, циклоспорины, глюкокортикоиды, альдостерон, дексаметазон и блокаторы кальциевых каналов. Активность Pgp объясняет, почему определённые вещества не накапливаются в головном мозге в достаточных количествах, чтобы оказывать влияние на ЦНС. Функция изменяется при воспалении, причём основным эффектом in vivo является подавление его транспортной активности. Так, индукция провоспалительными цитокинами (TNFα, IL-1β, IL-6, IL-2, IFNγ) ведёт к снижению экспрессии мРНК Pgp в эндотелиоцитах ЦНС, его синтеза и активности [30].
Воспаление также влияет на influx-транспортёры. Так, белок интерлейкин-6 (IL-6), секретируемый астроцитами, стимулирует активность котранспортёра Na-K-Cl, а его чрезмерная активация приводит к развитию отёка мозга [31].
Специальный интерес представляют эффекты, оказываемые на функции ГЭБ липополисахаридом (ЛПС). Известно, что на эндотелиоцитах ГЭБ экспрессируется рецептор TLR4, напрямую опосредующий эффекты ЛПС. В первичной культуре эндотелиоцитов ЛПС вызывает нарушение гематоэнцефалического барьера, связанное с дисфункцией белков плотных контактов, а в высоких дозах индуцирует их апоптоз [32]. Эффекты ЛПС на разрушение ГЭБ смягчаются ингибитором циклооксигеназы индометацином [33], а значит, циклооксигеназы участвуют в механизмах ЛПС-индуцированного разрушения ГЭБ.
Получены противоречивые данные о влиянии муральных и глиальных клеток нейроваскулярной единицы на реакцию эндотелиоцитов на ЛПС. Культивирование эндотелиоцитов головного мозга крупного рогатого скота совместно с астроцитами крыс оказывало протективный эффект на ЛПС-индуцированное разрушение эндотелиоцитов. Такого эффекта не наблюдалось при совместном культивировании эндотелиоцитов мышей с астроцитами и перицитами [34].
Реагируют ли ГЭБ и эндотелиоциты на базовые уровни ЛПС в кровотоке? Известно, что физиологическая концентрация ЛПС в крови (выявленная при помощи ЛАЛ-теста) составляет до 1 EU/мл, и ЛПС может играть роль в физиологической регуляции функций ГЭБ. По данным авторов работы [35], которые провели анализ результатов 74 исследований, посвящённых изучению эффектов ЛПС на ГЭБ, в 60% случаев сообщалось о его повреждающем действии. Эффекты ЛПС на ГЭБ зависят от дозы липополисахарида, кратности его введения, вида экспериментального животного, его пола, возраста и т.д. Авторы построили модель логистической регрессии, в которой учли эти экспериментальные факторы и установили, что значимым предиктором среди них является только вид экспериментального животного. Так, повреждающие изменения ГЭБ у мышей в 4 раза более вероятны, чем у крыс. Доза ЛПС, напротив, не была значимым фактором. В большинстве исследований использовалась септическая доза ЛПС, что затрудняет обобщение полученных результатов для менее тяжёлых инфекций, распространённых у людей.
Транспорт иммуноактивных веществ через ГЭБ. Основная функция ГЭБ – предотвращение проникновения из крови в мозг веществ, которые могут иметь нейротоксические эффекты. К таким молекулам относят цитокины и хемокины, однако многие из них регулируемо транспортируются через ГЭБ в направлении от крови к мозгу. Различия между поступлением в ЦНС нейроактивных веществ через нерегулируемую “утечку” и посредством регулируемого транспорта в данном случае – ключевой вопрос: в первом случае происходит дисфункция ГЭБ, что может привести к нейротоксичности; во втором – ЦНС, по-видимому, регулирует транспортные свойства ГЭБ в соответствии со своими потребностями.
О механизмах транспорта цитокинов через ГЭБ и о факторах, влияющих на этот транспорт, известно немного. Например, цитокин-индуцированный хемоаттрактант нейтрофилов CINC1 проникает в мозг по ненасыщаемому механизму, предположительно по механизму трансклеточной диффузии. Хемокин CCL2 транспортируется по кавеоло-зависимому пути. Белком-переносчиком TNF в ГЭБ, вероятно, является его рецептор, так как у мышей с нокаутом этого рецептора не происходит транспорта TNF через ГЭБ, но нарушения транспорта для эпидермального фактора роста, IL-1 и их рецепторов не отмечается [36]. Большинство исследований транспорта цитокинов проведено на мышах, но продемонстрировано, что транспорт IL-1α и IL-6 через ГЭБ осуществляется в эмбриогенезе у крыс, а IL-1β и IL-6 – у эмбрионов овцы [37], что свидетельствует об экспрессии переносчиков цитокинов в ГЭБ на ранних стадиях развития и у разных видов животных.
TNF-α, транспортирующийся через ГЭБ при системном воспалении, стимулирует клетки микроглии, способствуя развитию нейровоспаления и повышению секреции TNF, что приводит к апоптозу дофаминергических клеток в чёрной субстанции (substantia nigra). Такая модель двойного действия цитокинов периферической крови и клеток ЦНС может в целом применяться к цитокинам, которые проходят через ГЭБ при воспалении [38].
Интересно, что скорость транспорта цитокинов неоднородна по всему мозгу. Так, клетки циркумвентрикулярных органов транспортируют примерно в 7 раз больше цитокинов, чем соседние области [39]. Такой быстрый обмен информацией между иммунной и нервной системами обеспечивает возможность реализации регуляторной функции этих органов.
Транспорт клеток иммунной системы через ГЭБ. ЦНС считается иммунопривилегированным органом, поэтому в физиологических условиях транспорт клеток иммунной системы через ГЭБ минимален и строго регулируется. В норме мононуклеарные клетки попадают в мозг во время эмбрионального развития и становятся резидентными иммунокомпетентными клетками – микроглией. Однако при патологических процессах – инфекции, травме – лейкоциты периферической крови под действитем цитокиновых сигналов рекрутируются в мозг, а ГЭБ имеет особые механизмы надзора и рекрутирования лейкоцитов в ткань мозга.
Поступление лейкоцитов в спинномозговую жидкость и паренхиму головного мозга через ГЭБ зависит от многоступенчатого процесса экстравазации, который варьируется в зависимости от базальной экспрессии молекул адгезии. Типичный процесс экстравазации лейкоцитов в ткани включает начальный захват циркулирующих лейкоцитов, их “якорение”, “катание” по эндотелию и последующий диапедез. Прикрепление лейкоцитов к эндотелиальным клеткам обеспечивается повышенной экспрессией молекул адгезии VCAM, ICAM, интегринов. Начальные этапы захвата лейкоцитов опосредуются взаимодействиями селектинов на поверхности эндотелиальных клеток с гликопротеинами на поверхности лейкоцитов [40]. Периферические и менингеальные эндотелиальные клетки, а также эндотелиальные клетки сосудистого сплетения экспрессируют селектины, которые хранятся в тельцах Вибеля–Паладе. В ответ на медиаторы воспаления тельца Вибеля–Паладе мигрируют к люминальной поверхности эндотелиоцитов, обеспечивая быструю экспрессию селектинов на люминальной мембране. Эндотелиальные клетки паренхимы головного мозга не хранят селектины в тельцах Вибеля–Паладе и нуждаются в синтезе селектинов de novo для обеспечения захвата и миграции лейкоцитов. Диапедез лейкоцитов может осуществляться как параклеточным, так и трансцеллюлярным транспортом, затрагивая в основном белки плотных контактов и цитоскелета эндотелиоцитов. Активированная микроглия секретирует воспалительные факторы, усиливает экспрессию молекул адгезии в эндотелиоцитах и участвует в рекрутировании лейкоцитов ЦНС, повышая интенсивность воспалительного процесса [41].
Один из факторов, стимулирующих проникновение лейкоцитов в ЦНС, – провоспалительный цитокин IL-1β. При этом механизм проникновения в данном случае, вероятно, рецепторный: эндотелиоциты экспрессируют рецептор к IL-1 типа 1 (IL-1R1) в ЦНС. Нокдаун IL-1R1 в эндотелии отменяет приток лейкоцитов в ЦНС, индуцированный интрацеребровентрикулярно вводимым IL-1β. Интересно, что влияние IL-1β на миграцию лейкоцитов может быть ингибировано при системном воспалении. Системное введение ЛПС в течение 2 часов после интрацеребровентрикулярной инъекции IL-1β ингибирует рекрутирование лейкоцитов в ЦНС, предотвращая активацию селектинов на эндотелиоцитах [42].
Сложилось мнение, что перенос лейкоцитов в ЦНС при патологии осуществляется в основном в сосудистых сплетениях. При повреждении спинного мозга макрофаги рекрутируются к месту повреждения через сосудистое сплетение путём повышения экспрессии молекул адгезии на эпителиальных клетках сплетения. На модели окклюзии средней мозговой артерии показано, что около двух третей Т-клеток, которые инфильтрируют периинфарктную зону, рекрутируются из стромы сосудистого сплетения. В том же исследовании отмечено, что апикальная область стромы сосудистого сплетения физически связана с сосудами паренхимы головного мозга. Предполагается, что быстрая миграция Т-клеток из стромы сосудистого сплетения в мозолистое тело и периинфарктную зону связана с наличием прямого пути доставки Т-клеток в ткани ЦНС, в обход спинномозговой жидкости и мозговых барьеров [43].
В ряде исследований, посвящённых изучению механизмов возникновения и развития экспериментального аутоиммунного энцефаломиелита (ЭАЭ) у мышей продемонстрировано существование “нейронных шлюзов” (neural gateways), включающих симпатическую норадренергическую иннервацию местных кровеносных сосудов и регуляцию проникновения аутореактивных CD4+ Т-клеток в ЦНС. Эти исследования показали, что высвобождаемый симпатическими окончаниями в ответ на различные стимулы норадреналин усиливает NFkB-опосредованную транскрипцию провоспалительных генов в эндотелиальных клетках определённых сосудов, тем самым обеспечивая “ворота” для входа иммунных клеток в ЦНС. Например, так называемый рефлекс “gravity–gateway” способствует проникновению аутореактивных CD4+-клеток в дорсальные кровеносные сосуды, соответствующие пятому поясничному (L5) позвонку. Этот рефлекс является соматосимпатическим, опосредованным проприоцептивными сигналами, вызванными сокращениями мышц задних конечностей. Норадреналин, высвобождаемый симпатическими окончаниями на уровне L5, вызывает расширение кровеносных сосудов и усиливает сигналинг IL-6 и экспрессию хемокина CCL20, способствуя доступу активированных CD4+-клеток в ЦНС. Рефлекс “pain–gateway” – болевой рефлекс, вызываемый стимуляцией ноцицепторов задних конечностей, запускает высвобождение норадреналина и увеличивает экспрессию CX3CL1 и активацию резидентных моноцитов в вентральных сосудах, иннервируемых симпатическими нервами на уровне L5, что приводит к рецидиву экспериментального аллергического энцефаломиелита (ЭАЭ) [44].
Эти исследования также подтвердили наличие stress-gateway-рефлекса, вовлекающего паравентрикулярное ядро гипоталамуса и симпатические нервные волокна, иннервирующие сосуды пограничных областей третьего желудочка, таламуса и зубчатой извилины. Предположительно, этот рефлекс может вызывать опосредованное CCL5 накопление аутореактивнопатогенных CD4+ Т-клеток в этих структурах и косвенно запускать реакции со стороны волокон блуждающего нерва, способствующие повреждению кишечника у мышей с ЭАЭ [45].
Используя прижизненную двухфотонную микроскопию на модели ЭАЭ, научная группа Х. Векерле проследила процесс инвазии специфических к основному белку миелина Т-клеток (TMBP-клеток) в ЦНС. Экспериментальным животным вводили 5 × 103 TMBP-клеток для индукции ЭАЭ. Показано, что небольшое количество единичных TMBP-клеток проникает в лептоменингеальные сосуды в первый день после введения клеток. В последующие часы в сосуды оболочек проникает больше TMBP-клеток; они активно передвигаются внутри кровеносных сосудов, часто против кровотока. Это подобное ползанию движение преобладает в сосудах оболочек мозга, в то время как классическое для Т-клеток “катание” наблюдается в сосудах периферических органов. После “ползания” внутри сосудов оболочек Т-клетки проникают через стенку сосудов и продолжают мигрировать по их аблюминальной поверхности. Значительная часть Т-клеток устанавливает контакты с местными антигенпрезентирующими клетками, вследствие чего происходит стимуляция эффекторных Т-клеток к выработке провоспалительных медиаторов, последующей тканевой инвазии и образованию воспалительных инфильтратов [46]. Эти наблюдения подробно описывают начальные стадии иммуннологических реакций в ЦНС.
Секреция иммуноактивных веществ клетками ГЭБ. Клетки, которые образуют ГЭБ и сосудистые сплетения, постоянно взаимодействуют с другими клетками ЦНС. Концепция перекрёстных взаимодействий воплощена в термине нейроваскулярной единицы. Перекрёстные взаимодействия служат для информирования клеток ГЭБ о потребностях ЦНС и адаптации ею функций в конкретной ситуации. Многие из веществ, обеспечивающих это перекрёстное взаимодействие, являются нейроиммунными медиаторами, включая цитокины, простагландины, оксид азота и т.д. Таким образом, нейроиммунные медиаторы непосредственно участвуют в перекрёстных взаимодействиях нейроваскулярной единицы и формировании функциональной активности ГЭБ.
Эндотелиоциты ГЭБ секретируют различные цитокины. Конститутивная секреция цитокинов может модулироваться иммунными стимуляторами, такими как ЛПС, вирусы, вирусные белки, бактерии. Эндотелиоциты могут реагировать на эти сигналы, поступающие из ЦНС или крови. Совместное культивирование эндотелиоцитов с другими компонентами нейроваскулярной единицы, такими как астроциты и перициты, модулирует интенсивность секреции цитокинов. Секреция цитокинов является биполярной: они могут секретироваться с люминальной поверхности эндотелиоцита в кровь или с аблюминальной поверхности эндотелиоцита в ЦНС [47].
Возможность комбинации нескольких нейроиммунных осей означает, что эндотелиоцит может получать сигнал (например, с люминальной поверхности) и высвобождать цитокин с другой стороны (с аблюминальной стороны к мозгу). В этом случае информация с одной стороны ГЭБ передаётся на другую. Например, авторы работы [48] выяснили механизм, обеспечивающий иммунную регуляцию транспорта железа через ГЭБ с помощью астроцитарного церулоплазмина. ЛПС, действующий на люминальную поверхность эндотелиоцитов, вызывает секрецию IL-6 и IL-1β с их аблюминальной поверхности, которые, в свою очередь, стимулируют астроцитарную секрецию церулоплазмина, действующего на аблюминальную поверхность эндотелиоцитов, что облегчает перенос железа через ГЭБ [48].
Таким образом, ГЭБ представляет собой многокомпонентную систему, в которой каждое молекулярное и клеточное звено участвует в формировании защитного барьера для поддержания гомеостаза ЦНС. Нарушения в работе любого из этих звеньев представляет собой угрозу для проницаемости барьера, что в результате может привести к развитию патологического процесса. Дисфункции ГЭБ констатировали при многих заболеваниях: рассеянном склерозе, гипоксическом и ишемическом инсульте, болезнях Паркинсона и Альцгеймера, эпилепсии, опухолях головного мозга. Наблюдаемая дисфункция барьера может варьировать от лёгких и временных изменений его проницаемости до хронического разрушения барьера, связанного с перестройкой функциональной активности транспортёров и деградацией базальной мембраны. В большинстве случаев невозможно определить, является ли нарушение барьера причиной возникновения заболевания или результатом его прогрессирования. Тем не менее нарушение барьера часто способствует развитию патологии и её усилению.
При болезни Альцгеймера наблюдается дисфункция ГЭБ: существует гипотеза о двухстадийных сосудистых нарушениях, согласно которой сосудистая дисфункция играет ведущую роль на начальных этапах развития заболевания, приводя к ишемии-гипоксии и повреждению эндотелиоцитов на первой стадии. Во время второй стадии повреждение эндотелиоцитов и ГЭБ ведёт к отложению нейротоксических веществ в ЦНС, окислительному стрессу и нейровоспалению. Нейровоспаление и окислительный стресс усиливают активность β- и γ-секретаз, что в свою очередь способствуют образованию и постепенному накоплению амилоида Aβ. Кроме того, известна корреляция между дисфункцией ГЭБ и накоплением нейрофибриллярных клубков из гиперфосфорилированного τ-белка. Следовательно, ГЭБ может быть новой терапевтической мишенью для лечения болезни Альцгеймера [49].
При болезни Паркинсона также наблюдается повышенная проницаемость ГЭБ. Продемонстрировано, что избыточное накопление альфа-синуклеина способствует подавлению экспрессии белков плотных контактов ZO-1 и окклюдина. При этом у пациентов с болезнью Паркинсона выявлено снижение экспрессии и функциональной активности транспортёров ГЭБ, таких как GLUT-1 и Pgp. Дисфункция транспортёров при болезни Паркинсона вызывает снижение клиренса нейротоксических веществ, что способствует прогрессированию заболевания [50].
В спинномозговой жидкости пациентов с болезнью Паркинсона повышено количество факторов ангиогенеза [51]. Аберрантный ангиогенез приводит к образованию незрелой сосудистой сети с отсутствием плотных контактов, что имеет своим следствием проникновение в ЦНС нейротоксических веществ и потерю дофаминергических нейронов. Таким образом, повреждение ГЭБ ускоряет развитие нейродегенеративных заболеваний.
Нарушение ГЭБ и трансэндотелиальная миграция активированных лейкоцитов – одни из самых ранних цереброваскулярных аномалий, наблюдаемых в мозге пациентов с рассеянным склерозом. Показано, что у людей с наследственной предрасположенностью вирусные инфекции и токсины окружающей среды снижают иммунную толерантность и стимулируют высвобождение провоспалительных факторов, таких как IL-6 и NF-kB. Последние способствуют разрушению плотных контактов эндотелиоцитов, нарушают целостность ГЭБ и стимулируют трансмиграцию лейкоцитов. Экспрессия транспортёра Pgp также увеличивается и усиливает миграцию CD4+ и CD8+ Т-клеток, что усиливает нейровоспаление [52]. В условиях воспаления повышается и экспрессия селектинов эндотелиоцитов, что способствует адгезии лейкоцитов. Цитокины и хемокины активируют рецепторы адгезии эндотелия и усиливают последующую лейкоцитарную инфильтрацию. CD4+ Т-клетки идентифицируют белки миелиновой оболочки и активируют высвобождение провоспалительных факторов (включая IFN-γ, TNF-α, оксид азота и свободные радикалы), что приводит к демиелинизации. Нейтрофилы, моноциты и микроглия инфильтрируют паренхиму мозга и высвобождают внеклеточный глутамат, вызывая эксайтотоксичность и ускоряя дисфункцию ГЭБ, а это усиливает прогрессирование заболевания [53].
Исследования ГЭБ при заболевании COVID-19, вызываемом вирусом SARS-CoV-2, находятся в начальной стадии. Представляется, что вирус поражает сосудистую сеть многих систем и органов, в том числе головного мозга. Последствия COVID-19 включают в себя неврологические симптомы: головную боль, тошноту, головокружение, образование микротромбов и в редких случаях энцефалит. Изучение взаимодействия SARS-CoV-2 с клетками, формирующими ГЭБ, представляется весьма актуальным для понимания механизмов возникновения таких последствий. С использованием постмортальных материалов головного мозга больных COVID-19 показано, что белок ACE2, известный как мишень связывания спайкового белка SARS-CoV-2, повсеместно экспрессируется в сосудах различных калибров лобной коры. При этом экспрессия ACE2 повышается у пациентов с деменцией и гипертонией. В клеточной культуре первичных эндотелиальных клеток головного мозга человека (hBMVEC) также обнаруживается экспрессия ACE2. В условиях моделирования ГЭБ in vitro продемонстрировано, что спайковый белок S1 способствует потере целостности барьера, вызывая провоспалительную реакцию на эндотелиальных клетках головного мозга, что может способствовать изменению состояния функции ГЭБ [54].
Л. Пеллегрини с соавторами обнаружили экспрессию рецептора ACE2 в зрелых клетках хороидного сплетения, но не в нейронах или других типах клеток. Они доказали тропность SARS-CoV-2 к эпителиальным клеткам хороидного сплетения и обнаружили, что вирус практически не заражает нейроны или глию. Утверждается, что при повреждении эпителия SARS-CoV-2 происходит нарушение защитной функции ГЭБ, что может приводить к нейровоспалению [55]. Показано также, что радиоактивно меченый спайковый белок S1 преодолевает ГЭБ у мышей как при внутривенном, так и при интраназальном введении. Дальнейшие исследования подтвердили, что S1 проникает через ГЭБ с участием фермента ACE2 посредством адсорбционного рецептор-опосредованного трансцитоза [56]. Комплекс данных демонстрирует подверженность ГЭБ воздействию SARS-CoV-2, что необходимо для понимания механизмов как неврологических, так и психиатрических проявлений этой вирусной инфекции.
* * *
Таким образом, очевидно, что нарушения функций гематоэнцефалического барьера играют существенную роль в механизмах развития нейровоспаления и нейродегенерации. Сочетание барьерной функции со способностью реагировать на иммуноактивные вещества и секретировать их позволяет ГЭБ осуществлять взаимодействие нервной и иммунной систем, транслируя сигналы в двух направлениях. Эти взаимодействия не постоянны и модулируются микроокружением ГЭБ. Такая модуляция требует перекрёстных взаимодействий компонентов барьера и нейроваскулярной единицы. ГЭБ представляет собой динамическую регуляторную систему, которая участвует как в превращении ЦНС в иммунопривилегированный орган, так и в обеспечении двунаправленной связи между иммунной системой и мозгом.
Список литературы
Bolton S.J., Anthony D.C., Perry V.H. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo // Neuroscience. 1998. № 4 (86). P. 1245–1257.
Кондратьев А.Н., Ценципер Л.М. Глимфатическая система мозга: строение и практическая значимость // Анестезиология и реаниматология. 2019. № 6. С. 72–80.
Muoio V., Persson P.B., Sendeski M.M. The neurovascular unit – concept review // Acta Physiologica (Oxford, England). 2014. № 4 (210). P. 790–798.
Tietz S., Engelhardt B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions // The Journal of Cell Biology. 2015. № 4 (209). P. 493–506.
Daneman R., Prat A. The Blood-Brain Barrier // Cold Spring Harbor Perspectives in Biology. 2015. № 1 (7): a020412.
Löscher W., Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family // NeuroRx: The Journal of the American Society for Experimental NeuroTherapeutics. 2005. № 1 (2). P. 86–98.
Sanchez-Covarrubias L., Slosky L.M., Thompson B.J. et al. Transporters at CNS Barrier Sites: Obstacles or Opportunities for Drug Delivery? // Current pharmaceutical design. 2014. № 10 (20). P. 1422–1449.
Ayloo S., Gu C. Transcytosis at the blood-brain barrier // Current Opinion in Neurobiology. 2019. (57). P. 32–38.
Methner C., Mishra A., Golgotiu K. et al. Pericyte constriction underlies capillary derecruitment during hyperemia in the setting of arterial stenosis // American Journal of Physiology – Heart and Circulatory Physiology. 2019. № 2 (317): H255–H263.
Sweeney M., Foldes G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease // Frontiers in Cardiovascular Medicine. 2018. № 5:154. https://doi.org/10.3389/fcvm.2018.00154
Shepro D., Morel N.M. Pericyte physiology // FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1993. № 11 (7). P. 1031–1038.
Muramatsu R., Yamashita T. Pericyte function in the physiological central nervous system // Neuroscience Research. 2014. (81–82). P. 38–41.
Sorokin L. The impact of the extracellular matrix on inflammation // Nature Reviews. Immunology. 2010. № 10 (10). P. 712–723.
Thomsen M.S., Routhe L.J., Moos T. The vascular basement membrane in the healthy and pathological brain // Journal of Cerebral Blood Flow & Metabolism. 2017. № 10 (37). P. 3300–3317.
Wolburg H., Wolburg-Buchholz K., Fallier-Becker P. Structure and functions of aquaporin-4-based orthogonal arrays of particles // International Review of Cell and Molecular Biology. 2011. (287). P. 1–41.
Abbott N. J., Rönnbäck L., Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier // Nature Reviews. Neuroscience. 2006. № 1 (7). P. 41–53.
Lapenna A., De Palma M., Lewis C.E. Perivascular macrophages in health and disease // Nature Reviews. Immunology. 2018. № 11 (18). P. 689–702.
Ajami B., Bennett J.L., Krieger C. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life // Nature Neuroscience. 2007. № 12 (10). P. 1538–1543.
Pun P.B.L., Lu J., Moochhala S. Involvement of ROS in BBB dysfunction // Free Radical Research. 2009. № 4 (43). P. 348–364.
Mimee A., Smith P.M., Ferguson A.V. Circumventricular organs: targets for integration of circulating fluid and energy balance signals? // Physiology & Behavior. 2013. (121). P. 96–102.
Erickson M.A., Banks W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions // Pharmacological Reviews. 2018. № 2 (70). P. 278–314.
Chow B.W., Kaplan L., Granger A.J. et al. Caveolae in CNS arterioles mediate neurovascular coupling // Nature. 2020. № 7797 (579). P. 106–110.
Luissint A.-C., Artus C., Glacial F. et al. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation // Fluids and barriers of the CNS. 2012. № 1 (9): 23.
Abbott N.J. Inflammatory mediators and modulation of blood-brain barrier permeability // Cellular and Molecular Neurobiology. 2000. № 2 (20). P. 131–147.
Alluri H., Grimsley M., Anasooya Shaji C. et al. Attenuation of Blood-Brain Barrier Breakdown and Hyperpermeability by Calpain Inhibition // The Journal of Biological Chemistry. 2016. № 53 (291). P. 26958–26969.
Podjaski C., Alvarez J.I., Bourbonniere L. et al. Netrin 1 regulates blood-brain barrier function and neuroinflammation // Brain: A Journal of Neurology. 2015. № 6 (138). P. 1598–1612.
Armao D., Kornfeld M., Estrada E.Y. et al. Neutral proteases and disruption of the blood-brain barrier in rat // Brain Research. 1997. № 2 (767). P. 259–264.
Rempe R.G., Hartz A.M., Bauer B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers // Journal of Cerebral Blood Flow & Metabolism. 2016. № 9 (36). P. 1481–1507.
Guo L., Zhang H., Hou Y. Plasmalemma vesicle-associated protein: A crucial component of vascular homeostasis // Experimental and Therapeutic Medicine. 2016. № 3 (12). P. 1639–1644.
Fernandez C., Buyse M., German-Fattal M., Gimenez F. Influence of the pro-inflammatory cytokines on P-glycoprotein expression and functionality // Journal of Pharmacy & Pharmaceutical Sciences: A Publication of the Canadian Society for Pharmaceutical Sciences, Societe Canadienne Des Sciences Pharmaceutiques. 2004. № 3 (7). P. 359–371.
Sun D., Lytle C., O’Donnell M. E. IL-6 secreted by astroglial cells regulates Na-K-Cl cotransport in brain microvessel endothelial cells // The American Journal of Physiology. 1997. № 6 (272). P. C1829–1835.
Cardoso F.L., Kittel A., Veszelka S. et al. Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells // PloS One. 2012. № 5 (7): e35919.
Wang H., Sun J., Goldstein H. Human immunodeficiency virus type 1 infection increases the in vivo capacity of peripheral monocytes to cross the blood-brain barrier into the brain and the in vivo sensitivity of the blood-brain barrier to disruption by lipopolysaccharide // Journal of Virology. 2008. № 15 (82). P. 7591–7600.
Banks W.A., Gray A.M., Erickson M.A. et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit // Journal of Neuroinflammation. 2015.12:223.
Varatharaj A., Galea I. The blood-brain barrier in systemic inflammation // Brain, Behavior, and Immunity. 2017. 60:1–12.
Pan W., Kastin A.J. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice // Experimental Neurology. 2002. № 2 (174). P. 193–200.
Wang Y., Jin S., Sonobe Y. et al. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes // PloS One. 2014. № 10 (9): e110024.
Joshi G., Aluise C.D., Cole M.P. et al. Alterations in brain antioxidant enzymes and redox proteomic identification of oxidized brain proteins induced by the anti-cancer drug adriamycin: implications for oxidative stress-mediated chemobrain // Neuroscience. 2010. № 3 (166). P. 796–807.
Yarlagadda A., Alfson E., Clayton A.H. The Blood Brain Barrier and the Role of Cytokines in Neuropsychiatry // Psychiatry (Edgmont). 2009. № 11 (6). P. 18–22.
Vestweber D. How leukocytes cross the vascular endothelium // Nature Reviews. Immunology. 2015. № 11 (15). P. 692–704.
Reale M., Iarlori C., Thomas A. et al. Peripheral cytokines profile in Parkinson’s disease // Brain, Behavior, and Immunity. 2009. № 1 (23). P. 55–63.
Ching S., Zhang H., Lai W. et al. Peripheral injection of lipopolysaccharide prevents brain recruitment of leukocytes induced by central injection of interleukin-1 // Neuroscience. 2006. № 2 (137). P. 717–726.
Llovera G., Benakis C., Enzmann G. et al. The choroid plexus is a key cerebral invasion route for T cells after stroke // Acta Neuropathologica. 2017. № 6 (134). P. 851–868.
Kamimura D., Ohki T., Arima Y., Murakami M. Gateway reflex: neural activation-mediated immune cell gateways in the central nervous system // International Immunology. 2018. № 7 (30). C. 281–289.
Arima Y., Harada M., Kamimura D. et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier // Cell. 2012. № 3 (148). P. 447–457.
Bartholomäus I., Kawakami N., Odoardi F. et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions // Nature. 2009. № 7269 (462). P. 94–98.
Erickson M.A., Banks W.A. Neuroimmune Axes of the Blood–Brain Barriers and Blood–Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions // Pharmacological Reviews. 2018. № 2 (70). P. 278–314.
McCarthy R.C., Kosman D.J. Activation of C6 glioblastoma cell ceruloplasmin expression by neighboring human brain endothelia-derived interleukins in an in vitro blood-brain barrier model system // Cell communication and signaling: CCS. 2014. (12): 65.
Cai Z., Qiao P.F., Wan C.Q. et al. Role of Blood-Brain Barrier in Alzheimer’s Disease // Journal of Alzheimer’s disease: JAD. 2018. № 4 (63). P. 1223–1234.
Pan Y., Nicolazzo J.A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics // Advanced Drug Delivery Reviews. 2018. (135). P. 62–74.
Janelidze S., Lindqvist D., Francardo V. et al. Increased CSF biomarkers of angiogenesis in Parkinson disease // Neurology. 2015. № 21 (85). P. 1834–1842.
Kooij G., Kroon J., Paul D. et al. P-glycoprotein regulates trafficking of CD8(+) T cells to the brain parenchyma // Acta Neuropathologica. 2014. № 5 (127). P. 699–711.
Macrez R., Stys P.K., Vivien D. et al. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities // The Lancet Neurology. 2016. № 10 (15). P. 1089–1102.
Buzhdygan T.P., DeOre B.J., Baldwin-Leclair A. et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier // Neurobiol Dis. 2020. 146: 105131. https://doi.org/10.1016/j.nbd.2020.105131
Pellegrini L., Albecka A., Mallery D.L. et al. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids // Cell Stem Cell. 2020. № 27 (6). P. 951–961.
Rhea E.M., Logsdon A.F., Hansen K.M. et al. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice // Nat Neurosci. 2021. № 24 (3). P. 368–378.
Дополнительные материалы отсутствуют.