

Высокомолекулярные соединения (серия А), 2019, T. 61, № 6, стр. 485-490
Комплексы анионных холестеринсодержащих липосом и катионных хитозановых микрочастиц
А. А. Ефимова a, *, К. С. Трошева a, Е. А. Красников a, Г. Г. Кривцов b, А. А. Ярославов a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1–3, Россия
b Научно-исследовательский институт вакцин и сывороток им. И.И. Мечникова
105064 Москва, М. Казенный пер., 5а, Россия
* E-mail: ephimova@genebee.msu.su
Поступила в редакцию 24.05.2019
После доработки 26.06.2019
Принята к публикации 08.07.2019
Аннотация
Описано получение мультилипосомальных комплексов адсорбцией анионных холестеринсодержащих липосом на поверхности катионных хитозановых микрочастиц. Использование липосом с мольной долей холестерина в липосомальной мембране, равной 0.3, формирует комплекс, в котором каждая микрочастица связывает 150 интактных (неразрушенных) липосом. Размер комплексных частиц (250–400 нм) позволяет рекомендовать их в качестве потенциального носителя лекарственных веществ, способного проникать в клетки по механизму пассивного транспорта.
ВВЕДЕНИЕ
Среди многочисленных систем доставки биологически активных веществ особое место занимают бислойные липидные везикулы (липосомы) [1, 2]. Уникальное строение липосом позволяет инкапсулировать в них вещества различной природы: гидрофобные и амфифильные в липосомальную мембрану, гидрофильные во внутреннюю водную полость липосом [3–7]. Иммобилизация нескольких десятков анионных липосом на поверхности катионного коллоидного носителя позволяет сконцентрировать их в небольшом объеме и тем самым повысить эффективность захвата инкапсулированного лекарства клетками [8–10].
Электростатическое связывание липосом на поверхности полипептидных везикул [11], полилактидных мицелл [8, 10] и хитозановых частиц [12, 13] приводит к получению биодеградируемых мультилипосомальных носителей, которые могут быть выведены из организма после выполнения ими транспортной функции. Для создания таких конструкций были использованы липосомы, сформированные из электронейтрального и отрицательно заряженного липидов [8, 10, 12, 13]. Вместе с тем традиционные липосомы, рекомендованные в качестве контейнеров лекарств, содержат в своем составе холестерин [14], повышающий стабильность липосом в биологическом окружении [15], при этом мольная доля холестерина может достигать 0.4–0.5 [16–18].
Показано, что встраивание холестерина в липосомальную мембрану уменьшает подвижность алкильных цепей липидных молекул [19, 20], что приводит к формированию дефектов в анионных липосомах после их связывания с линейным катионным полимером [21]. Вопрос о сохранении целостности анионных липосом, адсорбированных на катионном полимерном носителе, является ключевым, так как определяет возможное биомедицинское применение мультилипосомальных конъюгатов.
В настоящей работе описана адсорбция анионных липосом с разным содержанием холестерина на поверхности катионных микрочастиц хитозана, природного биодеградируемого полимера [22, 23]. Особое внимание уделено целостности адсорбированных липосом, а также cоставу и размеру полученных конструкций.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Фосфатидилхолин (ДОФХ)
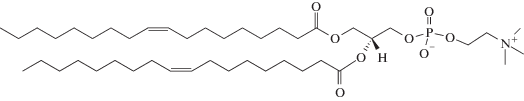
дифосфатидилглицерол (кардиолипин, КЛ2–)
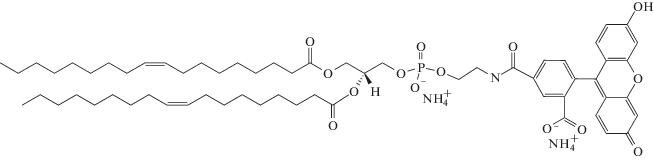
1,2-диолеоил-глицеро-3-фосфоэтаноламин-N-(карбокси-флуоресцеин) (ДОФЭ-КФ)
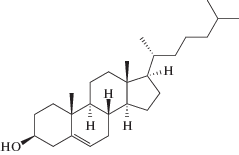
и холестерин (Хол) (“Avanti”)
использовали без дополнительной очистки.
Малые моноламелярные липосомы получали методом озвучивания [24]. Для этого смешивали необходимое количество растворов липидов и холестерина в хлороформе и удаляли органический растворитель на вакуумном роторном испарителе при 50°С. Липидную пленку диспергировали в соответствующем буферном растворе и дополнительно гомогенизировали ультразвуком частоты 22 кГц в течение 400 с (2 × 200 с) в непрерывном режиме при постоянном охлаждении водой. Использовали ультразвуковой диспергатор “Cole-Parmer 4710”. Полученные липосомы отделяли от титановой пыли на центрифуге “J-11 Beckman” в течение 5 мин при скорости 10 000 об/мин. Мольная доля отрицательно заряженных полярных “головок” составляла νКЛ = 2[КЛ2–]/(2[КЛ2–] + [ДОФХ]) (молекулы КЛ2– несут две анионные группы) 0.1. Мольная доля холестерина варьировалась и составляла 0.1, 0.2, 0.3 и 0.4.
Липосомы со встроенной в бислой флуоресцентной меткой получали добавлением к смеси растворов липидов ДОФЭ-КФ (0.1 мас. % от общего количества липидов).
Липосомы с заключенным во внутренний объем хлоридом натрия готовили, диспергируя липидную пленку в 10-2 М ТРИС-буферном растворе, дополнительно содержавшем 1М NaCl. Полученную суспензию диализовали в течение 1.5 ч против 10–2 М ТРИС-буфера, который меняли каждые 30 мин. Во всех экспериментах использовали свежеприготовленные липосомы, размеры которых находились в пределах 30–50 нм (по данным светорассеяния).
Хитозан с Mw = 30 × 103 и степенью деацетилирования 85% был получен деацетилированием хитина [25, 26]. Суспензию микрочастиц (микрогелей) хитозана получали смешением равных объемов 0.005 М водного раствора хитозана и 0.005 М водного раствора сульфата натрия [27]. Смесь перемешивали в течение 15 мин и далее диализовали против 10–2 М ацетатного буфера (рН 5.5) в течение 3 ч. Буфер меняли каждые 45 мин. Ацетатный буфер с рН 5.5 обеспечивал максимальный положительный заряд микрочастиц хитозана. Масса отсечения диализных мешков (Sigma-Aldrich) составляла 12.5 × 103.
Воду очищали двойной перегонкой с последующим пропусканием через систему “Milli-Q” фирмы “Миллипор” (США). Удельная электропроводность очищенной воды 0.6 мкСм/см.
Методы
Средний гидродинамический диаметр и электрофоретическую подвижность (ЭФП) частиц определяли методом квазиупругого светорассеяния на приборе “Brookhaven 90 Plus”. Измерения проводили в термостатируемой ячейке при 20°С.
pH растворов оценивали с помощью pH-метра “Hanna 210”. В качестве измерительного электрода использовали стеклянный электрод HI 1131B.
Интенсивность флуоресценции определяли на спектрофлуориметре “F-4000” (“Hitachi”) в термостатируемой ячейке, использовали кварцевые кюветы шириной 1 см.
Электропроводность растворов регистрировали на кондуктометре CDM 83 (“Radiometer”). Для измерений использовали цилиндрическую полиэтиленовую кювету, объем пробы составлял 1 мл.
Осаждение комплексов микрочастиц хитозана и анионных липосом проводилось на центрифуге J2-21 (“Beckman”) в течение 40 минут со скоростью вращения ротора 18 000 об/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Хитозановые частицы получали в ацетатном буфере с рН 5.5 взаимодействием хитозана с М = = 30 × 103 с сульфат-анионами (подробнее см. в Экспериментальной части). ЭФП частиц была равна +1.6 ± 0.1 (мкм/с)/(В/см). Очевидно, что положительный заряд микрочастиц определялся зарядами, привнесенными протонированными аминогруппами хитозановых макромолекул, не участвовавшими в образовании солевых связей с сульфат-анионами. Размер (гидродинамический диаметр) частиц составлял 225 ± 20 нм и лишь незначительно (на 5%) увеличивался при замене ацетатного буфера на физиологический раствор с концентрацией NaCl = 0.15 моль/л.
На поверхности хитозановых микрочастиц адсорбировали анионные липосомы ДОФХ/КЛ2–/Хол с мольной долей анионных КЛ2– “головок” νКЛ = = 0.1 и мольной долей холестерина νХол, которая изменялась от 0.1 до 0.4. Адсорбцию проводили в буфере рН 5.5, где оба компонента несли противоположные по знаку заряды: микрочастицы – положительный, липосомы – отрицательный. После формирования комплекса хитозан–липосома внешний раствор меняли на ТРИС буфер с рН 7, следуя описанной ранее процедуре [12]; замена буфера не влияла на степень связывания липосом с микрочастицами [13]. Полученные таким образом суспензии анализировали методом микроэлектрофореза, измеряя ЭФП частиц комплекса.
На рис. 1 представлены кривые, отражающие зависимость ЭФП комплекса от концентрации липосом с мольной долей холестерина 0.1–0.4. Во всех случаях увеличение концентрации липосом сопровождалось нейтрализацией заряда микрочастиц и их последующей перезарядкой. При одинаковой форме кривых нейтрализующие концентрации липосом (Липнейтр) различались в 2 раза: они составляли 0.2 мг/мл для липосом с νХол = = 0.1 и 0.2 (кривая 1) и 0.4 мг/мл для липосом с νХол = 0.3 и 0.4 (кривая 2). Такое различие отражало разную форму участия анионных липидов в формировании солевых связей с катионными группами хитозана. В литературе описано изменение фазового состояния липидного бислоя (липидной мембраны) при встраивании в него холестерина [19, 20]. Увеличение содержания холестерина ограничивает латеральную (в пределах каждого монослоя) и трасмембранную (между монослоями) подвижность липидных молекул, что отражает переход мембраны из жидкокристаллического (жидкого) состояния в гелевое (твердое) [21]. В твердых липосомах с равномерным распределением анионного липида между монослоями в образовании электростатического комплекса с катионным полимером участвуют только анионные липиды, расположенные на внешней стороне липосомальной мембраны (половина от общего количества анионных липипдов в мембране). В отличие от этого, связывание поликатиона с жидкими липосомами сопровождается переходом анионных липидов с внутреннего монослоя на внешний (индуцированным флип-флопом), в результате чего все анионные липиды участвуют в образовании солевых связей с поликатионом [24]. При таком механизме для нейтрализации заряда поликатиона требуется вдвое большее количество твердых липосом по сравнению с жидкими (при одинаковом содержании анионного липида в мембранах обоих типов липосом). Именно это различие мы наблюдаем в случае электрофоретического титрования микрочастиц катионного хитозана анионными липосомами ДОФХ/КЛ2–/Хол с разным содержанием холестерина (рис. 1), что позволяет говорить об индуцированном хитозаном флип-флопе в “жидких” липосомах с νХол = 0.1 и 0.2 (кривая 1), а также об отсутствии флип-флопа в “твердых” липосомах с νХол = 0.3 и 0.4 (кривая 2).
Рис. 1.
Зависимость ЭФП частиц комплекса хитозан–липосома от концентрации липосом. νХол = 0.1 (а), 0.2 (б), 0.3 (в) и 0.4 (г). Здесь и на рис. 2–4: липосомы ДОФХ/КЛ2-/Хол, концентрация хитозана 2.5 ×10–2 мг/мл, 10–2 M ТРИС-буфер, pH 7.
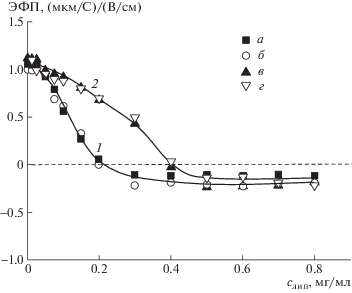
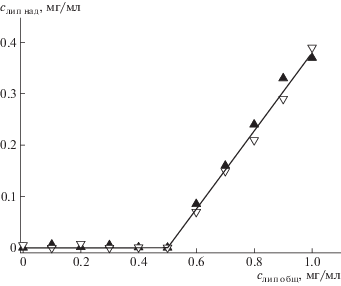
Таким образом, увеличение доли холестерина νХол в мембране до 0.3 вдвое повышает предельное количество липосом Nмакс, которое может связаться с каждой микрочастицей хитозана. Этот результат представляет большой интерес, поскольку позволяет заметно увеличить емкость мультилипосомального контейнера простым повышением содержания холестерина в липосомальной мембране. Мы оценили Nмакс для комплекса микрочастиц хитозана и твердых липосом с νХол = 0.3 и 0.4, использовав липосомы со встроенным в мембрану флуоресцентно меченным липидом ДОФЭ-КФ. Для оценки Nмакс брали соотношение, приведенное в работе [12]. Суспензию хитозановых микрочастиц смешивали с суспензией флуоресцентно меченных липосом, отделяли комплекс хитозан–липосома центрифугированием и измеряли интенсивность флуоресценции надосадочной жидкости. Последнюю пересчитывали в концентрацию липосом, использовав соответствующую калибровочную кривую, и представляли в виде зависимости концентрации свободных (не связанных с хитозаном) липосом от общей концентрации липосом в системе. Как следует из данных рис. 2, оба типа твердых липосом связываются с хитозановыми микрочастицами до концентрации 0.5 мг/мл, что соответствует комплексу с Nмакс = 150. Иными словами, каждая микрочастица хитозана диаметром 225 нм может в пределе связать 150 твердых анионных липосом диаметром 40 нм.
Рис. 2.
Зависимость концентрации липосом в надосадочной жидкости после отделения комплекса хитозан–липосома от общей концентрации липосом. νХол = 0.3 (темные точки) и 0.4 (светлые).
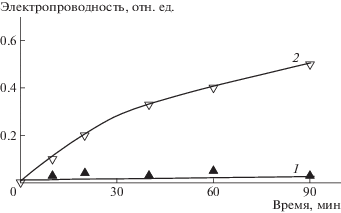
Критически важным является сохранение целостности липосом в составе мультилипосомального контейнера. Ответ на этот вопрос был получен с помощью метода кондуктометрии. Липосомы ДОФХ/КЛ2–/Хол с νХол = 0.3 и 0.4, внутренний объем которых был заполнен 1 М раствором соли (NaCl), адсорбировали на поверхности хитозановых микрочастиц и следили за изменением электропроводности суспензии. Появление дефектов в липосомальной мембране сопровождалось вытеканием соли в окружающий раствор и возрастанием электропроводности. Из результатов, представленных на рис. 3, следует, что из липосом с νХол = 0.3 соль не вытекала, т.е. они сохраняли свою целостность после адсорбции (кривая 1), в то время как липосомы с νХол = 0.4 теряли 20% соли в течение первых 20 мин и 50% соли через 1.5 ч после формирования комплекса (кривая 2).
Рис. 3.
Кинетика изменения относительной электропроводности комплекса хитозан–липосомы. νХол = 0.3 (темные точки) и 0.4 (светлые). Липосомы загружены 1М NaCl.
Для оценки размера мультилипосомальных контейнеров использовали метод динамического светорассеяния. На рис. 4 приведены результаты для комплекса хитозановых микрочастиц и липосом с νХол = 0.3. Именно эти липосомы обеспечивали формирование комплекса с максимальным содержанием интактных (неразрушенных) липосом. Размер комплекса увеличивался по мере заполнения поверхности микрочастиц липосомами и достигал максимального значения 420 нм в области полной нейтрализации заряда микрочастиц: ср. данные рис. 1 (кривую 2) и данные рис. 4. Дальнейшее повышение концентрации липосом приводило к уменьшению размера комплекса до 310 нм, очевидно, вследствие появления на поверхности микрочастиц избыточного отрицательного заряда, привнесенного адсорбированными липосомами.
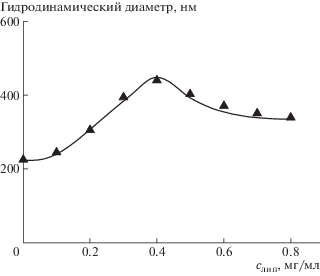
Согласно литературным данным, размер частиц носителя 250–400 нм обеспечивает его проникновение в клетки по механизму пассивного транспорта через увеличенные поры в кровеносных капиллярах в местах воспаления [28]. Представленные в работе результаты указывают на то, что электростатические комплексы хитозановых микрочастиц и твердых анионных липосом с мольной долей холестерина 0.3 удовлетворяют этому размерному требованию. В большинстве случаев размер частиц комплекса не превышает 400 нм; такие мультилипосомальные частицы представляют интерес для иммобилизации и доставки лекарственных веществ.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 19-03-00314).
Список литературы
Bozzuto G., Molinari A. // Int. J. Nanomed. 2015. V. 10. P. 975.
Burgess P., Hutt P.B., Farokhzad O.C., Langer R., Minick S., Zale S. // Nat. Biotechnol. 2010. V. 28. P. 1267.
Bozzuto G., Molinari A. // Int. J. Nanomed. 2015. V. 10. P. 975.
Knapp C.M., Whitehead K.A. // Exert. Opin. Drug Deliv. 2014. V. 11. P. 1923.
Song J., Jang J. // Adv. Colloid Interface Sci. 2014. V. 203. P. 37.
Cavalieri F., Tortora M., Stringaro A., Colone M., Baldassarri L. // J. Hosp Infect. 2014. V. 88. № 4. P. 183.
Lehner R., Wang X., Marsch S., Hunziker P. // Nanomedicine. 2013. V. 9. P. 742.
Yaroslavov A.A., Efimova A.A., Sybachin A.V., Chvalun S.N., Kulebyakina A.I., Kozlova E.V. // RSC Adv. 2015. V. 5. P. 31460.
Kuhn P., Eyer K., Robinson T., Schmidt F.I., Mercer J., Dittrich P.S. // Integr. Biol. 2012. V. 4. P. 1550.
Yaroslavov A.A., Efimova A.A., Rudenskaya G.N., Melik-Nubarov N.S., Grozdova I.D., Ezhov A.A., Chvalun S.N., Kulebyakina A.I., Razuvaeva E.V. // Mendeleev Commun. 2017. V. 27. P. 299.
Yaroslavov A.A., Zaborova O.V., Sybachin A.V., Kalashnikova I.V., Kesselman E., Schmidt J., Talmon Y., Rodriguez A.R., Deming T.J. // RSC Adv. 2015. V. 5. P. 98678.
Efimova A.A., Mulashkin F.D., Rudenskaya G.N., Evtushenko E.G., Orlov V.N., Melik-Nubarov N.S., Krivtsov G.G., Yaroslavov A.A. // Polymer Science B. 2018. V. 60. № 1. P. 84.
Yaroslavov A.A., Efimova A.A., Mulashkin F.D., Rudenskaya G.N., Krivtsov G.G. // Mendeleev Commun. 2018. V. 28. № 2. P. 140.
Bouaoud C., Lebouille J.G., Mendes E., De Braal H.E., Meesters G.M. // J. Liposome Res. 2016. V. 26. P. 103.
Bhardwaj U., Burgess D. // Int. J. Pharm. 2010. V. 388. P. 181.
Bulbake U., Doppalapudi S., Kommineni N., Khan W. // Pharmaceutics. 2017. V. 9. P. 12.
Sotiriosrigatos E.D., Viliotou V., Stathopoulos J.G. // Oncol. Rep. 2005. V. 13. P. 589.
Zhang J.A., Xuan T., Parmar M., Ma L., Ugwu S., Ali S., Ahmad I. // Int. J. Pharm. 2004. V. 270. P. 93.
Chong P.L., Zhu W., Venegas B. // Biochim. Biophys. Acta. 2009. V. 1788. P. 2.
Keller S.L., Anderson T.G., McConnell H.M. // Biophys. J. 2000. V. 79. P. 2033.
Yaroslavov A.A., Efimova A.A., Kostenko S.N. // Polymer Science A. 2012. V. 54. № 4. P. 264.
Song J., Suh C.H., Park Y.B., Lee S.H., Yoo N.C., Lee J.D., Kim K.H., Lee S.K. // Eur. J. Nucl. Med. 2001. V. 28. P. 489.
Zhang J., Xia W., Liu P., Cheng Q., Tahi T., Gu W., Li B. // Mar. Drugs. 2010. V. 8. P. 1962.
Efimova A.A., Sybachin A.V., Yaroslavov A.A. // Polymer Science C. 2011. V. 53. № 1. P. 89.
Bernkop-Schnürch A., Dünnhaupt S. // Eur. J. Pharm. Biopharm. 2012. V. 81. P. 463.
Маркушин С.Г., Коптяева И.Б., Гендон Ю.З., Васильев Ю.М., Кривцов Г.Г., Акопова И.И. // Журн. микробиологии, эпидемиологии и иммунобиологии. 2009. Т. 2. С. 40.
Al-Remawi M.M.A. // Am. J. Appl. Sci. 2012. V. 9. № 7. P. 1091
Borbás E., Sinkó B., Tsinman O., Tsinman K., Kiserdei É., Démuth B., Balogh A., Bodák B., Domokos A., Dargó G., Balogh G.T., Nagy Z.K. // Mol. Pharmaceut. 2016. V. 13. P. 3816.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)