

Высокомолекулярные соединения (серия А), 2019, T. 61, № 3, стр. 232-243
МАГНИТОЧУВСТВИТЕЛЬНЫЕ МУЛЬТИЛИПОСОМАЛЬНЫЕ КОНТЕЙНЕРЫ ДЛЯ ИММОБИЛИЗАЦИИ И КОНТРОЛИРУЕМОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
В. В. Спиридонов a, *, И. Г. Панова a, А. В. Сыбачин a, В. В. Кузнецов a, М. И. Афанасов a, Ю. А. Алёхина b, Н. С. Мелик-Нубаров a, А. А. Ярославов a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, Россия
b Московский государственный университет имени М.В. Ломоносова. Физический факультет
119991 Москва, Ленинские горы, Россия
* E-mail: vasya_spiridonov@mail.ru
Поступила в редакцию 08.11.2018
После доработки 10.12.2018
Принята к публикации 24.12.2018
Аннотация
Представлены мультилипосомальные контейнеры, образованные путем последовательной адсорбции катионного полипептида поли-L-лизина и анионных липосом на поверхность анионных магнитных микрогелей – продуктов синтеза γ-Fe2O3 (маггемита) в матрице анионного природного полисахарида, смешанной Na/Ca соли альгиновой кислоты. Состав и морфология магниточувствительных микрогелей подтверждены данными рентгенофазового анализа, ИК-фурье-спектроскопии, Мессбауэровской спектроскопии, магнитометрии, просвечивающей электронной микроскопии. Размер частиц мультилипосомальных контейнеров с предельным содержанием липосом в физиологическом растворе около 400 нм. Они не цитотоксичны вплоть до концентрации поли-L-лизина 600 мкМ. Показано, что данные объекты, заполненные противоопухолевым антибиотиком – доксорубицином, способны перемещаться в суспензии под действием внешнего магнитного поля, при этом целостность липосом в составе контейнера сохраняется. Полученные контейнеры представляют интерес для контролируемой доставки биологически активных (лекарственных) веществ.
ВВЕДЕНИЕ
Инкапсулирование лекарственного вещества в наноразмерном носителе позволяет решить несколько принципиальных вопросов, связанных с перспективой лекарства в биологическом окружении. Это касается длительности циркуляции лекарства в кровотоке, направленного транспорта лекарства к целевым клеткам и органам, его контролируемого высвобождения в адресной зоне, предотвращения преждевременного разложения лекарства ферментами, повышения биодоступности малорастворимых лекарств [1–4]. В качестве носителей можно использовать полимерные гидрогели и мицеллы, дендримеры, мезопористые наночастицы оксида кремния, металлические наночастицы, наноэмульсии типа “масло в воде” [5–11]. Особый интерес для инкапсулирования лекарств представляют сферические везикулы, бислойная липидная мембрана которых отделяет внутренний водный объем частицы от окружающего водного раствора [12]. Такие везикулы (иначе называемые липосомами) биосовместимы, биодеградируемы и способны инкапсулировать гидрофильные и гидрофобные вещества [12, 13]; размер и поверхностный заряд липосом легко поддаются контролю [13]. Для получения липосом разработаны простые методические приемы, в том числе ультразвуковая обработка смешанной водно-липидной суспензии и пропускание суспензии через фильтр с калиброванным размером пор [14]. Липосомальные формы лекарств продемонстрировали свою эффективность при лечении ряда тяжелых заболеваний [15–17].
Существенным ограничением традиционных липосом с диаметром от 50 до 100 нм является их малый внутренний объем, определяющий предельную емкость липосомального контейнера [18]. Увеличить эффективный объем контейнера можно, концентрируя малые липосомы с инкапсулированным лекарством в небольшом объеме путем связывания их с поверхностью коллоидного носителя [19–22]: полимерных микросфер с привитыми поликатионными цепями (“сферических поликатионных щеток”), полилактидных мицелл, хитозановых микрогелей. Такой прием позволяет связать несколько десятков интактных липосом на каждой коллоидной частице [23, 24], при этом цитотоксичность полученных мультилипосомальных контейнеров мало отличается от цитотоксичности исходных липосом [19, 25].
Для повышения эффективности терапевтического действия липосомального лекарства его “нацеливают” на определенные (воспаленные) области организма. Поверхность липосом модифицируют “узнающими” (векторными) молекулами, специфически взаимодействующими с комплементарными рецепторами на поверхности клеток-мишеней [26]. Другой подход состоит в том, чтобы сделать липосомы чувствительными к внешнему воздействию, так называемому “стимулу” (температуре, микроволновому излучению, магнитному полю), адресно воздействующему на воспаленную область [27–32].
В настоящей работе представлены мультилипосомальные контейнеры, образованные путем электростатического связывания анионных липосом на поверхности биодеградируемого полимерного микрогеля с инкапсулированными наночастицами маггемита, γ-Fe2O3. Встраивание маггемита в микрогель придает мультилипосомальным контейнерам чувствительность к внешнему магнитному полю, что позволяет концентрировать их в области, задаваемой положением внешнего магнитного устройства.
Исследована природа взаимодействия полимера с наночастицами маггемита, а также агрегативная устойчивость и цитотоксичность мультилипосомальных контейнеров; получен магнитоуправляемый мультилипосомальный контейнер, заполненный противоопухолевым антибиотиком – доксорубицином. Описанный подход демонстрирует возможности использования мультилипосомальных контейнеров для инкапсулирования и контролируемого транспорта биологически активных веществ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе применяли реагенты – соль Мора (NH4)2Fe(SO4)2 ⋅ 6H2O и хлороформ квалификации х.ч., серную кислоту квалификации о.с.ч. – все производства “Реахим” (Россия); альгинат Na 95%–Ca 5% (“ISP”, Великобритания), который очищали проточным диализом и выделяли с помощью лиофильной сушки. Использовали гипофосфит натрия моногидрат NaH2PO2 ⋅ H2O (“VKLaborundFeinchemikalien”, Германия) и гидроксид натрия квалификации ч.д.а. (“Riedel-deHaen”, Чехия), поли-L-лизин гидробромид (ПЛ) и доксорубицин гидрохлорид фирмы “Sigma-Аldrich” (США); а также липиды: диолеоилфосфатидилхолин (ФХ), пальмитоилолеоилфосфатидилсерин (ФС), диолеоилфосфоэтаноламин-N-(лиссамин родамин-Б-сульфонил) аммоний (Л*) – все фирмы “Avanti” (США).
Получение магнитных микрогелей
Синтез композиционного материала из альгината Na/Ca и наночастиц маггемита осуществляли по следующей методике. Предварительно 100 мг альгината Na/Ca растворяли в 5 мл воды, затем к полученному раствору добавляли по каплям 1 мл водного раствора, содержавшего от 35 до 300 мг соли Мора. К этой смеси быстро приливали 1 мл водного раствора NaOH (18–90 мг/мл), интенсивно перемешивали и по каплям добавляли 1 мл водного раствора гипофосфита натрия (14–100 мг/мл). Окончание реакции определяли по изменению цвета реакционной смеси с зеленого на темно-красный. Очищали полученный раствор от неорганических солей методом проточного диализа. Для выделения полимерного продукта (композита) использовали лиофильную сушку.
Содержание железа в композите определяли спектрофотометрически на спектрофотометре “Ultrospec-4050” (“LKB”, Швеция) при λ = 510 нм, используя значение коэффициента экстинкции ε, равное 1800 л/(моль см). Для этого к 5 мг композита последовательно прибавляли 10 мкл концентрированной соли H2SO4 и 10 мл 10%-го раствора сульфосалициловой кислоты, доводили рН раствора до двух.
Структурные исследования осуществляли методом просвечивающей электронной микроскопии (ПЭМ) на установке JEM-100B фирмы “JEML” (Германия), оснащенной приставкой для рентгенофазового анализа. Образцы готовили путем нанесения капли водного раствора композита на медную сетку, затем их изучали без предварительного контрастирования.
ИК-спектры образцов снимали на ИК-фурье-спектрометре “PerkinElmer Spectrum 100 FT-IR” в режиме измерения поглощения с использованием приставки НПВО. Спектры регистрировали в диапазоне волновых чисел 650–2000 см–1.
Мессбауэровские спектры снимали на спектрометре электродинамического типа MS-1104ЕМ (Россия) с источникам 57Co(Rh), толщина поглотителя 0.12 мг/см2 (57Fe). Значения изомерных сдвигов приводили относительно α-Fe.
Получение анионных липосом
Сухие навески фосфатидилхолина и фосфатидилсерина растворяли в хлороформе. Рассчитанный объем растворов смешивали в круглодонной колбе так, чтобы суммарная масса липидов составляла 20 мг, мольную долю анионного липида α = [ФС]/([ФС] + [ФХ]) варьировали в интервале 10–30 мол. %. Затем на роторном испарителе Laborota-4000 (“Heidolph”, Германия) отгоняли органический растворитель при 37°С и скорости вращения 120 об/мин.
Липосомы получали методом озвучивания [33]. Сформированную тонкую липидную пленку диспергировали в 2 мл буферного раствора 10–2 моль/л Трис с pH 7 при помощи ультразвукового гомогенизатора Cole-Parmer CP-750 с частотой 20 кГц и амплитудой 20% в течение 600 с (2 × 300 с) при постоянном охлаждении водопроводной водой.
Для удаления титановой пыли липосомальные суспензии центрифугировали в течение 5 мин на центрифуге “Minispin “Eppendorf” при 12000 об/мин. Суспензии моноламелярных липосом с концентрацией липидов 10 мг/мл использовали в течение суток. Размер липосом контролировали методом динамического светорассеивания на установке “Brookhaven-90 Plus” (“Brookhaven”, США). Средний диаметр везикул составлял ~50–70 нм.
Для флуоресцентно меченых липосом в процессе формирования липидной пленки к смеси растворов липидов добавляли раствор флуоресцентно меченого диолеоилфосфоэтаноламин-N-(лиссамин родамин-Б-сульфонил) аммония, Л* (0.09 мас. % от общего содержания липидов).
Приготовление липосом, заполненных раствором электролита
Анионные липосомы получали по методике, описанной выше, однако вместо буфера Трис использовали 2 мл 1 моль/л раствора NaCl в буферном растворе 10–3 моль/л Трис с pH 7. Для удаления хлорида натрия из внешнего раствора проводили диализ липосомальной суспензии в течение 4 ч относительно используемого буфера.
Приготовление анионных липосом, заполненных доксорубицином
Липосомы, заполненные доксорубицином, получали методом активной загрузки [33]. Предварительно готовили рН-градиентные липосомы с “внутренним” ацетатным буфером с рН 4 и “внешним” буфером Трис с pH 7, которые добавляли к раствору доксорубицина в буфере Трис с рН 7. Включение доксорубицина в липосомы контролировали по изменению флуоресценции системы.
Интенсивность флуоресценции растворов регистрировали на спектрофлуориметре F-3000 фирмы “Hitachi” (Япония) в термостатируемой ячейке в кварцевых кюветах шириной 1 см.
Электропроводность растворов измеряли с помощью кондуктометра “CDM-83 Radiometer” (Дания). Значения pH растворов оценивали pH-метром pH 210 фирмы “Hanna” с использованием стеклянного электрода марки HI-1131B.
Исследование цитотоксичности
Цитотоксичность нанокомпозитов и комплексов на их основе исследовали на культурах клеток аденокарциномы молочной железы человека MCF-7. Количество выживших клеток определяли по изменению окраски раствора красителя тетразолиевого синего, который способен восстанавливаться в митохондриях с образованием нерастворимого в воде продукта (формазана), имеющего фиолетовый цвет. Число жизнеспособных клеток пропорционально количеству формазана, определяемому спектрофотометрически [15]. Оптическую плотность раствора в лунках с клетками измеряли при λ = 550 нм на фотометре Multiscan (“Titertek”, США). Количество живых клеток рассчитывали по отношению оптической плотности в лунке с заданной концентрацией к оптической плотности в контроле. Каждое значение находили усреднением трех параллельных измерений.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Мультилипосомальные контейнеры были получены послойной электростатической адсорбцией поли-L-лизин гидробромида и анионных липосом на поверхности микрогеля, сформированного из альгината Na/Ca с инкапсулированными наночастицами маггемита. Преимуществом такого подхода является использование биодеградируемого анионного микрогеля в качестве “ядра” будущего мультилипосомального контейнера и биодеградируемой пары липосома–полилизин, выполняющей роль “опушки”.
Получение и свойства нанокомпозитов Na/Ca альгинат-маггемит
Для образования композитного микрогеля применялась ранее описанная методика [34], в которой анионный полисахарид карбоксиметилцеллюлоза, составляющий основу композита, был заменен на альгинат. Ее достоинство в том, что синтез наночастиц γ-Fe2O3 из соли Мора протекает при комнатной температуре в одну стадию и не требует инертной среды [35]. Матрица альгината стабилизирует образующиеся наночастицы в результате электростатического взаимодействия карбоксильных групп полимера с ионами Fe3+ на поверхности маггемита [36].
Мольное отношение N исходных компонентов: соли Мора и мономерных звеньев альгината варьировали от 0.25 до 2.0. В пределах этого диапазона ионы железа в реакционной системе оказывались как в недостатке, так и в избытке по отношению к числу мономерных звеньев альгинита. Во всех случаях были представлены водорастворимые продукты, которые после диализа и лиофильной сушки превращались в красно-коричневые волокнистые материалы. Образцы растворяли в концентрированной серной кислоте, определяли концентрацию железа в растворе спектрофотометрически и пересчитывали в массовую долю железа в продукте φ. Из данных табл. 1 следует, что массовая доля железа в композите увеличивалась с ростом значения N.
Таблица 1.
Состав реакционной смеси и содержание железа в композитах
| [Альгинат Na/Ca], осново-моль/л | [(NH4)2Fe(SO4)2 ⋅ ⋅ 6H2O], моль/л | N | φ |
|---|---|---|---|
| 0.06 | 0.015 | 0.25 | 0.03 |
| 0.06 | 0.03 | 0.5 | 0.05 |
| 0.06 | 0.06 | 1.0 | 0.10 |
| 0.06 | 0.12 | 2.0 | 0.18 |
Для визуализации неорганических частиц в составе композита и анализа их структурных характеристик был использован метод ПЭМ. На ПЭМ-изображениях всех полученных композитов присутствуют контрастные частицы со средним диаметром ~10–12 нм. В качестве примера на рис. 1 представлена микрофотография образца с φ = 0.18 и его электронограмма. Последняя дает возможность рассчитать межплоскостные расстояния кристаллических решеток неорганических наночастиц (табл. 2). Сравнение полученных данных с представленными в таблице значениями для эталонных образцов железосодержащих соединений [37, 38] показало, что экспериментальные значения соответствуют кристаллической структуре магнитных оксидов железа: маггемита (γ-Fe2O3) и/или магнетита (Fe3O4).
Рис. 1.
Микрофотография композита с φ = 0.18 (а) и электронограмма наночастиц оксида железа в составе композита (б).

Окончательный вывод о природе наночастиц был сделан по результатам, полученным методом Мессбауэровской спектроскопии: 57Fe спектр композита с φ = 0.18 представляет собой суперпозицию двух секстетов магнитной сверхтонкой структуры и квадрупольного дублета (рис. 2). Параметры секстетов (изомерный сдвиг δА = 0.41 ± ± 0.01 мм/c, магнитное поле на ядре 57Fe HA = = 497 ± 3 кЭ, ширина линии на полувысоте WA = = 0.75 ± 0.03 мм/c, спектральный вклад SA = 38%; δB = 0.46 ± 0.01 мм/c, HB = 522 ± 3 кЭ, WA = 0.54 ± ± 0.03 мм/c, SB = 55%) и дублета (δ = 0.41 ± ± 0.01 мм/c, квадрупольное расщепление ΔЕ = = 0.72 ± 0.02 мм/c, WA = 0.54 ± 0.02 мм/c, S = 7%) типичны для наночастиц γ-Fe2O3, проявляющих суперпарамагнитные свойства. При этом в спектре отсутствует секстет, который отвечает за “промежуточную” степень окисления Fe2+/Fe3+ (δ > 0.6 мм/с), что указывает на отсутствие в образце примеси Fe3O4. Квадрупольный дублет в спектре означает, что исследуемый образец содержит фракцию очень мелких частиц (предположительно менее 3–4 нм) с частотой суперпарамагнитной релаксации магнитных моментов катионов при 78 К выше частоты ларморовой прецессии ядерного спина.
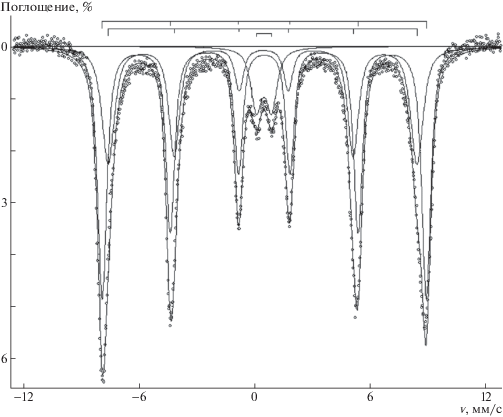
Таким образом, по данным Мессбауэровской спектроскопии, представленный композит содержит единственную форму оксида железа – маггемит γ-Fe2O3.
Взаимодействие наночастиц маггемита с полимерной матрицей изучали методом ИК-фурье-спектроскопии. На рис. 3 представлены ИК-фурье-спектры исходного альгината и композитов с различным содержанием γ-Fe2O3. Полоса при 1730 см–1 относится к валентным колебаниям С=О фрагмента карбоксильной группы и отражает его участие в образовании внутри- и межмолекулярных водородных связей [39]. Полосы при 1600 и 1420 см–1 в спектре альгината связаны с валентными антисимметричными и симметричными колебаниями карбоксильной группы полианиона. Наконец, полосы при 1220 и 1025 см–1 обусловлены валентными колебаниями фрагмента полисахарида С–ОН и валентными колебаниями фрагмента полисахарида С–О–С соответственно [39].
Рис. 3.
ИК-фурье-спектры альгината (1) и композитов альгинат–маггемит (2–5); φ = 0.05 (2), 0.07 (3), 0.15 (4) и 0.26 (5).
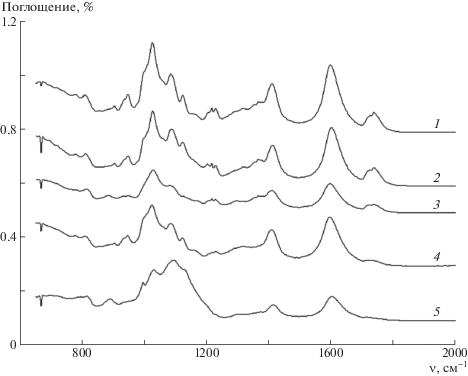
Повышение доли маггемита приводит к прогрессивному уменьшению интенсивности полосы в спектрах композита при 1730 см–1. Полосы при 1600, 1025 и 1420 см–1 сохраняются, однако их интенсивность становится значительно ниже; полоса при 1220 см–1 практически исчезает. Эти изменения свидельствуют о том, что по мере возрастания доли маггемита в композиционном образце система водородных связей разрушается, и все бóльшее количество функциональных групп полимера образует электростатические и координационные связи с ионами Fe3+ наночастиц маггемита.
Формирование комплексов наночастиц маггемита с полимерной матрицей обеспечивает их агрегативную стабильность в водном окружении. Лиофилизированные образцы композитов растворяли в буферном растворе Трис с рН 7 и определяли их средний гидродинамический диаметр методом динамического светорассеяния и электрофоретическую подвижность (ЭФП) методом лазерного микроэлектрофореза. Результаты измерений для растворов композитных микрогелей и исходного альгината приведены на рис. 4.
Рис. 4.
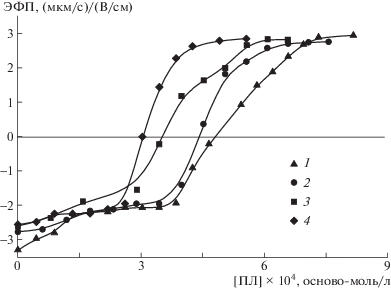
Зависимость ЭФП микрогелей магнитных композитов альгинат–маггемит от концентрации полилизина с φ = 0.05 (1), 0.07 (2), 0.15 (3), 0.26 (4). Здесь и на рис. 5–7, 10 [АЛГ] = 2.5 × 10–4 осново-моль/л; буферный раствор 10–2 моль/л Трис pH 7.
Альгинат в водном растворе представляет собой гель со средним диаметром частиц 550 нм, трехмерная сетчатая структура которого обусловлена ионными связями между карбоксильными группами полимера и катионами Ca2+ [40]. Формирование наночастиц маггемита в объеме геля и их электростатическое и координационное взаимодействие с функциональными группами альгината сопровождается контракцией микрогеля; при этом размер композита с максимальным содержанием маггемита уменьшается до 240 нм. Таким образом, альгинат “подшивается” дважды: сначала ионами Ca2+, затем наночастицами. На первом этапе результатом сшивки является формирование микрогелевых частиц полумикронного размера. На втором этапе сшивание приводит к дополнительному двукратному уменьшению размера микрогеля и формированию конъюгата.
Электрофоретическая подвижность микрогеля при связывании маггемита меняется незначительно: от –3.0 для исходного альгината до –2.6 (мкм/с)/(В/см) для композита с φ = 0.26 (табл. 3). Отрицательный заряд на поверхности композитных частиц обеспечивает их агрегативную стабильность в водном растворе в течение нескольких месяцев.
Таблица 3.
Гидродинамический диаметр и ЭФП альгинатного микрогеля и композитов в буферном растворе 10–3 моль/л Трис с рН 7
| Массовая доля маггемита в образце | Диаметр, нм | ЭФП, (мкм/с)/(В/см) |
|---|---|---|
| 0 | 550 | –3.3 |
| 0.05 | 260 | –3.0 |
| 0.07 | 250 | –2.8 |
| 0.15 | 220 | –2.8 |
| 0.26 | 240 | –2.6 |
Особый интерес представляют магнитные свойства композитных микрогелей, которые определяют возможность использования внешних магнитных полей для направленного транспорта таких объектов. В табл. 4 суммированы результаты исследования намагниченности, коэрцитивной силы и времени релаксации двух композитов с φ = 0.15 и 0.26.
Таблица 4.
Магнитные характеристики композитов альгинат–маггемит
| Массовая доля маггемита в образце | Намагниченность насыщения, сгс м/г | Коэрцитивная сила, Oe | Время релаксации, с |
|---|---|---|---|
| 0.15 | 1.3 × 10–2 | 42 | 1.5 |
| 0.26 | 3.2 ×10–2 | 34 | 1.0 |
Значения намагниченности насыщения полученных образцов типичные для ферромагнитиков. Невысокие величины коэрцитивных сил и времен релаксации указывают на то, что наночастицы в составе композита относятся к классу магнитомягких материалов, которые быстро намагничиваются и теряют магнитные свойства при снятии магнитного поля. Эта способность оказывается полезной при конструировании магнитоуправлемых средств доставки лекарственных веществ.
Получение и свойства мультилипосомального контейнера
Выше отмечалось, что анионные микрогели, содержащие магнитные наночастицы, несут поверхностный отрицательный заряд. Для связывания с микрогелем анионных липосом его поверхность предварительно модифицировали катионным полипептидом полилизином. За его адсорбцией на поверхности магнитного композитного микрогеля следили, контролируя электрофоретическую подвижность и размер частиц в системе.
Добавление раствора полилизина к водным суспензиям композитных микрогелей сопровождается уменьшением заряда микрогелевых частиц и перезарядкой поверхности при высоких значениях концентрации поликатиона (рис. 4). Нейтрализация заряда микрогелей достигается при разной концентрации добавленного поликатиона [ПЛ]ЭФП=0, которая прогрессивно уменьшается с ростом массовой доли φ маггемита в композите. Такой ход зависимостей [ПЛ]ЭФП = 0 – φ объясняется тем, что с ростом φ увеличивается количество карбоксильных групп альгината, вовлеченных в комплексообразование с наночастицами маггемита, и, следовательно, уменьшается количество свободных групп альгината, способных электростатически связываться с полилизином.
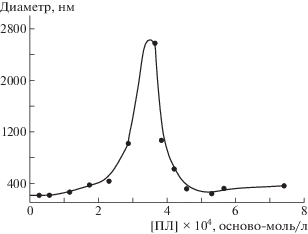
Нейтрализация заряда коллоидных частиц противоположно заряженным полимером сопровождается их агрегацией и последующим уменьшением размера частиц в системе в избытке полимера [41, 42]. Взаимодействие анионного магнитного композита с полилизином подчиняется этой же схеме. На рис. 5 представлена типичная зависимость размера частиц комплекса композит–полилизин от концентрации поликатиона. Добавление полилизина к суспензии магнитного композита с φ = 0.15 приводит вначале к росту размера частиц в системе, который затем уменьшается до размера близкого к исходному. Максимальный размер комплекса композит–полилизин наблюдается при полной нейтрализации заряда микрогеля поликатионом. Стабилизация размера комплекса в избытке поликатиона связана с появлением на его поверхности положительного заряда, привнесенного адсорбированным полимером. Диаметр агрегативно стабильных частиц положительно заряженного комплекса композит–полилизин составляет –300 нм.
Рис. 5.
Зависимость гидродинамического диаметра магнитного композита альгинат–маггемит с φ = 0.15 от концентрации полилизина.
Дальнейшие эксперименты проводили с комплексом, полученным путем исчерпывающей адсорбции полилизина на поверхности композитного микрогеля с массовой долей маггемита 0.15. Это условие реализуется при добавлении 130 мкл 10–2 осново-моль/л раствора полилизина к 2.5 мл раствора композита (φ = 0.15) с концентрацией альгината 2.5 × 10–4 осново-моль/л. Этот комплекс обладает приемлемыми магнитными свойствами, а водная суспензия такого комплекса сохраняет стабильность в течение длительного времени.
Мультилипосомальный контейнер получали адсорбируя липосомы, сформированные из смеси анионного ФС и электронейтрального ФХ, на поверхности комплекса композит–полилизин; мольную долю α фосфатидилхолина в липосомах варьировали от 10 до 30%. Связывание анионных липосом с комплексом композит–полилизин сопровождается вначале нейтрализацией заряда комплекса, а затем, появлением на его поверхности отрицательного заряда при больших значениях концентрации липосом (рис. 6). Как и ожидалось, чем выше содержание анионного липида в липосомальной мембране, тем меньше требовалось липосом для нейтрализации заряда комплекса.
Рис. 6.
Зависимость ЭФП комплекса композит–полилизин с φ = 0.15 от концентрации ФС–ФХ липосом; α = 10 (1), 20 (2) и 30% (3); [ПЛ] = 5 × 10–4 осново-моль/л.
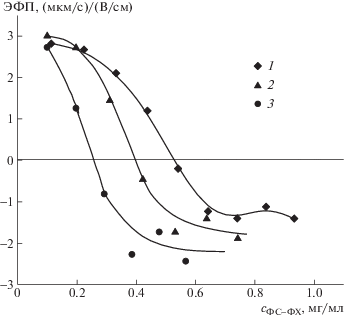
Простая геометрическая оценка максимальной плотности заполнения поверхности комплекса липосомами показала, что на каждой 300 нм частице комплекса может разместиться до 100 липосом диаметром 50 нм. В дополнение к этой оценке следует принять во внимание электростатическую природу адсорбции анионных ФС–ФХ липосом на поверхности комплекса, которая сопровождается нейтрализацией заряда комплекса зарядами адсорбированных липосом. Такой механизм означает, что плотность заполнения поверхности комплекса липосомами должна уменьшаться с ростом содержания анионного липида в липосомальной мембране. Именно это и наблюдали в эксперименте по электрофоретическому титрованию комплекса суспензиями липосом с разными значениями α (рис. 6).
Принципиальный вопрос касается целостности липосом в составе мультилипосомального контейнера. Для ответа на этот вопрос использовали липосомы c α = 10, 20 и 30%, внутренний объем которых был заполнен раствором 1 моль/л NaCl. Липосомы адсорбировали на поверхности комплекса и регистрировали электропроводность Ω полученных суспензий. Во всех случаях значения Ω оставались неизменными по крайней мере в течение 6 ч после формирования мультилипосомального контейнера, что свидетельствует о сохранении целостности липосом после их связывания в нем.
Другое важное требование к синтезированным мультилипосомальным контейнерам – устойчивость к диссоциации на исходные компоненты в водно-солевых растворах, в частности в физиологическом растворе с [NaCl] = 0.15 моль/л. Возможную диссоциацию мультилипосомального контейнера контролировали, используя липосомы со встроенным в мембрану флуоресцентно меченым липидом Л*. Известно, что связывание меченых липосом с катионным полимером приводит к тушению флуоресценции метки, а последующая диссоциация комплекса к ее возгоранию [43]. В экспериментах данной работы адсорбция трех типов меченых липосом с α =10, 20 и 30% на поверхности комплекса уменьшала интенсивность флуоресценции метки (рис. 7а). Последующее добавление раствора соли к суспензиям мультилипосомального контейнера вызывает различные эффекты в зависимости от значения α.
Рис. 7.
Зависимости интенсивности флуоресценции метки от концентрации альгината при добавлении суспензии комплекса композит–полилизин с φ = 0.15 к суспензии меченых липосом (а) и концентрации соли при добавлении раствора NaCl к суспензии мультилипосомального контейнера при [ПЛ] = 5 × × 10–4 осново-моль/л (б); α = 10 (1), 20 (2) и 30% (3).
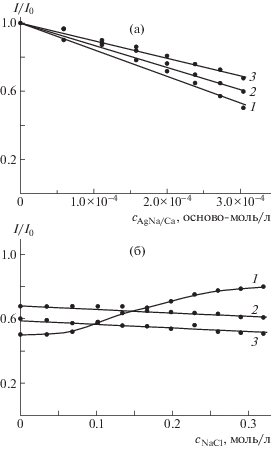
Возгорание флуоресценции в суспензии мультилипосомального контейнера из липосом с α = = 10% начинается при концентрации соли равной 0.075 моль/л (рис. 7б, кривая 1). Полное возгорание флуоресценции метки, т.е. 100% диссоциация мультилипосомального контейнера на ФС–ФХ липосомы и катионный комплекс, регистрируется при [NaCl] = 0.25 моль/л. Напротив, в суспензиях мультилипосомальных контейнеров, сформированных липосомами с α = 20 и 30% (кривые 2 и 3), флуоресценция не восстанавливается в физиологическом растворе, что означает отсутствие диссоциации мультилипосомальных контейнеров в этих условиях.
Добавление ФС–ФХ липосом к суспензии 300 нм комплекса приводит к росту размера мультилипосомальных контейнеров до значения в несколько микрон, который регистрируется при полной нейтрализации заряда комплекса адсорбированными липосомами. Дальнейшее увеличение концентрации липосом влечет их дополнительную (избыточную) адсорбцию, что придает мультилипосомальным контейнерам отрицательный заряд и уменьшает их размер до 400 нм.
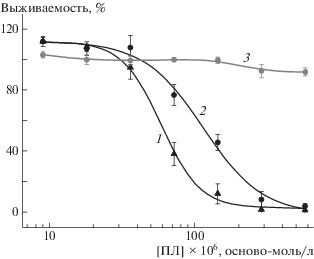
Цитотоксичность мультилипосомальных контейнеров определяли по выживаемости чувствительных к лекарствам клеток аденокарциномы молочной железы человека MCF-7. На рис. 8 представлены зависимости выживаемости клеток от концентрации индивидуального полилизина и полилизина в составе комплекса и мультилипосомальных контейнеров с липосомами с α = 20%. В качестве количественной меры цитотоксичности взята концентрация добавленного полилизина, при которой регистрируется 50% выживаемость клеток (В-50). Бóльшее значение В-50 соответствует меньшей цитотоксичности образца. Так, для полилизина В-50 она составляет 65 × × 10–6 осново-моль/л, это означает, что положительно заряженный полимер является довольно токсичным веществом (кривая 1). Связывание полилизина с анионным композитным микрогелем понижает цитотоксичность полученного комплекса в 2 раза, т.е. до значения В-50, равного 135 × 10–6 осново-моль/л (кривая 2), несмотря на то что его заряд остается положительным. Наконец, тройной комплекс микрогель–полилизин–липосома (мультилипосомальный контейнер) не показывает цитотоксичности во всем интервале исследованных значений концентрации полилизина вплоть до 600 × 10–6 осново-моль/л (кривая 3).
Рис. 8.
Зависимость выживаемости клеток MCF-7 от концентрации полилизина (1), а также полилизина в составе комплекса (2) и мультилипосомального контейнера (3) с φ = 0.15; α = 20% (3).
Способность заполненного лекарством мультилипосомального контейнера реагировать на приложение внешнего магнитного поля была продемонстрирована в ходе следующего эксперимента. Так, ФС–ФХ липосомы (α = 20%), нагруженные противоопухолевым антибиотиком доксорубицином, были адсорбированы на поверхности катионного комплекса, сформированного из магнитного микрогеля и полилизина φ = 0.15. Встраивание доксорубицина в липосомы сопровождается заметным тушением его флуоресценции при λисп = 555 нм (λвозб = 490 нм) [20, 44]. Их взаимодействие с комплексом не приводит к возгоранию флуоресценции доксорубицина, что означает отсутствие вытекания лекарства из липосом в окружающий раствор [33, 44] и подтверждает сделанный ранее вывод о сохранении целостности анионных липосом, нагруженных доксорубицином, после формирования мультилипосомального контейнера.
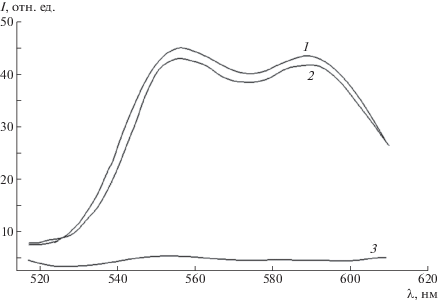
В отсутствие внешнего магнитного поля водный раствор мультилипосомальных контейнеров, заполненных доксорубицином, представляет собой жидкость оранжевого цвета из-за присутствия в них окрашенных наночастиц маггемита γ-Fe2O3 (рис. 9а). Когда рядом с кюветой, заполненной этим раствором, ставили постоянный магнит, то частицы магнитного микрогеля начинали перемещаться в направлении источника магнитного поля и концентрироваться на стенке кюветы (рис. 9б). Далее, удерживая магнит, с помощью шприца удаляли супернатант, добавляли в кювету 2.5 мл буферного раствора 10–2 моль/л Трис и редиспергировали осадок многократным встряхиванием кюветы. Липосомы разрушали добавлением избытка Тритона Х-100 и регистрировали спектр флуоресценции полученного раствора (рис. 10, кривая 1). Для сравнения на рисунке показан спектр флуоресценции суспензии исходных липосом той же концентрации, но разрушенных действием Тритона Х-100 (кривая 2), и спектр флуоресценции супернатанта (кривая 3). Полученные результаты позволяют сделать вывод о том, что практически весь доксорубицин сохраняется в составе мультилипосомального контейнера после его иммобилизации на стенке кюветы. Описанный подход представляет интерес для получения магнитоуправляемых конструкций для направленной доставки лекарственных веществ.
Рис. 9.
Концентрирование частиц мультилипосомального контейнера с доксорубицином на стенке кюветы с помощью внешнего магнитного поля при φ = 0.15 и α = 20%. Пояснения в тексте.

Рис. 10.
Спектры флуоресценции суспензии, полученной после отделения мультилипосомального контейнера (заполненного доксорубицином) с помощью магнита, редиспергирования осадка и разрушения липосом добавлением Тритона Х-100 (1); суспензии исходных липосом, разрушенных добавлением Тритона Х-100 (2) и супернатанта после отделения мультилипосомального контейнера (3). φ = 0.15, α = 20%, [ПЛ] = 5 × 10–4 осново-моль/л, 0.6 мг/мл липосом, [доксорубицин] = 3 × 10–4 моль/л.
ЗАКЛЮЧЕНИЕ
Таким образом, электростатическое связывание анионных липосом с полисахаридным микрогелем, поверхность которого покрыта катионным полипептидом полилизином, позволяет получить мультилипосомальный контейнер диаметром 400 нм. Целостность липосом в контейнере сохраняется; контейнер не диссоциирует на исходные компоненты в физиологическом растворе.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-03-01024).
Список литературы
Xiong F., Tian J., Hu K., Zheng X., Sun J., Yan C., Yao J., Song L., Zhang Y., Gu N. // Nanoscale. 2016. V. 8. № 39. P. 17085.
Ulbrich K., Hola K., Subr V., Bakandritsos A., Tucek J., Zboril R. // Chem. Rev. 2016. V. 116. № 9. P. 5338.
Amidon S., Brown J.E., Dave V.S. // Aaps Pharmscitech. 2015. V. 16. № 4. P. 731.
Khadka P., Ro J., Kim H., Kim I., Kim J.T., Kim H., Cho J.M., Yun G., Lee J. // Asian J. Pharm. 2014. V. 9. № 6. P. 304.
Li J., Mooney D.J. // Nat. Rev. Mater. 2016. V. 1. № 12. P. 16071.
Ahmad Z., Shah A., Siddiq M., Kraatz H.B. // RSC Adv. 2014. V. 4. № 33. P. 17028.
Caminade A.M., Turrin C.O. // J. Mater. Chem. B. 2014. V. 2. № 26. P. 4055.
Wang Y., Zhao Q., Han N., Bai L., Li J., Liu J., Li J., Che E., Hu L., Zhang Q., Jiang T., Wang S. // Nanomedicine. 2015. V. 11. № 2. P. 313.
Zheng H., Zhang Y., Liu L., Wan W., Guo P., Nyström A.M., Zou X. // J. Am. Chem. Soc. 2016. V. 138. № 3. P. 962.
Fornaguera C., Dols-Perez A., Caldero G., Garcia-Celma M.J., Camarasa J., Solans C. // J. Control. Release. 2015. V. 211. P. 134.
Jaiswal M., Dudhe R., Sharma P.K. // Biotech. 2015. V. 5. № 2. P. 123.
Sercombe L., Veerati T., Moheimani F., Wu S.Y., Sood A.K., Hua S. // Front. Pharmacol. 2015. V. 6. P. 286.
Pattni B.S., Chupin V.V., Torchilin V.P. // Chem. Rev. 2015. V. 115. № 19. V. 10938.
Woodle M., Papahadjopoulos D. // Meth. Enzym. 1989. V. 171. P. 193.
Kim H., Britton G.L., Peng T., Holland C.K., McPherson D.D., Huang S.L. // Int. J. Nanomed. 2014. V. 9. P. 155.
Bartneck M., Scheyda K.M., Warzecha K.T., Rizzo L.Y., Hittatiya K., Luedde T., Storm G., Trautwein C., Lammers T., Tacke F. // Biomaterials. 2015. V. 37. P. 367.
Mu L.M., Ju R.J., Liu R., Bu Y.Z., Zhang J.Y., Li X.Q., Lu W.L. // Adv. Drug Deliv. Rev. 2017. V. 115. P. 46.
Stylianopoulos T., Jain R.K. // Nanomedicine. 2015. V. 11. № 8. P. 1893.
Yaroslavov A.A., Sybachin A.V., Zaborova O.V., Zezin A.B., Talmon Y., Ballauff M., Menger F.M. // Adv. Colloid Int. Sci. 2015. V. 226. P. 54.
Sybachin A.V., Khlynina P.O., Spiridonov V.V., Panova I.G., Melik-Nubarov N.S., Yaroslavov A.A. // Polym. Int. 2018. V. 67. № 10. P. 1352.
Ding J., Wang J. // J. Drug. 2017. V. 1. № 2. P. 14.
Ponomarev V.S., Kozhikhova K.V., Shulepov I.D., Tokareva M.I., Ivantsova M.N., Mironov, M.A. // Russ. Chem. Bull. 2014. V. 63. № 7. V. 1619.
Yaroslavov A.A., Sybachin A.V., Zaborova O.V., Migulin V.A., Samoshin V.V., Ballauff M., Kesselman E., Schmidt J., Talmon Y., Menger F.M. // Nanoscale. 2015. V. 7. № 5. P. 1635.
Yaroslavov A.A., Efimova A.A., Sybachin A.V., Chva-lun S.N., Kulebyakina A.I., Kozlova E.V. // RSC Adv. 2015. V. 5. № 40. P. 31460.
Yaroslavov A.A., Sybachin A.V., Zaborova O.V., Pergushov D.V., Zezin A.B., Melik-Nubarov N.S., Plamper F.A., Müller A.H., Menger F.M. // Macromol. Biosci. 2014. V. 14. P. 491.
Wang T., Hartner W.C., Gillespie J.W., Praveen K.P., Yang S., Mei L.A., Petrenko V.A., Torchilin V.P. // Nanomedicine. 2014. V. 10. № 2. P. 421.
Zhao Y., Ren W., Zhong T., Zhang S., Huang D., Guo Y., Yao X., Wang W.-Q., Zhang X., Zhang Q. // J. Control Release. 2016. V. 222. P. 56.
Qiu F., Mhanna R., Zhang L., Ding Y., Fujita S., Nelson B.J. // Sens. Actuators Chem. B. 2014. V. 196. P. 676.
Di Corato R., Béalle G., Kolosnjaj-Tabi J., Espinosa A., Clement O., Silva A.K., Menager C., Wilhelm C. // ACS Nano. 2015. V. 9. № 3. P. 2904.
Veremeeva P.N., Grishina I.V., Lapteva V.L., Yaroslavov A.A., Sybachin A.V., Palyulin V.A., Zefirov N.S. // Mendel. Commun. 2014. V. 24. P. 152.
Hardiansyah A., Huang L.Y., Yang M.C., Liu T.Y., Tsai S.C., Yang C.Y., Lin C.H. // Nanoscale Res. Lett. 2014. V. 9. № 1. P. 497.
Zhang K., Liu M., Tong X., Sun N., Zhou L., Cao Y., Pei R. // Biomacromolecules. 2015. V. 16. № 9. P. 2618.
Yaroslavov A.A., Panova I.G., Sybachin A.V., Spiridonov V.V., Zezin A.B., Mergel O., Gelissen A., Tiwari R., Plamper F., Richtering W., Menger F.M. // Nanomedicine. 2017. V. 13. P. 1491.
Spiridonov V.V., Panova I.G., Makarova L.A., Afanasov M.I., Zezin S.B., Sybachin A.V., Yaroslavov A.A. // Carb. Polym. 2017. V. 177. P. 269.
Janot R., Guérard D. // J. Alloys Compd. 2002. V. 333. № 1. V. 302.
Maccarini M., Atrei A., Innocenti C., Barbucci R. // Colloid. Sur. A. 2014. V. 462. P. 107.
Topchieva I.N., Spiridonov V.V., Zakharov A.N., Afanasov M.I., Mironov A.V., Perov N.S., Semisalova A.S. // Mendel. Commun. 2015. V. 25. P. 145.
Guivar J.A.R., Martínez A.I., Anaya A.O., Valladares L.D.L.S., Félix L.L., Dominguez A.B. // Adv. Nanopart. 2014. V. 3. № 03. P. 114.
Spiridonov V.V., Panova I.G., Afanasov M.I., Zezin S.B., Sybachin A.V., Yaroslavov A.A. // Polymer Science B. 2018. V. 60. № 1. P. 116.
Xin Y., Bligh M.W., Kinsela A.S., Waite T.D. // J. Membrane Sci. 2016. V. 497. P. 289.
Panova I.G., Sybachin A.V., Spiridonov V.V., Kydralieva K., Jorobekova S., Zezin A.B., Yaroslavov A.A. // Geoderma. 2017. V. 307. P. 91.
Siyawamwaya M., Choonara Y.E., Bijukumar D., Kumar P., Du Toit L.C., Pillay V.A. // Int. J. Mater. Po. 2015. V. 64. № 18. P. 955.
Yaroslavov A.A., Efimova A.A., Sybachin A.V., Izumrudov V.A., Samoshin V.V., Potemkin I.I. // Colloid J. 2011. V. 73. P. 430.
Kitaeva M.V., Melik-Nubarov N.S., Menger F.M., Yaroslavov A.A. // Langmuir. 2004. V. 20. P. 6796.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)