

Высокомолекулярные соединения (серия А), 2019, T. 61, № 6, стр. 569-576
КОМПЛЕКСЫ АМФИФИЛЬНЫХ МОЛЕКУЛЯРНЫХ ЩЕТОК С ПОЛИИМИДНОЙ ОСНОВНОЙ ЦЕПЬЮ С ПОЛИ-N-ВИНИЛАМИДАМИ В СЕЛЕКТИВНЫХ РАСТВОРИТЕЛЯХ
Т. Н. Некрасова a, *, В. Д. Паутов a, Т. Д. Ананьева a, Т. К. Мелешко a, И. В. Иванов a, А. В. Якиманский a, b
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр. 31, Россия
b Институт химии Санкт-Петербургского государственного университета
198504 Санкт-Петербург, Петродворец, Университетский пр., 26, Россия
* E-mail: polar@imc.macro.ru
Поступила в редакцию 23.04.2019
После доработки 07.06.2019
Принята к публикации 20.06.2019
Аннотация
Методом поляризованной люминесценции при стационарном возбуждении изучено взаимодействие молекулярной щетки, состоящей из основной полиимидной цепи и люминесцентно меченых боковых цепей полиметакриловой кислоты, с поли-N-виниламидами (поли-N-винилпирролидон и поли-N-винилкапролактам) различной молекулярной массы в органических растворителях разного термодинамического качества по отношению к цепным блокам полимеров: в селективном (метанол) и в общем (диметилформамид). Понижение подвижности привитых цепей полиметакриловой кислоты с ростом молекулярной массы поли-N-виниламида, наблюдаемое и в селективном, и в общем растворителе, свидетельствовало о переходе от “рыхлой” к более плотной структуре интерполимерного комплекса. На основе анализа наносекундных времен релаксации, характеризующих подвижность участков привитых цепей полиметакриловой кислоты и отражающих изменение внутри- и межмолекулярных взаимодействий, показано, что в диметилформамиде формирование интерполимерных комплексов приводит к более “рыхлой” структуре.
BВЕДЕНИЕ
Интерполимерные комплексы (ИПК), образованные при взаимодействии макромолекул комплементарного химического строения за счет водородных связей, электростатических взаимодействий, ван-дер-ваальсовых или диполь-дипольных взаимодействий, переноса электрона в комплексах с переносом заряда, приобретают новые свойства, отличные от свойств их компонентов. Благодаря этому, они широко используются для создания новых композиционных материалов, применяемых в биотехнологии, медицине, мембранных технологиях, сельском хозяйстве и т.д. Исследование ИПК, образованных линейными поликарбоновыми кислотами с неионогенными водорастворимыми полимерами, и определение перспектив их применения началось в 60-х годах прошлого столетия с пионерских работ В.А. Кабанова, К. Abe и их коллег [1, 2]. Интерес к изучению подобных интерполимерных комплексов не ослабевает и в настоящее время [3, 4].
Активное развитие исследований в области ИПК на основе поликарбоновых кислот обусловлено перспективностью разработки на их основе новых материалов для практического применения. Послойная сборка в интерполимерные полиэлектролитные комплексы с использованием поли(мет)акриловых кислот является эффективным способом получения полимерных пленочных мембран для первапорационного разделения водно-органических смесей [5, 6], мембран для процессов диализа [7], микро- и нанокапсул с управляемой структурой для инкапсулирования лекарственных средств и их адресной доставки [8–10]. Большое число исследований посвящено подобным полиэлектролитным послойным материалам [3, 11, 12]. Новым направлением в этой области является изучение влияния архитектуры полимеров на формирование ИПК. Например, при сопоставлении полиэлектролитного комплексообразования линейного и звездообразного полимеров одного химического строения показано, что близкое расположение полимерных лучей в последнем усиливает комплексообразование [13].
Вместе с тем, все больший интерес в новейшей литературе привлекает комплексообразование полимеров за счет водородных связей [14–16], позволяющее формировать более сложные макромолекулярные структуры. На их основе получают самоорганизующиеся полимерные материалы [17, 18], способные к самовосстановлению [19], эффективные носители лекарственных препаратов с контролируемой скоростью их высвобождения [20, 21]. Поли(мет)акриловая кислота (ПМАК) способна образовывать ИПК с водородными связями со многими полимерами (ПЭО, ПВС, полиакриламид, простые эфиры целлюлозы, поли-N-виниламиды (ПВАМ)). Среди них такие ПВАМ, как поли-N-винилпирролидон (ПВП) и поли-N-винилкапролактам (ПВК), которые обладают высокой комплексообразующей способностью, обусловленной наличием боковой протоноакцепторной группы >N–C=O, хорошей совместимостью со многими полимерами, растворимостью в воде и в органических растворителях. Благодаря этому они широко используются в биотехнологии, медицине, мембранных технологиях в качестве модифицирующих добавок, а также при создании композиционных материалов [22–24]. В связи с активным синтезом полимеров нового поколения сложной архитектуры сополимеров (разветвленные, блок-, графт- и звездообразные), в том числе разветвленных привитых сополимеров (молекулярных полимерных щеток) с боковыми цепями ПМАК, несомненный интерес представляет исследование их комплексообразования с этими ПВАМ.
Согласно результатам теоретического моделирования и некоторым экспериментальным данным, сложная архитектура полимеров во многом обусловливает нетривиальную многоуровневую самоорганизацию разветвленных сополимерных макромолекул в растворе в структурные образования разной формы (сферы, везикулы, слоистая структура) [25, 26], обладающие разными свойствами. Показано, что на процессы самоорганизации влияют как внутренние параметры сополимеров (разветвленность, степень ветвления, длина и плотность прививки цепей), так и внешние условия, например природа растворителя. В случае ИПК для их формирования, помимо химической природы и молекулярной массы [3] полимерных компонентов, также важную роль играет природа растворителя [27]. Образование ИПК наблюдается не только в воде, но и в ряде полярных органических растворителей (ДМФА, N-метилпирролидон, метанол, этанол). При переходе от воды к органическому растворителю возможно изменение конформации макромолекул, образование сольватокомплексов с одним из компонентов, препятствующее образованию ИПК.
Взаимодействие комплементарных макромолекул в растворах изучают различными методами [28]. Среди них метод поляризованной люминесценции позволяет установить особенности структурной организации ИПК, в том числе сформированных линейными и привитыми полимерными цепями. Определяемые с его помощью наносекундные времена релаксации характеризуют внутримолекулярную подвижность участков цепи взаимодействующих макромолекул и являются чувствительным индикатором структурной организации ИПК. Ранее в работе [29] с помощью поляризованной люминесценции было исследовано взаимодействие между цепями полиметакриловой кислоты, привитыми к полиимиду (ПИ–прив–ПМАК), и макромолекулами линейного ПВП в воде и ДМФА. Показано, что формирование интерполимерных комплексов [ПВП] : : [ПИ–прив–ПМАК] сопровождается уменьшением подвижности обеих взаимодействующих макромолекул. При этом подвижности участков их макромолекул оказались выше подвижности таких участков в ИПК, образованных линейными цепями ПМАК и ПВП. Это указывает на большую дефектность (“рыхлость”) интерполимерных комплексов, сформированных при взаимодействии привитых цепей ПМАК с ПВП. Однако подвижность входящих в состав ИПК макромолекул различной молекулярной массы в растворителях разной природы остается мало изученной.
В данной работе исследованы релаксационные свойства, характеризующие внутримолекулярную подвижность цепей ПМАК, регулярно привитых к полиимиду, с линейными макромолекулами ПВП и ПВК различной молекулярной массы в метаноле, который является селективным растворителем, и в общем для основной ПИ цепи и боковых ПМАК цепей сополимера растворителе ДМФА.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез регулярно привитого сополиимида ПИ–прив–ПМАК
Регулярно привитой сополиимид ПИ-прив-ПМАК:
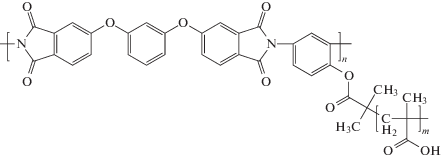
синтезировали в результате последовательного проведения четырех стадий [30, 31].
На первой стадии синтезировали исходный гидроксилсодержащий ПИ на основе 2,4-диаминофенола и 4,4'-(1,3-фенилен-диокси)бисфталевого ангидрида.
Во время второй стадии функционализировали ПИ 2-бром-изобутиратными группами для мультицентрового полиимидного макроинициатора (ПИМИ) с этими группами в каждом повторяющемся звене ПИ.
На третьей стадии получали привитой сополиимид с боковыми цепями поли(трет-бутилметакрилата) (ПИ–прив–ПТБМА) путем контролируемой радикальной полимеризации с переносом атома (ATRP) трет-бутилметакрилата на ПИМИ.
И, наконец, во время четвертой стадии проводили кислотный гидролиз сложноэфирных групп боковых цепей сополимера-прекурсора ПИ–прив–ПТБМА в условиях, обеспечивающих 100% конверсию.
Для осуществления контролируемой радикальной полимеризации применяли методику ATRP с использованием активаторов, генерируемых электронным переносом (Activators Generated by Electron Transfer, AGET ATRP) с использованием медь-содержащего активатора и 2-этилгексаноата олова в качестве восстановителя. Согласно данным ГПХ средняя степень поликонденсации основной ПИ цепи составляла n = 52 (Mn = 31.2 × 103, Ð = 2.5), средняя степень полимеризации регулярно привитых боковых цепей ПМАК m = 110 (Ð = 1.6). Рассчитанная на основе этих параметров и значений конверсии мономера (из данных гравиметрии) плотность прививки боковых цепей составляла 100%.
Синтез молекулярных щеток, содержащих ПИ основную цепь и люминесцентно меченые регулярно привитые цепи полиметакриловой кислоты (ПИ–прив–ПМАК*), осуществляли по методике, описанной в работе [32].
Люминесцентные метки антраценовой структуры ковалентно присоединены к звеньям ПМАК в количестве 1 люминесцентная метка на 490 звеньев ПМАК.
ПВП и ПВК получали радикальной полимеризацией. Использовали фракции ПВП с М = 100 × × 103 (ПВП100) и ПВК с М = 120 × 103 (ПВК120) и М = 12 × 103 (ПВК12), а также ПВП с М = 10 × 103 (ПВП10) фирмы “Fluka”.
Соотношение взаимодействующих компонентов в растворе характеризовали величиной β = = [ПВАМ] : [ПИ–прив–ПМАК*] (концентрации приведены в молях мономерных звеньев).
Взаимодействие ПИ–прив–ПМАК* с ПВП и ПВК изучали методом поляризованной люминесценции. Измерения поляризации люминесценции P раствора люминесцентно меченого сополимера ПИ–прив–ПМАК* при взаимодействии с ПВАМ проводили на установке, описанной в работе [33] и совмещенной с персональным компьютером для автоматической регистрации и обработки экспериментальных данных. Наносекундные времена релаксации τВМП, характеризующие внутримолекулярную подвижность участков привитых цепей ПМАК, определяли, используя соотношение:
(1)
${{\tau }_{{{\text{ВМП}}}}} = {\text{ }}(1{\text{/}}P_{0}^{'} + 1{\text{/}}3)3{{\tau }_{{{\text{фл}}}}}{\text{/}}(1{\text{/}}P--1{\text{/}}P_{0}^{'}),$Рис. 1.
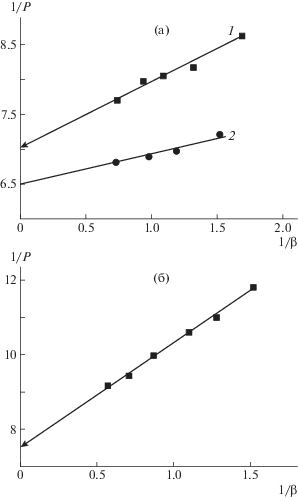
Зависимости обратной величины поляризации люминесценции раствора 1/Р от 1/β для раствора, содержащего [ПВП] : [ПИ–прив–ПМАК*] (1) и [ПВК] : [ПИ–прив–ПМАК*] (2), в метаноле (а) и ДМФА (б).
Для корректного сопоставления времен релаксации, полученных в растворителях с разной вязкостью, их приводили к одному значению параметра Tпр/ηпр = 335, соответствующему значению Т/η воды при 298 К, с помощью соотношения
учитывая, что при фиксированной температуре время релаксации линейно зависит от вязкости растворителя (при условии отсутствия конформационных превращений). Концентрация меченого полимера в растворе не превышала спол = = 0.3 мг/мл.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В сополимерах ПИ–прив–ПМАК* основная ПИ цепь гидрофобна, ее изолированный аналог – ПИ на основе 2,4-диаминофенола и 4,4'-(1,3-фенилен-диокси)бисфталевого ангидрида – растворим в ДМФА и нерастворим в метаноле. Линейные макромолекулы ПМАК представляют собой дифильный полимер, содержащий в каждом мономерном звене неполярную группу α-CH3 и полярную группу СООН. Наличие двух типов групп определяет зависимость конформационных свойств ПМАК от природы растворителя [22]. В воде в неионизованном состоянии (0.002 н HCl, θ-растворитель, 30°С) в макромолекулах ПМАК образуются стабилизированные гидрофобными взаимодействиями неполярные домены, взаимодействие между которыми приводит к компактизации макромолекулярного клубка. Эта структура разрушается при добавлении алифатических спиртов и ДМФА. Метанол для ПИ–прив–ПМАК является селективным растворителем – θ-растворителем для ПМАК (26°С) и осадителем для ПИ.
Ранее в работе [29] было показано, что в сополимерах ПИ–прив–ПМАК* с контурной длиной основной цепи ~32 нм (n = 16, длина повторяющегося звена основной ПИ цепи составляет ~2 нм) τВМП, характеризующие подвижность привитых цепей ПМАК*, в метаноле уменьшаются до 15 нс по сравнению с водой, что указывает на ослабление гидрофобных взаимодействий. Так как влияние длины основной цепи на релаксационные свойства привитых цепей ранее не изучалось, то для исследуемого в данной работе образца ПИ–прив–ПМАК* с контурной длиной основной цепи ~104 нм (n = 52) были определены времена релаксации. Обнаружено, что с таким увеличением длины основной цепи при сохранении средней степени полимеризации боковых цепей m ~ 100 и близкой плотности прививки значения τВМП уменьшаются до 10 нс. Возможно, это связано с различной подвижностью участков привитых цепей ПМАК, находящихся на разном расстоянии от точки прививки. В этом случае при взаимодействии люминесцирующей метки 9-антрилдиазометана с карбоксильными группами сначала реагируют наиболее подвижные участки цепи. Ранее [35] влияние структурно-кинетической неоднородности участков макромолекул сополимера на реакционную способность функциональных групп сополимера было исследовано на примере сополимеров стирола и α-метилстирола. Показано, что реакционная способность функциональных групп выше в тех сополимерах, участки цепей которых обладают высокой подвижностью [35]. Кроме того, в полужестких макромолекулах (основная цепь) с ростом числа звеньев возможен переход от вытянутой конформации макромолекулы к гауссову клубку [36], что также обусловливает изменение плотности звеньев кислоты, и, соответственно, уменьшение их заторможенности по мере удаления от точки прививки.
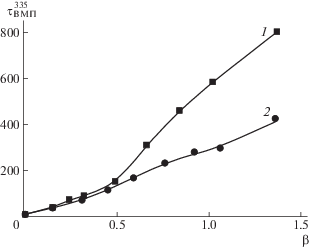
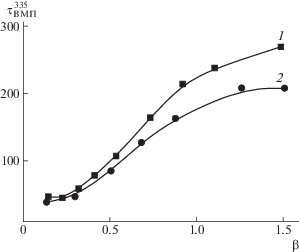
На рис. 2 приведено изменение τВМП335 привитых цепей ПМАК* в ПИ–прив–ПМАК* при взаимодействии в метаноле с ПВК120 и ПВП100. Видно, что с ростом β значения τВМП335 увеличиваются как для системы [ПВП100]/[ПИ–прив–ПМАК*], так и для [ПВК120]/[ПИ–прив–ПМАК*], что отражает уменьшение подвижности (возрастание заторможенности) участков привитых цепей ПМАК*, вызванное взаимодействием компонентов системы и формированием ИПК. С увеличением β до ~0.5 значения τВМП335 практически совпадают для обеих систем. При β > 0.5, значения τВМП335 для системы [ПВК120]/[ПИ–прив–ПМАК*] растут быстрее по сравнению с τВМП335 для [ПВП100]/[ПИ–прив–ПМАК*]. Например, при β = 1 значения τВМП335 = 535 и 229 нс соответственно. Такой характер зависимости τВМП335 от β, по-видимому, отражает различие в структуре образующихся ИПК. С ростом значения β происходит уменьшение степени сольватации взаимодействующих цепей за счет образования системы кооперативных водородных связей между протонодонорными и протоноакцепторными группами. Формирующаяся структура ИПК дополнительно стабилизирована лиофобными взаимодействиями реагирующих макромолекул. Объемные заместители в макромолекулах ПВК сильнее влияют на заторможенность цепей ПМАК*. Кроме этого, различие в значениях τВМП335 для ИПК [ПВП100]/[ПИ–прив–ПМАК*] и [ПВК120]/[ПИ–прив–ПМАК*] может быть также связано с наличием дефектности структуры: с различной длиной и конфигурацией петель, разным числом сбоев непрерывной последовательности связей.
Рис. 2.
Зависимость τВМП335 привитых цепей ПМАК* в ПИ–прив–ПМАК* при добавлении ПВК120 (1), ПВП100 (2) в метаноле.
Формирование ИПК в метаноле сопровождается изменением длительности люминесцентного свечения люминесцирующей метки τфл, присоединенной к ПМАК* (рис. 3).
Рис. 3.
Изменение длительности люминесцентного свечения τфл люминесцирующей метки при образовании ИПК ПИ–прив–ПМАК* с ПВК (1) и ПВП (2) в метаноле.
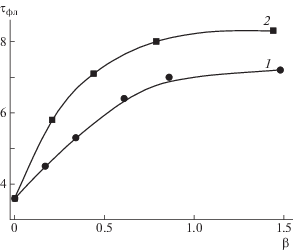
По мере формирования ИПК τфл увеличивается в 2.0–2.3 раза. Столь существенное изменение τфл указывает на увеличение жесткости окружения люминесцирующей метки и заторможенность ее движений (изменение τфл красителей при переходе от жидких растворов к замороженным) [37]. Это свидетельствует о том, что участок цепи, содержащий люминесцирующие метки, встраивается в ИПК. Высокочастотные движения люминесцирующих меток, вклад которых в спектр времен релаксации определяется параметром $1{\text{/}}P_{0}^{'}$, “замораживаются” и значение параметра уменьшаются от 14 до 7. При этом значения τфл для [ПВК120]/[ПИ–прив–ПМАК*], ниже, чем для [ПВП100]/[ПИ–прив–ПМАК*], что указывает на различный характер надмолекулярной организации ИПК.
В работе [38] показано, что структурно-динамические характеристики ИПК, образующихся при взаимодействии ПИ–прив–ПМАК с ПВАМ в воде, зависят от молекулярной массы ПВАМ. На рис. 4 приведено изменение τВМП335 ПИ–прив–ПМАК* при взаимодействии с низкомолекулярными ПВАМ ПВП10 и ПВК12 в метаноле. Сравнение данных рис. 4 и 2 показывает, что на всех стадиях формирования ИПК значения τВМП335 почти в 2 раза меньше, чем при формировании ИПК с высокомолекулярными ПВАМ.
Рис. 4.
Зависимость τВМП335 от β при взаимодействии ПИ–прив–ПМАК* с ПВК12 (1) и ПВП10 (2) в метаноле; СПИ–прив–ПМАК* = 0.3 мг/мл.
Степень полимеризации k цепей ПВП100 (ПВК120) nПВАМ ~ 1000 превышает значения m привитых цепей ПМАК mПМАК ~ 110 почти на порядок, в то время как для ПВП10 (ПВК12) значения nПВАМ ~ 90 сравнимы с mПМАК. Как и в случае ИПК [ПВАМ]/[ПИ–прив–ПМАК*] в воде, можно предположить, что в случае близких молекулярных масс привитых цепей ПМАК* и макромолекул ПВП (ПВК) взаимодействие происходит в пределах одной “ветви” молекулярной щетки (структура, изображение слева). С увеличением же молекулярной массы ПВП (ПВК) участки цепей ПВАМ, не включенные в ИПК со звеньями ПМАК в привитой цепи, могут контактировать либо с соседними, либо с удаленными ветвями ПМАК* по месту прививки (изображение справа):
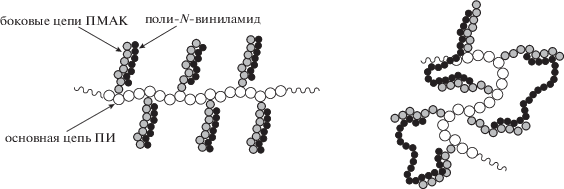
Это приводит к дополнительному усилению внутримолекулярных взаимодействий и компактизации макромолекулярного клубка. При больших β появление опалесценции указывает на усиление межмолекулярных взаимодействий и формирование ассоциатов.
Влияние ММ линейного полимера на формирование и свойства полиэлектролитных комплексов молекулярных щеток, а также липосом наблюдали в работах [39, 40].
В отличие от метанола, ДМФА является общим растворителем для обоих типов цепей сополимера ПИ–прив–ПМАК*. При смешении ПИ–прив–ПМАК* с ПВАМ в ДМФА, значения τВМП335 с ростом β увеличиваются (рис. 5) как для системы [ПВП100]/[ПИ–прив–ПМАК*], так и для [ПВК120]/[ПИ–прив–ПМАК*] (см. рис. 2), что указывает на создание ИПК. Особенности формирования ИПК, установленные в метаноле, проявляются и в ДМФА, а именно, влияние природы ПВАМ (рис. 5, кривые 1 и 2) и молекулярной массы (рис. 5, кривая 3). При этом, как и в метаноле, взаимодействие с ПВК влечет формирование ИПК, в которых цепи ПМАК* более заторможены, что, по-видимому, отражает бóльший вклад лиофобных взаимодействий в стабилизацию ИПК.
Рис. 5.
Изменение τВМП335 привитых цепей ПМАК* в ПИ–прив–ПМАК* при добавлении ПВК120 (1), ПВП100 (2) в ДМФА; 3 – зависимость приведена для ПВП с М = 68 × 103 из работы [29].
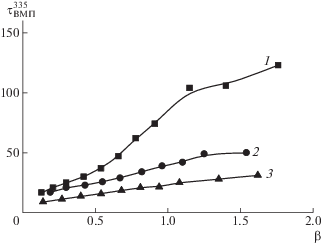
Однако значения τВМП335 в ДМФА при одинаковых значениях β существенно меньше соответствующих значений в метаноле при одинаковой молекулярной массе ПВАМ. Это указывает на формирование более “рыхлой” структуры ИПК, в которой подвижность цепей ПМАК менее заторможена.
Длительность свечения люминесцирующей метки, присоединенной к ПИ–прив–ПМАК*, при формировании ИПК в ДМФА практически не меняется, что также подтверждает образование более “рыхлой” дефектной структуры ИПК. Формирование более “рыхлой” структуры ИПК, образующейся в ДМФА, может быть связано с тем, что ДМФА, как и другие амидные растворители (ДМАА, N-метилпирролидон), формирует сольватокомплексы как со звеньями ПВАМ, так и полиимида [41]. Эти взаимодействия с ДМФА препятствуют образованию протяженных последовательностей между комплементарными звеньями полимеров. Конкуренция между контактами полимер-растворитель и полимер-полимер в ДМФА определяет особенности структурной организации ИПК, которая характеризуется меньшим числом связанных в ИПК цепей, меньшей длиной протяженных участков взаимодействующих цепей и большим числом петель. Заметим, что в случае линейной ПМАК при формировании ИПК с ПВП или ПВК в ДМФА и метаноле также была обнаружена более чем на порядок разница в значениях τВМП335 [42].
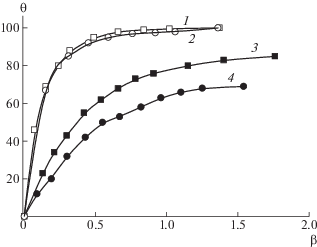
Для сравнительной оценки влияния растворителя на способность исследуемых полимеров образовывать ИПК была определена доля θ цепей ПМАК*, вошедших в комплекс:
(2)
$\theta {\text{ }} = {{Р}_{2}}(1{\text{ }}--{{Р}_{1}}{\text{/}}Р){\text{/}}({{Р}_{2}}--{{Р}_{1}}),$На рис. 6 приведено изменение доли связанных в ИПК цепей ПМАК при формировании ИПК для [ПВК120]/[ПИ–прив–ПМАК*] (кривые 1, 3) и [ПВП100]/[ПИ–прив–ПМАК*] (кривые 2, 4) в метаноле и ДМФА. Видно, что в метаноле цепи ПМАК* практически полностью включены в ИПК при β = 0.5, при этом подвижность цепей ПМАК в наносекундном диапазоне практически полностью заторможена, что связано не только с образованием протяженной последовательности Н-связей между карбоксильными группами ПМАК и карбонильными группами ПВАМ, но и с плотной упаковкой взаимодействующих цепей и формированием структуры ИПК типа глобулярной. В ДМФА в этих условиях в ИПК включено ~50% участков цепей ПМАК и их доля растет с ростом концентрации ПВАМ в растворе. Существенная разница зависимостей θ (β) и значений времен релаксации в метаноле и ДМФА указывает не только на различие в константах образования ИПК, но и на разную укладку взаимодействующих цепей в ИПК – более плотная в метаноле и “рыхлая” в ДМФА за счет образования прочных сольватокомплексов со звеньями образующих ИПК макромолекул.
ЗАКЛЮЧЕНИЕ
В ходе проведенного исследования с помощью метода поляризованной люминесценции количественно определены структурно-динамические характеристики интерполимерных комплексов разветвленного привитого сополимера (макромолекулярной щетки) ПИ–прив–ПМАК с линейными гомополимерными поли-N-виниламидами (поли-N-винилпирролидон и поли-N-винилкапролактам) разной молекулярной массы. Полученные значения характеристик подвижности люминесцентной метки в боковых цепях ПМАК привитого сополимера свидетельствуют о влиянии на структуру и стабильность комплексов не только природы полиамида и его ММ, но и разветвленной архитектуры щетки. В результате использования для формирования ИПК растворителей, обладающих разным термодинамическим сродством к цепным блокам полимерных компонентов, обнаружена также зависимость структуры и стабильности комплекса от природы блоков, составляющих щетку. Таким образом, в работе продемонстрированы новые дополнительные возможности для контролируемого регулирования свойств ИПК путем использования разветвленных сополимеров нового поколения с разнородными цепными блоками и целенаправленного выбора растворителя, обладающего разным термодинамическим качеством по отношению к этим блокам.
Работа выполнена в рамках гранта Правительства Российской Федерации для государственной поддержки научных исследований, проводимых под руководством ведущих ученых (договор № 14.W03.31.0022).
Список литературы
Кабанов В.А., Паписов И.М. // Высокомолек. соед. А. 1979. Т. 21. № 1. С. 234.
Tsuchida E., Abe K. // Adv. Polym. Sci. 1982. V. 45. P. 1.
Hydrogen-bonded Interpolymer Complexes. Formation, Structure and Applications / Eds. by V. Khutoryanskiy, G. Staikos. Singapore: World Scientific, 2009.
Ruiz-Rubio L., Laza J.M., Pérez L., Rioja N., Bilbao E. // Colloid. Polym. Sci. 2014. V. 292. № 2. P. 423.
Kononova S.V., Volod’ko A.V., Petrova V.A., Kruchinina E.V., Baklagina Y.G., Chusovitin E.A., Skorik Y.A. // Carbohydr. Polym. 2018. V. 181. P. 86.
Агеев E.П., Вихорева Г.A., Гальбрайх Л.С., Матушкина Н.Н., Чайка E.M., Яминский И.В. // Высокомолек. соед. A. 1998. Т. 40. С. 1198.
Zhumadilova G.T., Gazizov A.D., Bimendina L.A., Kudaibergenov S.E // Polymer. 2001. V. 42. P. 2985.
Zelikin A.N., Becker A.L., Johnston A.P.R., Wark K.L., Turatt F., Caruso F. // ACS Nano. 2007. V. 1. № 1. P. 63.
Lomova M.V., Ivanov I.V., German S.V., Meleshko T.K., Pavlov A.M., Inozemtseva O.A., Antipina M.N., Yakimansky A.V., Sukhorukov G.B., Gorin D.A. // J. Polym. Res. 2015. V. 22. № 10. P. 1.
Ibragimova A.R., Mirgorodskaya A.B., Vasilieva E.A., Khairutdinova E.I., Meleshko T.K., Ivanov I.V., Yakimansky A.V., Nizameev I.R., Kadirov M.K., Zakharova L.Y. // Coll. Surf. A. 2017. V. 526. P. 20.
Kulkarni A.D., Vanjari Yo.H., Sancheti K.H., Patel H.M., Belgamwar V.S., Surana S.J., Pardeshi C.V. // Artif. Cells, Blood Substitues, Immobilization Biotechnol. 2016. V. 44. № 7. P. 1615.
Kabanov V.A. // Russ. Chem. Rev. 2005. V. 74. № 1. P. 3.
Plamper F.A., Gelissen A.P., Timper J., Wolf A., Zezin A.B., Richtering W., Tenhu H., Simon U., Mayer J., Borisov O.V., Pergushov D.V. // Macromol. Rapid Commun. 2013. V. 34. № 10. P. 855.
Moustafine R.I., Viktorova A.S., Khutoryanskiy V.V. // Int. J. Pharm. 2019. V. 558. P. 53.
Hebbeker P.L., Steinschulte A.A., Schneider S., Okuda J., Möller M., Plamper F.A., Schneider S. // Macromolecules. 2016. V. 49. № 22. P. 8748.
Kharlampieva E., Sukhishvili S.A. // J. Macromol. Sci. C. 2006. V. 46. № 4. P. 377.
Xu W., Ledin P.A., Iatridi Z., Tsitsilianis C., Tsukruk V.V. // Angew. Chem., Int. Ed. 2016. V. 55. P. 4908.
Xu W., Steinschulte A.A., Plamper F.A., Korolovych V.F., Tsukruk V.V. // Chem. Mater. 2016. V. 28. № 3. P. 975.
Che S., Bi X., Sun L., Gao J., Huang P., Fan X., You Z., Wang Y. // ACS Appl. Mater. Int. 2016. V. 8. P. 20591.
Chun M.-K., Cho C.-S., Choi H.-K. // J. Controll. Release. 2002. V. 81. P. 327.
Khutoryanskiy V.V. // Int. J. Pharm. 2007. V. 334. P. 15.
Yoo J.E., Kim J.H., Kim Y., Kim C.K // J. Membr. Sci. 2003. V. 216. № 1–20. P. 95.
Mun G.A., Nurkeeva Z.S., Khutoryanskiy V.V., Bitekenova A.B. // Macromol. Rapid. Comm. 2000. V. 21. № 7. P. 381.
Кирш Ю.Э. Поли-N-винилпирролидон и другие поли-N-виниламиды. М.: Наука, 1998.
Zhulina E.B., Borisov O.V. // ACS Macromol. Lett. 2013. V. 2. P. 292.
Polotsky A.A., Kazakov A.D., Birshtein T.M. // Polymer. 2017. V. 130. P. 242.
Kononova S.V., Gubanova G.N., Romashkova K.A., Smirnova V.E., Popova E.N., Vlasova E.N., Kruchinina E.V., Gofman I.V., Saifutdinova I.F., Romanov D.P. // Polymer Science A. 2016. V. 58. № 3. P. 419.
Khutoryanskiy V.V., Smyslov R.Y., Yakimansky A.V. // Polymer Science A. 2018. V. 60. № 5. P. 553.
Pautov V.D., Nekrasova T.N., Anan’eva T.D., Meleshko T.K., Ilgach D.M., Yakimansky A.V. // Polymer Science A. 2013. V. 55. № 9. P. 535.
Yakimansky A.V., Meleshko T.K., Ilgach D.M., Bauman M.A., Anan’eva T.D., Klapshina L.G., Lermontova S.A., Balalaeva I.V., Douglas W.E. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 4267.
Meleshko T.K., Ivanov I.V., Kashina A.V., Bogorad N.N., Simonova M.A., Zakharova N.V., Filippov A.P., Yakimansky A.V. // Polymer Science B. 2018. V. 60. № 1. P. 35.
Pautov V.D., Nekrasova T.N., Anan’eva T.D., Meleshko T.K., Ilgach D.M., Yakimansky A.V. // Polymer Science A. 2013. V. 55. № 9. P. 526.
Temperature-Responsive Polymers: Chemistry, Properties and Applications / Eds. by V.V. Khutoryanskiy, T.K Georgiou. West Sussex: Wiley, 2018.
Anufrieva E.V., Birstein T.M., Nekrasova T.N., Ptitsyn O.B., Sheveleva T.V. // J. Polym. Sci., Polym. Symp. 1968. V. 16. P. 3519.
Краковяк М.Г., Ануфриева Е.В., Лущик В.Б., Громо-ва Р.А. // Высокомолек. соед. Б. 1978. Т. 20. № 2. С. 131.
Цветков В.Н. Жесткоцепные полимерные молекулы. Л.: Наука, 1986.
Теренин А.Н. Фотоника молекул красителей и родственных органических соединений. Л.: Наука, 1967.
Nekrasova T.N., Pautov V.D., Anan’eva T.D., Meleshko T.K., Ivanov I.V., Yakimansky A.V. // Polymer Science C. 2018. V. 60. № 1. P. 172.
Ivashkov O.V., Sybachin A.V., Efimova A.A., Pergushov D.V., Orlov V.N., Schmalz H., Yaroslavov A.A. // Chem. Phys. Chem. 2015. V. 16. № 13. P. 2849.
Бессонов М.И., Котон М.М., Кудрявцев В.В., Лайус Л.А. Полиимиды – класс термостойких полимеров. Л.: Наука, 1983.
Ануфриева Е.В., Koтoн M.M., Некрасова T.Н., Сазанов Ю.Н., Дауэнгауэр С.A., Лущик В.Б., Романова M.С., Спирина T.Н., Шевелева T.В. // Высокомолек. соед. А. 1988. Т. 30. № 8. С. 1773.
Ануфриева Е.В., Рамазанова M.Р., Краковяк M.Г., Лущик В.Б., Некрасова Н.Н., Шевелева T.В. // Высокомолек. соед. А. 1991. V. 33. № 6. P. 1180.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)