

Вестник РАН, 2022, T. 92, № 9, стр. 860-868
ФИЛОГЕНЕЗ МИКРОБА ЧУМЫ YERSINIA PESTIS: УНИКАЛЬНОСТЬ ЭВОЛЮЦИОННОЙ МОДЕЛИ
a Институт проблем экологии и эволюции им. А.Н. Северцова РАН
Москва, Россия
* E-mail: vvsuntsov@rambler.ru
Поступила в редакцию 20.04.2022
После доработки 27.05.2022
Принята к публикации 05.06.2022
- EDN: XTGSDE
- DOI: 10.31857/S0869587322090092
Аннотация
Возбудитель чумы – микроб Yersinia pestis – передаётся через укусы блох и поэтому занимает уникальное положение в семействе кишечных бактерий Enterobacteriaceae. Сложились два подхода к выяснению его происхождения и эволюции (филогении) – молекулярно-генетический (МГ) и экологический (ЭКО), которые базируются на разных эволюционных моделях и приводят к радикально различающимся выводам. МГ-подход прокламирует сальтационный принцип формирования этого патогена из клона псевдотуберкулёзного микроба Y. pseudotuberculosis 0:1b – возбудителя дальневосточной скарлатиноподобной лихорадки (ДСЛ) – путём встраивания, инактиваций, делеций и, реже, рекомбинаций генов и генетических структур, при этом в качестве базовой эволюционной модели принимает модель нейтральной эволюции нуклеотидных признаков-маркеров. Детали видообразовательного процесса МГ-подход не раскрывает. Согласно эволюционной ЭКО-модели, возбудитель чумы сформировался в процессе постепенных популяционно-генетических преобразований почти одновременно в трёх географических популяциях монгольского сурка (Marmota sibi-rica) из трёх различных клонов (популяций) возбудителя ДСЛ на Хэнтэе, Хангае и в Хархира-Тургенском горном массиве (Монголия). Триггером видообразовательного процесса послужил известный абиотический фактор – последнее (сартанское) максимальное похолодание климата в Центральной Азии 22–15 тыс. лет назад, вызвавшее глубокое промерзание грунта и изменение поведения блохи сурков Oropsylla silantiewi, которое, в свою очередь, привело к аберрантному травматическому заражению ДСЛ популяций монгольского сурка. Экологические факторы предписывают Y. pestis создание уникальной эволюционной модели.
Проблема возникновения и эволюции возбудителей инфекционных болезней остаётся одной из наиболее актуальных в современной инфектологии. Знание эволюционных механизмов имеет важное прагматическое значение для обеспечения контроля болезней, лечения, профилактики и прогнозирования возможных вспышек и эпидемий.
В последние полтора-два десятка лет при изучении возбудителей болезней, в том числе печально известной чумы, широко внедряются молекулярно-генетические (МГ) методы, которые во многих отношениях стали доминирующими. Современные МГ-методы изучения возбудителя чумы – микроба Yersinia pestis – сделали его диагностику по генетическим и молекулярным признакам в полной мере совершенной, позволяют охарактеризовать как конкретные генотипы (штаммы, клоны), так и геном вида Y. pestis в целом и его отдельные геноварианты (популяции/подвиды) [1–4]. К настоящему времени по биохимическим, генетическим и молекулярным признакам идентифицированы геноварианты возбудителя из большого числа природных очагов мира [5]. Расширяется исследование “археологической” ДНК, извлекаемой из останков (костей, зубов) жертв минувших пандемий [6, 7].
Сравнительный анализ современных и археологических геновариантов позволил предпринять попытку реконструкции всемирной истории интересующего нас инфекционного агента – его филогении и филогеографии [5–10]. Однако выводы МГ-филогенетических исследований пока вызывают оправданные сомнения, так как не поддаются интерпретации в соответствии с положениями синтетической теории эволюции и противоречат многочисленным фактам, представленным классическими естественно-научными направлениями – экологией, эпизоотологией, биогеографией, палеонтологией [11]. Несмотря на большое число созданных филогенетических схем, МГ-подход не даёт доверительного ответа на тривиальные вопросы: когда, где, каким образом и при каких условиях прошло формирование возбудителя чумы как вида Y. pestis. Одной из главных причин, вызывающих непреодолимые трудности в экологической и исторической интерпретации выводов МГ-подхода, помимо игнорирования классических данных и методических ошибок (например, некорректный выбор референтного штамма и/или внешней группы), видится использование для МГ-реконструкции возбудителя чумы неадекватной эволюционной модели, на основе которой проводится филогенетический анализ выбранных признаков (маркеров) [12]. Адекватная модель может быть разработана только путём интеграции всесторонних знаний о возбудителе чумы и его непосредственном предке – псевдотуберкулёзном микробе Y. pseudotuberculosis 0:1b (семейство Enterobacteriaceae), или, точнее, возбудителе дальневосточной скарлатиноподобной лихорадки (ДСЛ) [13].
Классическая модель видообразования Y. pestis. Согласно положениям классической версии теории природной очаговости чумы, вполне оформившейся к началу второй половины XX в., чуму считали очень древней инфекцией, возникшей в процессе коэволюции норовых грызунов (Rodentia), паразитирующих на них блох (Siphonaptera) и неких свободноживущих микроорганизмов или паразитических кишечных бактерий. Исходный хозяин возбудителя чумы не известен, предпочтение отдавали суркам (Marmotini) или песчанкам (Gerbillidae). Время возникновения возбудителя относили к широкому временно́му диапазону – олигоцену–плиоцену, от 30 до 5 млн лет назад. Местом происхождения считали или аридные районы Старого Света – родину песчанок, или Северную Америку – родину сурков. Широкое распространение очагов чумы в Старом и Новом свете связывали с древними межконтинентальными миграциями норовых грызунов – хозяев инфекции по сухопутным мостам – Берингийскому, Синайскому и Панамскому, периодически возникавшим в геологической истории. В настоящее время некоторые положения этой теории или полностью отвергнуты, или претерпели значительные изменения.
МГ-модель видообразования Y. pestis. В последние два-три десятилетия в медико-биологическую науку стала интенсивно внедряться МГ-методология исследований. В филогенетике возбудителей инфекционных болезней МГ-модели имеют две относительно самостоятельные составляющие: генетическую, имеющую отношение к адаптивным признакам, и молекулярную, относящуюся к филогенетическим признакам. В реконструкции филогении Y. pestis молекулярная модель стала базовой, задающей топологию филогенетического дерева, а генетическая – дополняющей её, придающей ей некоторое биологическое содержание и адаптационные детали.
Благодаря внедрению МГ-методологии в исследование возбудителя чумы удалось сделать два краеугольных открытия. Во-первых, изучением О-антигена было надёжно показано, что его прямым предком является возбудитель ДСЛ [14]. Таким образом, введение в филогенетику вида Y. pestis концепции внешней группы, которой оперирует МГ-методология для фиксации предковой формы, оказалось избыточным. Изучен регион доминантного распространения ДСЛ – обширные холодные пространства Северной Азии, Сибири, Дальнего Востока, Центральной Азии, она встречается в Японии и редко в Канаде [15, 16]. Был сформулирован первый базовый тезис: исходная популяция Y. pestis дивергировала от клона (популяции) Y. pseudotuberculosis 0:1b в некотором холодном регионе Азии.
Во-вторых, вопреки положениям классической теории природной очаговости чумы о геологической древности возбудителя, молекулярными методами была показана его эволюционная молодость: дивергенция исходной популяции чумного микроба от анцестрального клона ДСЛ имела место не ранее 30 тыс. лет назад [8–10]. Стал бесспорным второй базовый тезис: виновник чумы возник в конце плейстоцена или в голоцене в холодных районах Азии и естественным образом распространился только в Евразии, а на Африканский материк и в Новый Свет проник много позднее из-за хозяйственно-экономической деятельности человека. Здесь уместно отметить важный факт: рубеж плейстоцена и голоцена в Азии характеризовался максимальным (сартанским) похолоданием, наступившим 22–15 тыс. лет назад. В это время в Монголии впервые за последние десятки миллионов лет среднегодовая температура воздуха опустилась до – 6°С, грунт стал промерзать на глубину более 3 м, граница вечной мерзлоты проникла на юг до пустыни Гоби и достигла 42° с.ш., полностью охватив ареал монгольского сурка (Marmota sibirica) [17]. Как мы увидим ниже, похолодание в Азии, вызвавшее определённые эволюционные сдвиги в центральноазиатских биогеоценозах, должно было стать важным абиотическим компонентом эволюционной модели Y. pestis.
К сожалению, наряду с названными очевидными достижениями МГ-подход включает крайне деструктивную позицию – прокламирует сальтационное преобразование клона возбудителя ДСЛ в возбудителя чумы несколькими одномоментными генетическими актами – встраиванием в псевдотуберкулёзную клетку путём горизонтального переноса генов (ГПГ) из внешней среды или от других микроорганизмов сложных генетических структур – полифункциональных плазмид pFra и pPst, кодирующих специфические только для чумного микроба функции вирулентности, трансмиссии и коммуникации и острова высокой патогенности (HPI). При этом признаётся, что в геноме чумного микроба, в сравнении с псевдотуберкулёзным, произошли многочисленные и радикальные перестройки, вызвавшие изменения глубинных метаболических процессов, таких как обмен железа и кислорода [18, 19], что не согласуется с сальтационистскими взглядами.
В качестве базовой эволюционной МГ-модели, задающей топологию филогенетического дерева Y. pestis, принимают модель нейтральной эволюции, в соответствии с которой проводят статистический анализ нейтральных молекулярных признаков-маркеров (IS, DFR, VNTR, SNP, CRISPR и др.) [2–5]. Модель предполагает сначала возникновение из клона ДСЛ абстрактного общего предка MRCA (most recent common ancestor) [8, 20, 21], при этом гостально-векторная среда обитания и популяционные характеристики MRCA не обсуждаются. Безликий MRCA охарактеризован только статистически признаками-маркерами, не детерминированными функционально (экологическими функциями) (рис. 1). Остаётся непонятным, можно ли относить MRCA к уже состоявшемуся виду Y. pestis или это пока ещё переходная форма Y. pseudotuberculosis / Y. pestis. Таким образом, МГ-подход на филогенетическом дереве Y. pestis чётко фиксирует анцестральный вид, но уникальные особенности видообразовательного процесса не раскрывает, априори принимая всё внутривидовое разнообразие Y. pestis в качестве монофилетической (голофилетической) группы, исходящей из безликого единого предка MRCA.
Рис. 1.
Типичное филогенетическое дерево Yersinia pestis [21], топология которого задана статистическим анализом SNP-признаков
Генетические изменения (встраивание, делеции, инактивации генов) отчасти насыщают филогенетическую схему биологическим содержанием
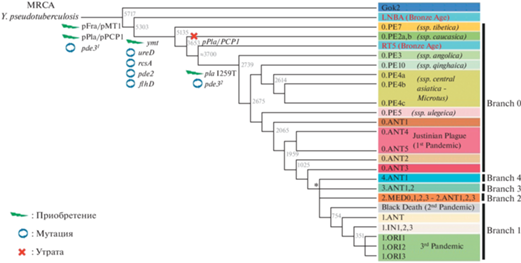
Полагают, что MRCA стал основателем наиболее древних “археологических” форм Gok2 (Готем, Швеция), LNBA (поздний неолит, бронзовый век, Европа–Азия) и других, существовавших в позднем неолите и бронзовом веке и охарактеризованных по фрагментам ДНК, извлечённым из останков человеческих жертв древних пандемий [6, 7, 21–23]. Фрагменты ДНК по нуклеотидной структуре оказались близкими к гомологичным генным структурам возбудителя псевдотуберкулёза 1-го серотипа и возбудителя чумы, относящегося к биовару Pestoides, циркулирующего в популяциях полёвок (Microtina) и монгольской пищухи (Ochotona pallasi pricei). Поэтому образцы “археологической” ДНК помещены в корень филогенетического дерева Y. pestis. В современных природных очагах чумы “археологические” ДНК-маркеры не выявлены. Отсюда пошло мнение, что древние возбудители чумы вымерли по неизвестным причинам, эволюционные процессы их формирования из MRCA не ясны. Таким образом, в МГ-подходе предполагается наличие объективных трудностей в выборе адекватной эволюционной модели.
Среди ныне существующих геновариантов, согласно молекулярным выводам, базальные позиции на филогенетическом дереве Y. pestis занимают геноварианты кластера 0.PE, образованного возбудителями биовара Pestoides, который включает штаммы, вирулентные для Microtina, но не вирулентные или слабовирулентные для сусликов, сурков, песчанок и человека. Набор активных генов у этих геновариантов богаче, чем у археологических находок [22, 24], что, казалось бы, соответствует идее о происхождении биовара Pestoides от археологических форм. На основании описанных молекулярных и генетических свойств экологически не детерминированные “археологические” находки ДНК чумного микроба из человеческих останков жертв древних пандемий считают принадлежащими вымершим переходным формам Y. pseudotuberculosis / Y. pestis. “Археологическую” ДНК чумного микроба из останков диких грызунов или синантропных крыс пока выявить и изучить не удалось.
Приведённые выше доводы разъясняют, почему в МГ-подходе не делается попыток более точно установить характеристики переходных форм Y. pseudotuberculosis / Y. pestis, которые могли бы пролить свет на механизм видообразовательного процесса и нестабильную переходную среду, где проходило преобразование популяции (клона) Y. pseudotuberculosis 0:1b в популяцию MRCA. Согласно МГ-подходу нестабильной переходной среды и соответствующих этой среде переходных форм попросту не существовало. Видообразование, как полагают, прошло сальтационным способом – молниеносным, в мгновение ока, внедрением микроба сразу в новую, уже существовавшую среду обитания “грызун–блоха”, то есть в новую экологическую нишу и новую адаптивную зону. Механизм видообразования связывают с генетико-статистическими нейтральными процессами (ГПГ, дрейф генов) без или с минимальным участием главных факторов эволюции – борьбы за существование и естественного отбора [12]. При этом ранние формы чумного микроба по неизвестным пока обстоятельствам вымерли, что уменьшает возможности раскрытия их свойств как переходных форм между предковой и производной и затрудняет создание эволюционной модели, адекватной процессу видообразования Y. pestis.
Оценивая МГ-эволюционную модель в целом, следует констатировать, что она, во-первых, не учитывает уникальность видообразовательного процесса при формировании популяции MRCA из клона (популяции) возбудителя ДСЛ и, во-вторых, априори придаёт многообразным геновариантам чумного микроба статус единой монофилетической (голофилетической) группы (не имеющей гомоплазийных проявлений).
Экологическая эволюционная модель филогении Y. pestis. Возбудитель чумы был открыт А. Йерсеном в Гонконге в 1894 г. в начале третьей пандемии. За прошедшие более чем 125 лет наукой накоплен огромный объём всесторонних знаний, позволяющих в той или иной мере реконструировать его историю. Во второй половине XX в. была сформулирована вполне стройная теория природной очаговости чумы. С началом нового тысячелетия эта теория была откорректирована МГ-новациями. Чума предстала как эволюционно молодая системная (“кровяная”) инфекция, возбудитель которой как вид Y. pestis сформировался на основе определённого клона (популяции) кишечного психрофильного возбудителя ДСЛ Y. pseudotuberculosis 0:1b. В связи с эволюционной молодостью возбудителя чумы имеется реальная возможность раскрыть популяционно-генетические процессы видообразования на основе знаний экологии популяций предкового псевдотуберкулёзного и производного чумного микробов [13]. Трудность вопроса состояла в отсутствии представлений об исходном хозяине: в популяциях какого вида фоновых норовых грызунов клон (популяция) возбудителя ДСЛ мог преобразоваться в популяцию возбудителя чумы? Экологические факты, позволяющие дать уверенный ответ на этот вопрос, были получены при изучении природных очагов чумы в Центральной Азии [13, 25–27].
Согласно экологическому сценарию, видообразование Y. pestis прошло в Центральной Азии в популяциях монгольского сурка под влиянием разновременно действовавших тривиальных физико-климатических факторов – нарастания аридности климата в продолжение кайнозоя и суровости климата во время последнего максимального (сартанского) похолодания в Азии. Аридность климата привела к формированию защитного поведения монгольского сурка, ставшего причиной интенсивного размножения псевдотуберкулёзного микроба в его пищеварительном тракте, точнее, только в ротовой полости, во время зимней спячки без проникновения в кишечный тракт и лимфо-миелоидный комплекс и (надо полагать) без возникновения инфекционного процесса. Суровость климата в сартанское время, 22–15 тыс. лет назад, вызвала сдвиги в поведении личинок сурочьей блохи Oropsylla silantiewi. В обычных условиях личинки блох являются детритофагами, питаются органическим субстратом в гнёздах хозяев. Но с началом сартанского похолодания и глубокого промерзания почвогрунтов до 3–4 м личинки O. silantiewi из промерзающей в зимне-весенние месяцы травяной выстилки зимовочных гнёзд сурка (–3 – –8°С) стали перемещаться в силу положительного термотаксиса на более тёплые тела спящих животных (5–37°С). При этом в поисках пищи часть личинок со стохастической закономерностью попадала в ротовую полость сурков, где они создавали на слизистой скарификации и питались кровью [25–27]. Так, смена способа питания блошиных личинок в холодный зимне-весенний период – переход от детритофагии к гематофагии – привела к уникальному травматическому (не традиционному алиментарному) способу заражения монгольского сурка ДСЛ. В свою очередь необычный способ массового заражения популяции спящих монгольских сурков ДСЛ стал причиной возникновения уникальной “кровяной” инфекции – чумы (рис. 2).
Рис. 2.
Этапы формирования трансмиссивной передачи чумы:
I – накопление возбудителя псевдотуберкулёза в ротовой полости готовящихся к спячке монгольских сурков как следствие аридизации ландшафтов Центральной Азии; II – аберрантное (травматическое) заражение сурков псевдотуберкулёзом и сепсис во время зимней спячки как следствие наступления сартанского максимального похолодания в Азии и перехода личинок сурковой блохи к факультативной гематофагии; возникновение переходной формы возбудителя Yersinia pseudotuberculosis /Yersinia pestis; III – адаптация переходной формы к персистированию в популяциях монгольского сурка, находящегося в активном состоянии; освоение новой экологической ниши и адаптивной зоны, становление нового вида Y. pestis (Тт – температура тела сурков)
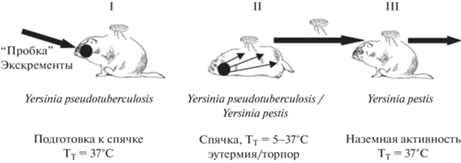
Причиной видообразования Y. pestis стал рекуррентный массовый механический вынос псевдотуберкулёзных микробных клеток в принципиально новую среду обитания (из ротовой полости в кровь), то есть в новую экологическую нишу и адаптивную зону. Э. Майр [28] назвал формирование новых популяций за счёт внепопуляционных мигрантов принципом основателя, а процесс формирования нового вида описал как перипатрическое видообразование. Подобное отпочкование нового дочернего вида от предкового посредством мигрантов, которое в природе обычно происходит с высокой эволюционной скоростью, В. Грант определил как “квантовое” видообразование [29]. Таким образом, при изучении видообразования и филогенеза возбудителя чумы базовая эволюционная модель должна соответствовать принципу перипатрического/квантового видообразования [30].
Чума не является сапрозоонозом. Важной экологической составляющей эволюционной модели Y. pestis выступает жизненный цикл: сохраняет ли микроб чумы адаптации к сапрофитическому существованию вне системы “грызун–блоха”, свойственные его псевдотуберкулёзному предку? В.В. Кутырев с соавторами пришли к выводу (по нашим представлениям – глубоко ошибочному!), что возбудитель чумы, как и его предок (возбудитель ДСЛ), по сути, является сапрозоонозным микроорганизмом, его жизненный цикл включает сапрофитическую фазу развития во внешней органике: через трансларвальные стадии почвенных нематод и блох микроб внедряется в трансмиссивный цикл “грызун–блоха–грызун” во время разлитых эпизоотий, и “в настоящее время уже нельзя отрицать очевидной роли членов почвенного биоценоза в инициации эпизоотий чумы” [31, с. 11]. Это сомнительное открытие российских чумологов, казалось бы, нашло поддержку в генетических фактах зарубежных исследователей. В соответствии с идеей “теллурической” чумы было высказано предположение, что возможность сапрофитического существования у микроба чумы связана с иным, чем у возбудителя ДСЛ, механизмом – горизонтальной одноактной аквизицией плазмиды pPst, кодирующей синтез пестицина [32]. Пестицин, как считают, обеспечивает существование возбудителя чумы в трупах и почве в межэпизоотический период. Распространяясь по всему трупу, пестицин позволяет патогену длительно сохраняться вне организма теплокровных хозяев и далее передаваться обратно в цикл “блоха–грызун” из трупа или почвы в хозяина или личинки блох.
Эволюционную модель “сапрозоонозной” или “теллурической” чумы нельзя считать общепринятой в МГ-подходе, но тем не менее при построении МГ-филогенетических схем Y. pestis в качестве базовых заимствуют модели, разработанные для других, не трансмиссивных инфекций, вызываемых микробами Escherichia coli, Mycobacterium tuberculosis, Salmonella typhi, Yersinia pseudotuberculosis и пр. [18, 33], жизненный цикл которых резко контрастирует с уникальным жизненным циклом Y. pestis. Это ставит под сомнение адекватность применяемых для реконструкции филогенеза Y. pestis МГ-моделей.
Иную, коммуникационную, роль в процессе видообразования микроба чумы отводит пестицину экологический сценарий. Плазмида pPst синтезирована на поздних стадиях видообразования [34]. Её роль, помимо регуляции синтеза фермента Pla, выполняющего функцию вирулентности, состоит в обеспечении популяционной стабильности и внутри- и межвидовой регуляции. Кроме гена pla pPst-плазмида несёт два других гена – pst и pim, образующих оперон, кодирующий функцию системы “токсин–антитоксин”, которая отвечает за регуляцию в стрессовых условиях. Такие системы известны для большого числа прокариот [35]. Назначение токсина (у чумного микроба это пестицин, Pst) – подавлять рост мутантных генотипов. Роль антитоксина (Pim) – нейтрализовать токсин. Нейтрализация осуществляется сложным взаимодействием двух белков, токсина и антитоксина. Когда клетка теряет оперон pst-pim, она обычно погибает. Поэтому в процессе видообразования клетки переходной популяции Y. pseudotuberculosis / Y. pestis всё в большей мере синтезировали пестицин и подавляли рост анцестральных псевдотуберкулёзных клеток и собственных более примитивных клеток, не синтезирующих или плохо синтезирующих пестицин [30]. Так в популяции Y. pseudotuberculosis / Y. pestis происходил отбор клеток, успешно синтезирующих пестицин. Роль пестицина как регулятора внутривидовых взаимоотношений в популяциях состоявшегося микроба чумы и межвидовых взаимодействий с предковым псевдотуберкулёзным микробом свидетельствует о сложном длительном процессе видообразования Y. pestis через переходные формы в нестабильной стрессовой переходной гостально-векторной среде “сурок–блоха”, но не об одноактном внедрении плазмиды pPst в псевдотуберкулёзную клетку путём ГПГ. Отсюда, в соответствии с экологическим сценарием, эволюционная модель Y. pestis не должна включать сапрофитическую компоненту и концепцию ГПГ.
Три района видообразования Y. pestis. Экологическими методами показано, что микроб чумы сформировался в паразитарной системе “монгольский сурок – блоха O. silantiewi”. При этом видообразование прошло параллельно, почти одновременно в трёх географических популяциях сурка [11, 36]. На Хэнтэе, в Забайкалье и в китайской провинции Внутренняя Монголия обитает подвид (географическая популяция) M. sibirica sibirica. Хангай и Центральную Монголию заселяет подвид M. sibirica caliginosus. В западных районах Монголии и в Тыве на Хархира-Турген-Монгун-Тайгинском горном поднятии обитает пока не описанный подвид монгольского сурка M. sibirica ssp. В этих географических популяциях монгольского сурка имеются “сурочьи” природные очаги чумы, в которых циркулируют разные геноварианты микроба (2.ANT3, 3.ANT2 и 4.ANT1). Видообразование, индуцированное последним максимальным (сартанским) похолоданием в Центральной Азии, прошло тритопно в сходных условиях в единой среде “M. sibirica – O. silantiewi”, но геноварианты возникли заметно разные. Причину этого можно видеть в генетических и молекулярных отличиях популяций возбудителя ДСЛ, циркулирующего в разных географических популяциях монгольского сурка, и случайных процессах, таких как дрейф генов переходной формы Y. pseudotuberculosis / Y. pestis в популяциях монгольского сурка [11].
Территориальная экспансия трёх исходных геновариантов из географических популяций монгольского сурка проходила самостоятельными параллельными маршрутами, из чего следует, что филогенетическое дерево Y. pestis имеет три корня, представленных геновариантами 2.ANT3, 3.ANT2 и 4.ANT1, и структура филогенетического дерева включает три самостоятельные голофилетические группы (рис. 3). В таком случае сходные параллельные мутации, создающие похожие признаки у представителей самостоятельных голофилетических групп, следует оценивать как гомоплазии. Гомоплазии не отражают наследственную изменчивость и не являются филогенетически информативными признаками, неверно истолковываются филогенетическими методами, и их следует исключать из МГ-филогенетического анализа.
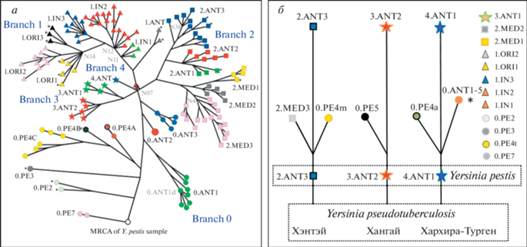
Таким образом, в МГ-подходе всё известное многообразие геновариантов возбудителя чумы, порождённое исходной формой MRCA, рассматривают как монофилетическую (голофилетическую) группу. Тем самым в реконструкцию истории Y. pestis закладывается принципиальная ошибка. Согласно экологическому сценарию, эволюционная модель должна иметь трёхкорневую топологию, включать три голофилетические группы, которые следует анализировать отдельно во избежание включения в филогенетический анализ случаев эволюционного параллелизма (гомоплазий).
* * *
Возбудитель чумы по биохимическим, генетическим и молекулярным признакам систематически относят к семейству кишечных бактерий Enterobacteriaceae, и в филогенетических построениях используют общепринятые эволюционные модели, характеризующие эволюционные процессы в популяциях возбудителей кишечных (или иных нетрансмиссивных) инфекций. В то же время этот патоген проявляет особые свойства, свидетельствующие о его уникальной эволюционной судьбе. Но эту уникальность не учитывают в МГ-методологии филогенетических реконструкций. Вид Y. pestis – действительно монофилетическая группа, но в широком понимании этого термина. Она имеет единого псевдотуберкулёзного предка – вид Y. pseudotuberculosis, предковый вид породил производный вид. Но дьявол кроется в деталях. Три различные популяции Y. pseudotuberculosis (ДСЛ) дали жизнь трём популяциям Y. pestis, и каждая из них основала самостоятельную голофилетическую группу действительно напрямую родственных геновариантов, то есть группу монофилетическую в узкой трактовке. Геноварианты из разных голофилетических групп не являются напрямую родственными, их родство опосредуется псевдотуберкулёзными предками. Налицо следующая ситуация: три основателя голофилетических групп, геноварианты 2.ANT3, 3.ANT2 и 4.ANT1, проявляют единство эволюционной тенденции, но не связанны единством происхождения (пара- или перипатрическое образование геновариантов, подвидов, географических популяций). Отсюда результат – МГ-филогении не интерпретируются с экологических (в широком понимании) позиций, убедительную интерпретацию истории чумного микроба на основе МГ-методологии создать не удаётся. Наоборот, экологические данные, в совокупности с генетическими и молекулярными, позволили предложить вполне оправданный ЭКО-сценарий происхождения и эволюции чумного микроба. Этот сценарий следует принять в качестве нулевой гипотезы при разработке адекватной эволюционной модели для реконструкции истории происхождения и эволюции вида Y. pestis. ЭКО-сценарий также может стать незаменимой вербальной эволюционной моделью для сравнения молекулярных и экологических методологий филогенетических исследований возбудителей многих других инфекционных болезней и создания эколого-молекулярного методологического синтеза.
Список литературы
Платонов М.Е., Евсеева В.В., Дентовская С.В., Анисимов А.П. Молекулярное типирование Yersinia pestis // Молекулярная генетика, микробиология и вирусология. 2013. № 2. С. 3–13.
Vogler A.J., Keim P., Wagner D.M. A review of methods for subtyping Yersinia pestis: From phenotypes to whole genome sequencing // Infect. Genet. Evol. 2016. V. 37. P. 21–36.
Вагайская А.С., Трунякова А.С., Дентовская С.В. Внутривидовая дифференциация Yersinia pestis: от фенотипа к полногеномному секвенированию // Бактериология. 2019. № 2. С. 42–54.
Кисличкина А.А., Платонов М.Е., Вагайская А.С. и др. Рациональная таксономия Yersinia pestis // Молекулярная генетика, микробиология и вирусология. 2019. № 2. С. 76–82.
Cui Y., Yu C., Yan Y. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis // PNAS. 2013. № 2. P. 577–582.
Spyrou M.A., Keller M., Tukhbatova R.I. et al. Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes // Nature Commun. 2019. V. 10:4470. https://doi.org/10.1038/s41467-019-12154-0
Rascovan N., Sjogren K.G., Kristiansen K. et al. Emergence and Spread of Basal Lineages of Yersinia pestis during the Neolithic Decline // Cell. 2019. № 176. P. 295–305.
Achtman M., Morelli G., Zhu P. et al. Microevolution and history of the plague bacillus, Yersinia pestis // PNAS. 2004. № 51. P. 17837–17842.
Morelli G., Song Y., Mazzoni C.J. et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity // Nature Genetics. 2010. № 12. P. 1140–1145.
Pisarenko S.V., Evchenko A.Yu., Kovalev D.A. et al. Yersinia pestis strains isolated in natural plague foci of Caucasus and Transcaucasia in the context of the global evolution of species // Genomics. 2021. V. 113. P. 1952–1961.
Сунцов В.В. Политопное видообразование микроба чумы Yersinia pestis как причина филогенетической трихотомии в географических популяциях монгольского сурка-тарбагана (Marmota sibirica) // Журнал общей биологии. 2021. № 6. С. 431–444.
Cui Y., Song Y. Chapter 6. Genome and Evolution of Yersinia pestis / R. Yang, A. Anisimov (eds.) // Yersinia pestis: Retrospective and Perspective. Advances in Experimental Medicine and Diology 918. Beijing: Springer Sceince-Business Media Dordrecht, 2016. P. 171–192.
Сунцов В.В. Происхождение чумы. Перспективы эколого-молекулярно-генетического синтеза // Вестник РАН. 2019. № 3. С. 260–269; Suntsov V.V. Origin of the Plague: Prospects of Ecological–Molecular–Genetic Synthesis // Herald of the Russian Academy of Sciences. 2019. № 3. P. 271–278.
Skurnik M., Peippo A., Ervela E. Characterization of the O-antigen gene cluster of Yersinia pseudotuberculosis and the cryptic O-antigen gene cluster of Yersinia pestis shows that the plague bacillus is most closely related to and has evolved from Y. pseudotuberculosis serotype O:1b // Mol. Microbiol. 2000. № 2. P. 316–330.
Fukushima H., Gomyoda M., Hashimoto N. et al. Putative origin of Yersinia pseudotuberculosis in western and eastern countries. A comparison of restriction endonuclease analysis of virulence plasmids // Int. J. Med. Microbiol. 1999. V. 288. P. 93–102.
Fukushima H., Matsuda Y., Seki R. et al. Geographical heterogeneity between Far Eastern and Western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains // J. Clin. Microbiol. 2001. № 10. P. 3541–3547.
Owen L.A., Richards B., Rhodes E.J. et al. Relict permafrost structures in the Gobi of Mongolia: age and signi-ficance // J. Quat. Sci. 1998. № 6. P. 539–547.
Chain P.S.G., Carniel E., Larimer F.W. et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis // PNAS. 2004. № 38. P. 13826–13831.
Willcocks S.J., Stabler R.A., Atkins H.S. et al. High-throughput analysis of Yersinia pseudotuberculosis gene essentiality in optimised in vitro conditions, and implications for the speciation of Yersinia pestis // BMC Microbiology. 2018. V. 18. Art. 46.
Achtman M. Insights from genomic comparisons of genetically monomorphic bacterial pathogens // Phil. Trans. R. Soc. B. 2012. № 367. P. 860–867.
Demeure C.E., Dussurget O., Fiol G.M. et al. Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics // Genes Immun. 2019. № 5. P. 357–370.
Rasmussen S., Allentoft M.E., Nielsen K. et al. Early divergent strains of Yersinia pestis in Eurasia 5.000 years ago // Cell. 2015. № 163:571e82.
Valtuena A.A., Neumann G.U., Spyrou M.A. et al. Stone Age Yersinia pestis genomes shed light on the early evolution, diversity, and ecology of plague // PNAS. 2022. № 17. e2116722119.
Sebbane F., Devalckenaere A., Foulon J. et al. Silencing and reactivation of urease in Yersinia pestis is determing by one G residue at the specific position in the ureD gene // Infect. Immun. 2001. V. 69. P. 170–176.
Сунцов В.В., Сунцова Н.И. Экологические аспекты эволюции микроба чумы Yersinia pestis и генезис природных очагов // Известия РАН. Серия биологическая. 2000. № 6. С. 645–657.
Сунцов В.В., Сунцова Н.И. Чума. Происхождение и эволюция эпизоотической системы (экологические, географические и социальные аспекты). М.: КМК, 2006.
Сунцов В.В. Исключительная роль специфической блохи сурков Oropsylla silantiewi (Ceratophyllidae: Siphonaptera) в видообразованиии микроба чумы – микроба Yersinia pestis // Паразитология. 2018. № 1. С. 3–18.
Майр Э. Популяции, виды и эволюция. М.: Мир, 1974.
Грант В. Эволюционный процесс. М.: Мир, 1991.
Сунцов В.В. “Квантовое” видообразование микроба чумы Yersinia pestis в гетероиммунной среде – популяциях гибернирующих сурков-тарбаганов (Marmota sibirica) // Сибирский экологический журнал. 2018. № 4. С. 379–394.
Кутырев В.В., Ерошенко Г.А., Попов Н.В. и др. Молекулярные механизмы взаимодействия возбудителя чумы с беспозвоночными животными // Молекулярная генетика, микробиология и вирусология. 2009. № 4. С. 6–13.
Easterday W.R., Kausrud K.L., Star B. et al. An additional step in the transmission of Yersinia pestis? // ISME Journal. 2012. V. 6. P. 231–236.
McNally A., Thomson N.R., Reuter S., Wren B.W. “Add, stir and reduce”: Yersinia spp. As model bacteria for pathogen evolution // Nat. Rev. Microbiol. 2016. Advance online publication. P. 1–15. https://doi.org/10.1038/nrmicro.2015.29
Сунцов В.В. Перспективы синтеза молекулярно-генетического и экологического подходов к проблеме видообразования микроба чумы Yersinia pestis // Успехи современной биологии. 2020. № 1. С. 43–57.
Кунин Е. Логика случая. О природе и происхождении биологической эволюции. М.: Центрполиграф, 2014.
Сунцов В.В. Гостальный аспект территориальной экспансии микроба чумы Yersinia pestis из популяций монгольского сурка-тарбагана (Marmota sibirica) // Зоологический журнал. 2020. № 11. С. 1307–1320.
Дополнительные материалы отсутствуют.