

Высокомолекулярные соединения (серия А), 2021, T. 63, № 4, стр. 249-262
НЕТКАНЫЕ МАТЕРИАЛЫ НА ОСНОВЕ УЛЬТРАТОНКИХ ВОЛОКОН ПОЛИ(3-ГИДРОКСИБУТИРАТА) С КОМПЛЕКСОМ ХЛОРИД ОЛОВА–ПОРФИРИН, ПОЛУЧЕННЫХ ЭЛЕКТРОФОРМОВАНИЕМ
С. Г. Карпова a, *, А. А. Ольхов b, c, А. Л. Жулькина c, А. А. Попов a, b, А. Л. Иорданский c
a Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
b Российский экономический университет им. Г.В. Плеханова
117997 Москва, Стремянный пер., 36, Россия
c Институт химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: karpova@sky.chph.ras.ru
Поступила в редакцию 18.11.2020
После доработки 29.01.2021
Принята к публикации 11.02.2021
Аннотация
Методами дифференциально-сканирующей калориметрии, электронного парамагнитного резонанса и сканирующей электронной микроскопии изучено влияние малых добавок (1–5%) комплекса хлорид олова–тетрафенилпорфирин на структуру и свойства ультратонких волокон на основе поли-3-гидроксибутирата. Показано, что при добавлении в волокна полимера порфиринового комплекса значительно возрастает энтальпия плавления и молекулярная подвижность, уменьшается концентрация зондового радикала в аморфных областях полимера. Экспозиция полимера в водной среде при 70°С приводит к резкому уменьшению энтальпии плавления, концентрации зондового радикала и росту молекулярной подвижности в композициях с содержанием порфиринового комплекса 5%. Отжиг волокон при 140°C вызывает рост энтальпии плавления и времени корреляции, а также снижение концентрации зондового радикала. Структура волокон сопоставлена со структурой ранее изученных ультратонких волокон с добавками комплексов разной стереоконфигурации.
ВВЕДЕНИЕ
Биодеградируемые биополимеры благодаря чрезвычайно широкому спектру применения весьма привлекательны в области биологии и медицины. Биоразлагаемые и биосовместимые полимеры бактериального происхождения, например поли-3-гидроксибутират (ПГБ), активно используют в качестве хирургических имплантатов в медицине, каркасов для клеточных культур в биоинженерии, новых лекарственных форм и систем в фармацевтике. Чтобы заменить традиционные синтетические полимеры биодеградируемыми, в ведущих научных организациях мира создают новые биополимеры, разрабатывают смесевые и композиционные полимерные материалы на основе природных полимеров и дисперсных наполнителей [1–3]. За последнее десятилетие в литературе появилось много работ по порфиринам и комплексам с металлами. Интерес к данному классу веществ объясняется уникальной геометрической и электронной структурой молекул. Нелинейно-оптические свойства и высокая поляризуемость делают металлопорфирины перспективными для применения в качестве активных сред оптической коммуникации [4, 5], хранения информации и обработки оптических и электрических сигналов [6, 7]. Комплексы переходных металлов, таких как железо, марганец, кобальт, с порфиринами считаются гомогенными катализаторами автоокисления ряда биогенных веществ. В этом процессе происходит промежуточное образование действенных форм кислорода – супероксидного анион-радикала, пероксидного и гидроксильного радикалов, пероксида водорода, цитостатическая активность которых хорошо известна. Указанные радикальные и ион-радикальные частицы вызывают окислительные деструктивные реакции в клетках, обусловливая бактерицидный эффект [8, 9].
Физико-химические свойства порфириновых металлокомплексов определяются химическим состоянием атома металла, а также пространственной геометрией атомарного окружения координационного центра. Благодаря своей пространственной структуре молекулы металлокомплексов порфиринов оказывают существенное воздействие на кристаллизацию и сегментальную ориентацию макромолекул полимеров при формировании композитных матриц на их основе.
Наиболее перспективными носителями функциональных низкомолекулярных веществ (частиц) можно назвать полимерные волокна наноразмерного диапазона. Одним из передовых методов получения таких волокон является электроформование полимерного раствора. Использование ряда природных полимеров, например поли-(3-гидроксибутирата) или полимолочной кислоты, создает дополнительные преимущества при разработке волоконных и матричных систем для экологических задач и медицины. Данные полимеры биосовместимы, одновременно проявляя свойства контролируемой биодеструкции без образования токсичных продуктов. Такие параметры волокон, как степень ориентации макромолекул, кристалличность, диаметр, геометрия поперечного сечения и пористость, определяют не только механические свойства фибриллярных материалов, но и их функциональные характеристики, наиболее важными из которых считаются скорость и механизм деградации макромолекул, сорбционная емкость лекарственного вещества, его диффузия. В предыдущих работах [10–16] было рассмотрено структурообразование волокнистых материалов на основе ПГБ, содержащих дипиридамол, хитозан, наночастицы диоксида титана и кремния, а также комплексов хлорид железа(III)–порфирин, цинк–порфирин, хлорид марганца–порфирин. Показано влияние комплексов FeCl–тетрафенилпорфирин, Zn–тетрафенилпорфирин, MnCl–тетрафенилпорфирин на структуру кристаллической и аморфной фаз волокон ПГБ. Наличие в них химически активных полярных функциональных групп способствует тому, что комплексы вступают в межмолекулярное взаимодействие с полимером, степень которого зависит от стереоконфигурации комплекса (пространственного расположения атомов Cl относительно плоскости комплекса). В результате такого взаимодействия может происходить как торможение, так и ускорение процессов кристаллизации, ориентации или релаксации полимерных макромолекул. Комплексы металлов с тетрафенилпорфиринами обладают уникальными фотокаталитическими и антимикробными свойствами [17, 18]. В связи с этим, создание нетканых высокопористых волокнистых биополимерных матриц для гигиены, терапии бактериальных инфекций и другого является практически важным направлением. Для формирования матриц с заданными свойствами необходимо установить взаимосвязь структуры комплексов, их концентрации в полимерном растворе со структурно-динамическими параметрами волокнистого материала. В работе [19] показано, что различия в физико-механических свойствах композиций связаны и с влиянием добавки на надмолекулярную структуру ПГБ, и с зависимостью формовочных свойств растворов от вида выбранного соединения порфирина и его концентрации.
Данные о структуре и свойствах материала на основе ПГБ и комплекса SnCl2–порфирин представляют интерес, так как этот комплекс применяется для создания гибридных структур для фотопереноса энергии и электрона, фотокатализаторов для разрушения органических токсикантов, фотосенсибилизаторов для медицинской диагностики и терапии, а также разработки фотоэлементов для водородной энергетики. Существенным преимуществом комплексов порфиринов с оловом(IV) можно назвать наличие у них двух экстралигандов, направленных по разные стороны от плоскости порфиринового макроцикла. Данная структурная особенность обусловливает значительно меньшую склонность таких комплексов к агрегации по сравнению с комплексами порфиринов с металлами в степени окисления +2 и +3. В зависимости от природы металла комплексы проявляют разную склонность к агрегации, что определяет их способность выступать в качестве образователя зародышей кристаллизации полимера [20–23].
Цель настоящей работы – получить данные о влиянии концентрации комплекса SnCl2–тетрафенилпорфирин на кристалличность и молекулярную динамику аморфных областей ПГБ, а также диаметр и геометрию поперечного сечения волокон на основе этого полимера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В данной работе использовали полигидроксибутират серии 16F, синтезированный микробиологическим методом компанией “Biomer ®” (Германия). Средневязкостная молекулярная масса ПГБ составляла 2.06 × 105, плотность ρ = = 1.248 г/см3, температура плавления Тпл = 176°С, степень кристалличности ~63%.
В качестве модифицирующего вещества для создания волокнистых матриксов применяли комплекс хлорида олова (SnCl2) с тетрафенилпорфирином (ТФП), полученный по методике [24] и предоставленный А.В. Лобановым (Институт химической физики РАН).
Формовочные растворы ПГБ и ПГБ–SnCl2–ТФП в хлороформе готовили при температуре 60°C с помощью магнитной мешалки. Концентрация ПГБ в растворе 7 мас. %, содержание SnCl2–ТФП составляло 1, 3 и 5% от массы ПГБ. Волокна получали методом электроформования на однокапиллярной лабораторной установке с диаметром капилляра 0.1 мм при напряжении электрического тока 12 кВ, расстоянии между электродами 18 см, электропроводности раствора 10 мкСм/см [25]. Диаметр волокон ~0.1–0.2 мм. В работе исследовали нетканый материал из этих волокон.
ЭПР-спектры (Х-диапазона) регистрировали на автоматизированном спектрометре ЭПР-В (Институт химической физики РАН). Микроволновая мощность в резонаторе не превышала 7 мВт, что позволяло избежать эффектов насыщения. При записи спектров амплитуда модуляции всегда была существенно меньше ширины резонансной линии и не больше 0.5 Гс. В качестве зонда использовали стабильный нитроксильный радикал ТЕМПО. Радикал вводили в волокна из газовой фазы при температуре 50°C.
Концентрация радикала в полимере не превышала 10–3 моль/л. Экспериментальные спектры спинового зонда в области медленных движений (τ > 10–10 с) анализировали в рамках модели изотропного броуновского вращения с помощью программы, описанной в работе [26]. При моделировании спектров применяли следующие главные значения g-тензора и тензора сверхтонкого взаимодействия радикала: gxx = 2.0096, gyy = 2.0066, gzz = 2.0025, Ахх = 7.0 Гc, Ayy = 5.0 Гc, Azz = 35.0 Гc. Величину Azz определяли экспериментально из ЭПР-спектров нитроксильного радикала в полимере, она не сильно отличалась от значения, приведенного в работе [27].
Времена корреляции вращения зонда τ в области быстрых вращений (5 × 10–11 < τ < 10–9 с) находили из спектров ЭПР по формуле [28]:
Исследование образцов методом ДСК осуществляли на приборе “DSC 204 F1” фирмы “Netzsch” в среде аргона при скорости нагревания 10 град/мин. Волокна после отжига в водной среде сушили в течение двух суток. Среднестатистическая ошибка измерения тепловых эффектов ±3%. Энтальпию плавления рассчитывали по программе “NETZSCH Proteus Thermal Analisis 4.8.4” по стандартной методике [29]. Пики разделяли с помощью программного обеспечения “NETZSCH Peak Separation 2006.01”. Вычисления проводили комбинированным методом Гаусса–Ньютона, в котором метод Marquardt соединен с оптимизацией длины шага итерации [30]. Ошибка составляла 2%.
Геометрию волокнистых материалов исследовали с помощью сканирующего электронного микроскопа “Hitachi TM-3000” (Япония) при ускоряющем напряжении 20 кВ. На поверхность образца нетканого волокнистого материала напыляли слой золота толщиной 100–200 Å.
Измерение влагопоглощения (сорбционной емкости) смесевых волокон осуществляли весовым методом (весы Мак-Бена с кварцевой спиралью). Чувствительность спирали 1.5 мм/мг. Из большого мата, полученного электроформованием, вырезали образец размером ~5 × 3 см и массой в пределах 60–100 мг. Толщину образца измеряли круговым индикатором (погрешность 1 микрон) с использованием покровных стекол для микроскопии. Прямое измерение толщины образца дает заниженное в несколько раз значение толщины, так как индентор индикатора вжимается в образец. Толщину вычисляли в разных точках по площади образца, разброс толщины 5–10%. Приготовленный образец подвешивали на кварцевую спираль и помещали в термостатированную вакуумную колонну установки. Для удаления воды, всегда содержащейся в пленке при естественной влажности в помещении, колонну вакуумировали в течение 1 ч с помощью масляного форвакуумного насоса при давлении 0.1 мм рт.ст. Затем колонну отсоединяли от насоса и соединяли с баллоном, содержащим пары воды при определенном давлении. Удлинение кварцевой спирали вследствие увеличения массы подвешенного образца регистрировали с помощью катетометра. После достижения сорбционного равновесия колонну отсоединяли от баллона напуска, а в баллоне повышали давление паров воды. Далее вновь соединяли колонну с баллоном напуска и регистрировали влагопоглощение при увеличенном давлении паров воды. Процедуру повторяли до достижения в баллоне напуска активности паров воды, равной 0.8–0.9.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние концентрации SnCl2–ТФП на геометрические параметры волокна
На рис. 1 представлены микрофотографии нетканых волокнистых материалов с различным содержанием комплекса. Как показано на рис. 1а, волокна ПГБ представляют собой чередование цилиндрических и веретенообразных структур. Наличие морфологической гетерогенности волокон обусловлено низкими значениями электропроводности и поверхностного натяжения полимерного формовочного раствора [30]. Средний диаметр цилиндрических участков составляет 2–3 мкм. Диаметр веретеноподобных участков колеблется около 10–15 мкм, их протяженность 20–30 мкм.
Рис. 1.
Микрофотографии волокнистых материалов из ПГБ (а) и смесей ПГБ с SnCl2–ТФП (б–г) при содержании SnCl2–ТФП 1 (б), 3 (в) и 5% (г).

При добавлении в раствор 1–3% SnCl2–ТФП веретеноподобные элементы в структуре волокна полностью исчезают (рис. 1б, 1в). Волокна выравниваются по толщине, и среднее значение их диаметра становится равным 5–6 мкм. Исчезновение веретеноподобных структур можно объяснить как увеличением электропроводности формовочного раствора, так и снижением его поверхностного натяжения при добавлении полярных комплексов, проявляющих также парамагнитные свойства. С ростом электропроводности раствора вытягивание капли раствора под действием электростатической силы происходит более интенсивно, что влечет за собой выравнивание волокон по толщине. Отсутствие существенного градиента по диаметру волокон свидетельствует о том, что электроформование протекает равномерно без расщепления первичной струи и образования дочерних струй, что было характерно, например, для волокон ПГБ с комплексами цинк–ТФП [15]. При дальнейшем увеличении концентрации SnCl2–ТФП в нетканом волокнистом материале формируются участки ленточного типа (рис. 1г). Ширина ленты колеблется в диапазоне 10–30 мкм. Диаметр цилиндрических участков остается практически неизменным. Как видно на микрофотографии, ленточные структуры образуют многочисленные спайки. Толщина лент лежит в диапазоне 1–5 мкм. Происхождение ленточных структур не совсем понятно. Возможно, с ростом концентрации комплексов SnCl2–ТФП происходит значительное падение поверхностного натяжения, в результате чего на выходе из сопла первичная струя полимерного раствора под действием гравитационной и электростатической сил преобразует свой профиль из цилиндрического в плоский.
Термофизические характеристики кристаллической фазы композиций ПГБ–SnCl2–ТФП после экспозиции в водной среде при 70°С
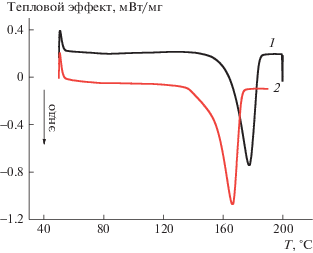
Рассмотрим влияние комплекса SnCl2–ТФП на кристаллическую фазу волокон ПГБ, используя метод ДСК (рис. 2). Следует отметить, что концентрация порфиринового комплекса в аморфных областях полимера значительно выше, чем заданные величины 1, 3 и 5%. Последние рассчитаны на всю массу образца, а частицы SnCl2–ТФП находятся только в аморфной прослойке, доля которой уменьшается с ростом концентрации комплекса в полимере. Так, в волокнах ПГБ с содержанием комплекса 5% его концентрация в аморфных областях будет составлять ∼15%. Термограммы как первого, так и второго сканирования характеризуются одиночными максимумами плавления. Термограмма волокна ПГБ, содержащего 1% SnCl2–ТФП, имеет правильную симметричную форму, что свидетельствует о достаточно однородной кристаллической фазе. Термограммы образцов ПГБ и ПГБ с содержанием комплекса SnCl2–ТФП 3 и 5% имеют несимметричную форму с низкотемпературным плечом, что обусловлено образованием линейных структур (рис. 3). Важно отметить, что энтальпия плавления ΔH, полученная методом ДСК, дает информацию как о доле кристаллической фазы, так и о линейных структурах (структуры из выпрямленных цепей с двумерным порядком).
Рис. 2.
Зависимость ∆Н от состава волокон: 1 – исходный образец, 2–4 – образцы после экспозиции в водной среде при 70°С в течение 30 (2), 120 (3) и 240 мин (4).
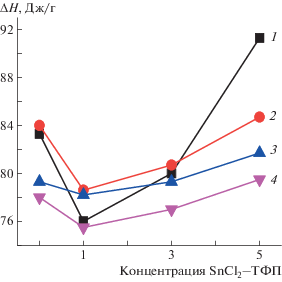
Экспериментальные данные по растворимости SnCl2–ТФП в хлороформе показывают, что между молекулами SnCl2–ТФП существует сильное межмолекулярное отталкивание. В результате, растворение комплекса происходит на молекулярном уровне. Металлокомплексы усиливают зародышеобразование полимера, что способствует резкому росту степени кристалличности волокон (см. рис. 2). В работе [14] показано, что наличие одного атома Cl в молекуле металокомплекса FeCl–ТФП обусловливает не столь сильное межмолекулярное отталкивание и в растворе, вследствие сегрегации комплексов образуются уже достаточно крупные частицы FeCl–ТФП. Последние, являясь зародышами кристаллизации, также предопределяют рост степени кристалличности. С ростом концентрации металлокомплекса размер частиц увеличивается и повышение энтальпии плавления замедляется. Распределение комплексов SnCl2–ТФП в растворителе на молекулярном уровне обеспечивает более резкий рост доли кристаллитов и линейных структур с увеличением концентрации добавки по сравнению с системой ПГБ–MnCl–ТФП. В то время как в волокнах с порфирином и комплексом цинк–порфирин взаимодействие между молекулами достаточно сильное, что способствует образованию крупных частиц. Как следствие, дополнительная кристаллизация при формовании волокна проявляется очень слабо.
При добавлении 1% металлокомплекса энтальпия плавления слабо снижается. При такой концентрации комплекса пик плавления имеет более совершенную форму, чем у волокон ПГБ и ПГБ с 3 и 5% комплекса, из-за наличия линейных систем. Энтальпия плавления с ростом концентрации металлокомплекса до 5% возрастает на 21%. Ранее для систем ПГБ–MnCl, FeCl–ТФП был обнаружен дисбаланс между данными ДСК и РСА ~7–10% [14, 16], что объясняется наличием линейных систем в полимерах.
Таким образом, наблюдаемый рост энтальпии плавления композиции ПГБ–SnCl2–ТФП обусловлен дополнительной кристаллизацией на молекулах металлокомплекса. Здесь этот эффект выражен более ярко, чем в композициях ПГБ–MnCl, Zn–ТФП.
При выдержке в воде происходит изменение молекулярной и надмолекулярной структуры полимера [31–34]. Необходимо было установить степень этих структурных изменений, поскольку ПГБ используется в медицинских целях и часто подвергается воздействию воды при повышенной температуре.
Данные по энтальпии плавления образцов ПГБ–SnCl2–ТФП, экспонированных в дистиллированной воде при 70°C в течение 30, 120 и 240 мин, представлены на рис. 2. Видно, что после 30 мин экспозиции в водной среде энтальпия плавления возрастает в волокнах ПГБ и ПГБ с содержанием SnCl2–ТФП 1 и 3%; при более длительной экспозиции (240 мин) она снижается относительно исходных полимеров (табл. 1). Вместе с тем, при содержании добавки в волокнах ПГБ 5% резко снижается ΔH уже после 30 мин экспозиции. Наиболее слабые изменения ΔH наблюдаются в волокнах с концентрацией комплекса 1%, так как именно в них кристаллиты являются наиболее совершенными, а следовательно, наименее подверженными водному воздействию. Ранее для пленочных образцов было показано [35], что смешение хитозана с ПГБ приводит к гидрофилизации системы. Благодаря высокому сродству воды к молекулам полисахарида, сорбционная емкость системы резко возрастает, что вызывает усиление пластифицирующего действия воды на ПГБ. В этом случае экспоненциально возрастают коэффициенты диффузии воды и низкомолекулярных соединений. По-видимому, аналогичные процессы происходят в волокнах на основе ПГБ с комплексами SnCl2–ТФП. Молекулы воды двояким образом воздействуют на структуру полимера: с одной стороны, они оказывают пластифицирующее действие, что позволяет кристаллитам увеличить свои размеры, с другой – при большой концентрации комплексов происходит разрушение торцевых поверхностей кристаллитов и линейных систем. После удаления воды в волокне остаются гидратированные комплексы, которые образуются между полярными группами ПГБ и металлокомплекса SnCl2–ТФП. В результате структура волокна становится более рыхлой, и энтальпия системы снижается.
Таблица 1.
Энтальпия и температура плавления ультратонких волокон смесей ПГБ с SnCl2–ТФП, исследованных методом ДСК
| Содержание SnCl2–ТФП, % | ΔH, Дж/г | Тпл, °С | ||
|---|---|---|---|---|
| первый скан | второй скан | первый скан | второй скан | |
| 0 | 83.3 | 87.8 | 175.6 | 156.1 |
| 1 | 76.0 | 77.0 | 177.4 | 169.7 |
| 3 | 80.0 | 81.9 | 175.1 | 170.2 |
| 5 | 91.4 | 76.9 | 167.4 | 159.0 |
| Отжиг при 140°С, в течение 2 ч | ||||
| 0 | 96.8 | 81.7 | 176.3 | 171.4 |
| 1 | 98.3 | 86.3 | 175.0 | 170.7 |
| 3 | 97.4 | 88.8 | 177.1 | 169.6 |
| 5 | 97.4 | 82.8 | 167.9 | 159.7 |
| Отжиг в водной среде при 70°С, в течение 2 ч | ||||
| 0 | 79.3 | 71.3 | 176.2 | 164.6 |
| 1 | 78.2 | 76.2 | 176.0 | 168.8 |
| 3 | 79.3 | 68.4 | 175.3 | 169.8 |
| 5 | 81.7 | 77.8 | 166.2 | 164.5 |
Резкое снижение ΔH в волокнах ПГБ с содержанием комплекса 5% уже после 30 мин экспозиции можно объяснить высокой долей торцевых поверхностей кристаллитов, линейных структур, а также большой концентрацией комплекса в полимере и тем, что при повышенной температуре в этих структурах образуются гидратированные комплексы. В волокнах ПГБ и ПГБ с содержанием добавки 1% пластифицирующий эффект воды преобладает и при небольших значениях времени экспозиции ΔH растет. Аналогичный эксперимент был выполнен для волокон ПГБ–порфирин [36]. Наблюдаются различия в поведении ΔH после экспозиции в водной среде, связанные, возможно, с размерами частиц комплекса. Самое большое отличие имеет место при концентрации комплекса в волокне 5%. Слабое изменение ΔH системы ПГБ–порфирин после экспозиции в водной среде вызвано сегрегацией частиц порфирина в частицы большого размера, в результате чего концентрация гидратированных комплексов меняется слабо.
На рис. 4 представлены изотермы сорбции нетканых волокнистых материалов ПГБ–SnCl2–ТФП различного состава при температуре 25°С. Из изотерм видно, что данные материалы имеют сравнительно низкую сорбционную способность. При одном и том же значении активности паров воды, например при 0.35, сорбционная емкость составляет ~0.03 мас. % у ПГБ и ~0.09 и 0.17 мас. % у ПГБ с содержанием SnCl2–ТФП 3 и 5% соответственно. Увеличение сорбционной емкости связано в основном с изменением плотности аморфных областей. Чем выше концентрация SnCl2–ТФП в смесевых волокнах, тем больше сорбционная емкость материала.
Рис. 4.
Изотермы сорбции нетканых волокнистых материалов ПГБ–SnCl2–ТФП при температуре 25°С. Содержание SnCl2–ТФП 0 (1), 3 (2) и 5% (3).
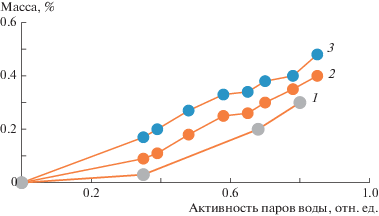
На сорбционную емкость нетканых волокнистых материалов влияют два фактора: величина удельной поверхности и степень гидрофильности волокон. В процессе сорбции пары воды конденсируются на поверхности полимерных волокон, проникая затем в объем нетканого материала. При этом пустоты между волокнами остаются свободными. Если бы вода конденсировалась в пустотах, то влагопоглощение составляло бы сотни процентов.
Динамические характеристики аморфной фазы ультратонких волокон ПГБ–SnCl2–ТФП после экспозиции в водной среде при 70°С
Структура аморфных областей в значительной степени определяется долей кристаллической фазы в волокне. Поэтому при добавлении малых значений концентрации комплекса меняется морфология волокна, степень кристалличности ПГБ, молекулярная динамика в аморфных областях. Молекулярная подвижность в аморфных областях изучена методом ЭПР с использованием стабильного нитроксильного радикала ТЕМПО.
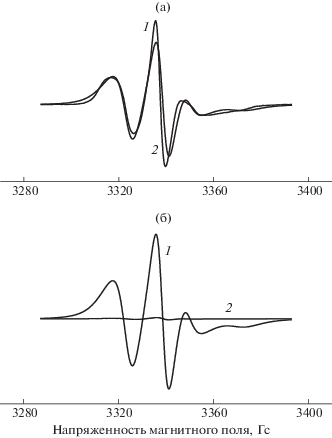
Получали ЭПР-спектры образцов ПГБ–SnCl2–ТФП, экспонированных в дистилллированной воде при 70°C. Радикал вводили в волокна после отжига в водной среде (образец сушили в течение двух суток). ЭПР-спектры радикала в матрице ПГБ и в смесевых композициях представляют собой суперпозицию двух спектров, соответствующих двум популяциям радикалов с временем корреляции τ1 и τ2. Время τ1 характеризует молекулярную подвижность в более плотных аморфных областях (медленная составляющая), τ2 – подвижность в менее плотных областях (быстрая составляющая) (рис. 5).
Рис. 5.
Спектры ЭПР нитроксильного радикала ТЕМПО для образцов ПГБ–SnCl2–ТФП 1%: а – экспериментальные (1) и теоретические (2) спектры; б – медленная (1) и быстрая (2) составляющие спектра.
Структура аморфных областей в полимере имеет гетерогенный характер, представлен набор структур с разной плотностью упаковки цепей и, следовательно, с разной молекулярной динамикой. Разрешающая способность нашего прибора дает возможность оценить только среднюю подвижность в достаточно рыхлых и плотных областях с различающимися временами корреляции τ1 и τ2. На рис. 6 показана зависимость времени корреляции от концентрации комплекса при разном времени экспозиции в водной среде. С ростом концентрации SnCl2–ТФП время корреляции уменьшается. После экспозиции в водной среде до 90 мин (включительно) характер изменения τ практически не меняется и только при выдержке в 120 и 240 мин зависимости имеют более пологий вид. Важно, что с ростом времени экспозиции для всех изучаемых композиций время корреляции снижается. Несмотря на рост степени кристалличности в волокнах ПГБ и ПГБ с содержанием металлокомплекса 1% на начальном этапе экспозиции (в результате чего молекулярная подвижность должна снижаться), подвижность в аморфных областях растет. Гидратированные комплексы, остающиеся в волокне после удаления воды, разрыхляют аморфную структуру наряду с молекулами SnCl2–ТФП, и именно поэтому наблюдается снижение времени корреляции (рис. 6). С ростом времени выдержки волокна в водной среде доля гидратированных комплексов возрастает, плотность аморфных областей в большей степени уменьшается, что и дает вклад в снижение τ.
Рис. 6.
Зависимость времени корреляции τ от состава смесевой композиции после экспозиции в водной среде. Время экспозиции 0 (1), 30 (2), 60 (3), 90 (4) и 240 мин (5).
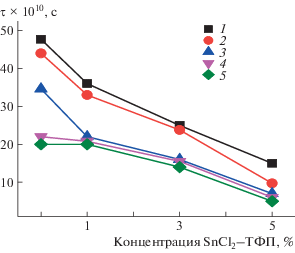
В системах ПГБ с комплексами FeCl, Zn, MnCl–ТФП время корреляции увеличивалось на начальном этапе экспозиции при добавлении комплекса (до 3%), что объяснялось ростом жесткости аморфной составляющей вследствие повышения доли кристаллических областей [14–16]. Необычную зависимость в настоящей работе можно объяснить тем, что комплекс SnCl2–ТФП распределяется в полимере на молекулярном уровне и эффект разуплотнения проявляется достаточно сильно, что выражается в в росте молекулярной подвижности. В то же время, комплексы FeCl, Zn, MnCl–ТФП образуют агломераты и слабее разрыхляют аморфные области.
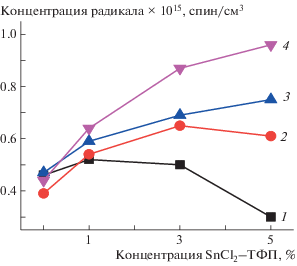
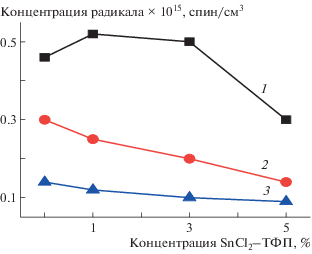
С ростом времени экспозиции в водной среде τ резко уменьшается, что связано с образованием гидратированных комплексов как с макромолекулами ПГБ, так и с металлокомплексами, которые разрыхляют аморфную структуру (рис. 6). Так как локальная концентрация комплекса в образцах со средним содержанием SnCl2–ТФП 5% составляет ∼12%, эффект разуплотнения в них самый высокий, о чем свидетельствуют данные по концентрации радикала в образцах (рис. 7). Равновесная концентрация радикала, адсорбированного в одинаковых по массе образцах изучаемых композиций, была рассчитана с использованием программного обеспечения фирмы “Bruker (winer)”. На рис. 7 представлены зависимости концентрации радикала от состава волокон для серии времен экспозиции. Видно, что рост концентрации комплекса в ПГБ приводит к уменьшению концентрации радикала в полимере, что обусловлено ростом содержания кристаллических и линейных областей. Вместе с тем, экспозиция в водной среде обусловливает все более высокую концентрацию радикала в полимере с ростом времени выдержки (за исключением ПГБ). Прослеживается закономерность в изменении энтальпии плавления и концентрации радикала при изменении времени экспозиции в водной среде (рис. 2 и 6), небольшие различия при содержании SnCl2–ТФП 1% и максимальные различия при содержании SnCl2–ТФП 5%.
Рис. 7.
Зависимость концентрации радикала от состава композиции после экспозиции в водной среде в течение 0 (1), 30 (2), 90 (3) и 240 мин (4).
С использованием программы NLSL аналогично процедуре, описанной в работе [26], из спектров были оценены соотношения концентрации различных по плотности областей и рассчитано время корреляции в плотных и рыхлых областях. С ростом концентрации комплекса время корреляции в плотных областях незначительно уменьшается, после экспозиции в водной среде изменения времени корреляции проявляются сильнее (табл. 2).
Таблица 2.
Время корреляции τ в плотных аморфных областях и отношение доли плотных к доле рыхлых областей a/а* в волокнах ПГБ–SnCl2–ТФП
| Содержание SnCl2–ТФП, % | τ × 1010, с | a/а* | ||||
|---|---|---|---|---|---|---|
| Исходное волокно | Отожженное волокно | Исходное волокно | Отожженное волокно | |||
| при 140°C | при 70°C в Н2О | при 140°C | при 70°C в Н2О | |||
| 0 | 64 | 77 | 69 | 87 | 89 | 88 |
| 1 | 63 | 76 | 64 | 73 | 89 | 83 |
| 3 | 61 | 74 | 55 | 65 | 79 | 51 |
| 5 | 55 | 70 | 38 | 48 | 61 | 38 |
Таким образом, температура и выдержка в водной среде достаточно сильно влияют на структуру межкристаллитных областей изучаемых волокон. Происходит довольно резкое снижение τ, а следовательно, рост молекулярной подвижности в аморфных областях, с увеличением концентрации металлокомплекса. После экспозиции в водной среде при 70°С в течение 30, 90, 120 и 240 мин прослеживается резкое снижение времени корреляции, что можно объяснить образованием гидратированных комплексов в большой концентрации, которые разрыхляют аморфную и кристаллическую структуру волокон. Вода пластифицирует полимер, цепи с высокой степенью выпрямленности встраиваются в линейные структуры, и фиксируемая ДСК кристалличность повышается, но только в полимерах ПГБ и ПГБ с содержанием SnCl2–ТФП 1% после 30 мин выдержки. Наиболее резкое снижение энтальпии плавления и времени корреляции при добавлении 5% комплекса в ПГБ можно трактовать сильным взаимодействием между молекулами воды и молекулами комплекса и их высокой локальной концентрацией, что приводит к разуплотнению торцевых поверхностей кристаллитов, линейных структур и аморфных областей. Еще одним фактором, влияющим на снижение энтальпии плавления и времени корреляции после экспозиции, может служить более рыхлая структура аморфных областей в волокнах с высоким содержанием металлокомплекса (3 и 5%). В них содержание воды резко возрастает (вследствие разуплотнения аморфной фазы молекулами металлокомплекса) и все бóльшая доля торцевых поверхностей, линейных структур, аморфных областей, молекул SnCl2–ТФП контактирует с молекулами воды. После удаления воды в волокне остаются гидратированные комплексы, которые разрыхляют торцевую поверхность кристаллитов, линейных систем и аморфную структуру, и молекулярная подвижность возрастает. С ростом времени выдержки полимера в водной среде доля гидратированных комплексов растет и плотность аморфных областей все сильнее понижается, что способствует снижению τ. Гидрофилизация изучаемых систем зависит как от состава композиции, так и от длительности экспозиции. Сорбционная емкость благодаря высокому сродству воды к молекулам металлокомплекса резко возрастает по сравнению с таковой у ПГБ. Хорошо известно, что вода, проникая в полимерную матрицу, может влиять как на физико-химические и механические свойства полимеров, так и на потоки третьего низкомолекулярного компонента. В работе [31] показано, что под воздействием воды происходит деградация пленок смесей ПГБ с этиленпропиленовым каучуком, меняется структура цепей, понижается содержание выпрямленных конформеров.
Термофизические характеристики кристаллической фазы композиций ПГБ–SnCl2–ТФП после отжига при 140°С
Отжиг композиций ПГБ–SnCl2–ТФП при 140°C оказывает влияние на структуру полимера, а значит и на его термофизические характеристики. На рис. 8 представлена зависимость ΔH от концентрации комплекса. Наиболее сильно эффект температурного отжига проявляется в волокнах ПГБ и ПГБ с содержанием комплекса 1%. После 120 мин отжига энтальпия в таких волокнах возрастает на 30%, в то время как в волокнах с концентрацией комплекса 5% – только на 6%. Это объясняется высокой концентрацией мелкодисперсной добавки, которая вносит дефекты, особенно существенные при температуре плавления торцевых поверхностей кристаллитов, дефектных кристаллитов и линейных систем. При резком охлаждении образца молекулы комплекса остаются в этих структурах и деформируют их. В результате энтальпия увеличивается не столь значительно как в волокнах ПГБ и ПГБ с 1% SnCl2–ТФП. Характер зависимостей ΔH от времени отжига в волокнах ПГБ–Zn, MnCl–ТФП практически такой же, только энтальпия плавления волокна выше.
Рис. 8.
Зависимость ∆Н от состава волокон после отжига при 140°С в течение 0 (1), 30 (2), 120 (3) и 240 мин (4).
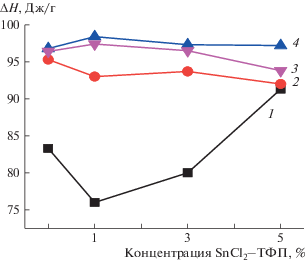
Таким образом, отжиг волоконного материала при 140°С приводит к росту энтальпии плавления волокон ПГБ с содержанием SnCl2–ТФП 0, 1 и 3% уже после 30 мин, в то время как при концентрации комплекса 5% изменения ΔH не столь велики, что обусловлено разрыхляющим действием комплекса. Дальнейшая выдержка материала при указанной температуре (до 240 мин) сопровождается незначительным повышением энтальпии плавления.
Динамические характеристики аморфной фазы ультратонких волокон ПГБ–SnCl2–ТФП после отжига при 140°С
Совершенствование кристаллической фазы и особенно дополнительная кристаллизация при отжиге происходят при участии проходных полимерных молекул. Поэтому следует ожидать изменения динамики вращения зонда τ, распределенного в межкристаллическом пространстве полимера и меняющего частоту вращения (время корреляции) в ответ на изменение сегментальной подвижности молекул композиции. Более того, при сравнении подвижностей в полимере появляется дополнительная возможность определить влияние добавки на устойчивость и динамику межкристаллической структуры.
Температурное воздействие позволяет резко интенсифицировать сегментальную подвижность и привести систему к более термодинамически равновесному состоянию. Наблюдаемые изменения молекулярной динамики радикала и сегментальной подвижности макромолекул полимера при отжиге образцов объясняются процессами доориентации и уплотнением цепей. Растет доля предельно выпрямленных макромолекул. Это связано с тем, что сегменты со степенью выпрямленности k больше критического значения (k* = = h/L = $\sqrt 2 $/β, где h – расстояние между концами цепей, L – длина цепи, β – число сегментов [35]) при отжиге стремятся принять предельно выпрямленную конфигурацию. Таким образом, при повышенных значениях температуры наблюдается увеличение степени кристалличности и доли линейных структур в волокне, а также рост доли аморфной фазы с высокой степенью выпрямленности цепей. Как результат, чаще всего концентрация радикала уменьшается, и он сосредоточивается в наиболее рыхлых участках аморфной фазы, повышается подвижность радикала, что свидетельствует о росте подвижности макромолекул полимера, так как проникновение радикала в плотные аморфные прослойки затруднено.
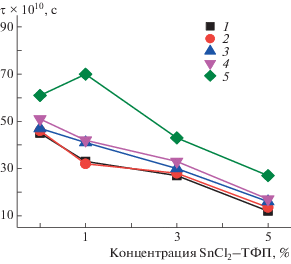
Однако в нашем случае наличие молекул SnCl2–ТФП препятствует сближению макромолекул и эффект уплотнения проявляется слабее. Действительно, время термического отжига при 140°C изучаемых композиций оказывает незначительное влияние на молекулярную динамику вращения радикала. В предыдущих исследованиях [14, 16] показано, что отжиг ПГБ с FeCl–ТФП или MnCl–ТФП при 140°С приводит к резкому снижению времени корреляции, что объясняется низкой концентрацией радикала в плотных областях полимера, доля и плотность которых резко возрастала с увеличением времени отжига. В настоящей работе видно слабое увеличение подвижности радикала с ростом времени отжига, что объясняется сильным разрыхляющим действием SnCl2–ТФП, вследствие чего концентрация радикала в плотных областях полимера достаточно высока. Среднее значение τ в результате снижается плавно (рис. 9).
Рис. 9.
Зависимость времени корреляции τ от состава композиции при отжиге при 140°С. Время экспозиции 0 (1), 30 (2), 60 (3), 90 (4) и 240 мин (5).
Данные по концентрации радикала в композиции показывают, что при увеличении времени отжига зависимости концентрации радикала от содержания SnCl2–ТФП становятся все более пологими, и содержание радикала резко уменьшается с увеличением времени отжига. Между поведением термофизических параметров и изменением концентрации радикала наблюдается определенное соответствие. Скачкообразный рост содержания кристаллитов и линейных структур после отжига сопровождается значительным уменьшением концентрации радикала и все более пологим характером зависимости концентрации радикала от содержания SnCl2–ТФП с ростом времени отжига (рис. 10), так как в наиболее плотных аморфных областях концентрация радикала невелика. Наиболее слабые изменения ΔH и концентрации радикала с ростом времени отжига наблюдаются в полимере с содержанием добавки 5%, а самые значительные – при концентрации металлокомплекса 1%.
Рис. 10.
Зависимость концентрации радикала от состава композиции при отжиге при 140°С в течение 0 (1), 30 (2) и 240 мин (3).
В табл. 2 приведены рассчитанные путем математической обработки спектров времена корреляции в плотных и рыхлых областях волокна. Время корреляции во всех образцах после отжига при 140°С выше, чем в исходных образцах.
Таким образом, сорбционная способность волокна по отношению к парамагнитному радикалу снижается с ростом времени отжига при 140°С вследствие роста доли кристаллических и линейных структур. Наиболее ярко изменения τ и концентрации радикала проявляются при содержании металлокомплекса 1%, наиболее слабые изменения видны при концентрации добавки 5%. Это обусловлено более совершенной кристаллической структурой и более высоким содержанием кристаллитов в волоконе ПГБ с 5% металлокомплекса, который является зародышем кристаллизации полимера.
Воздействие озонолиза на динамику вращения радикала в волокнах композиции ПГБ–SnCl2–ТФП
Изучение влияния озона на материалы и изделия медицинского назначения представляет интерес ввиду того, что метод озонирования является эффективным способом стерилизации и обеззараживания медицинских изделий. Несмотря на повседневный контакт данного агрессивного соединения с полимерами, его влияние на их морфологические и динамические характеристики остается мало исследованным.
В работе изучали влияние озона на смесевые композиции при температуре 22°С (рис. 11). Озонолиз сложным образом влияет на структуру полимера. Одновременно протекает два процесса – с ростом молекулярной подвижности и с ее замедлением. Согласно данным работы [36], под действием озона в первую очередь деструктируют складки на торцевых поверхностях кристаллитов, так как они представляют наиболее напряженные участки полимерных цепей. В результате, они распрямляются и их подвижность замедляется. На начальном этапе озонолиза преобладает процесс, приводящий к росту жесткости аморфных областей вследствие самопроизвольной доориентации цепей. Доориентация происходит как в ходе усиления межмолекулярного взаимодействия из-за накопления полярных кислородсодержащих групп, так и вследствие распрямления макромолекул и после деструкции при высокой степени выпрямленности [14, 37–39]. С наиболее высокой скоростью окисляются петли на торцевых поверхностях кристаллитов, так как на этих участках наиболее высока концентрация напряжения. Разрыв петель сопровождается распрямлением сегментов, что замедляет молекулярную подвижность в этих областях. Параллельно с указанными процессами происходит также деструкция цепей в рыхлых аморфных областях, что приводит к росту молекулярной подвижности, и, как следствие, к уменьшению τ. При более глубоких степенях окисления преобладают именно такие процессы, что и приводит к снижению времени корреляции, а следовательно, к росту молекулярной подвижности цепей в ультратонких волокнах. В исследуемой системе мелкодисперсная добавка, сильно разрыхляющая структуру полимера, препятствует усилению межмолекулярного взаимодействия и появлению заметного вклада в изменение молекулярной динамики. Именно поэтому рост τ в полимере с добавкой SnCl2–ТФП на начальном участке окисления не столь значителен как наблюдалось ранее [14, 37–39].
Рис. 11.
Зависимость времени корреляции τ от длительности озонолиза для композиции, содержащей 0 (1), 1 (2), 3 (3) и 5% SnCl2–ТФП (4).
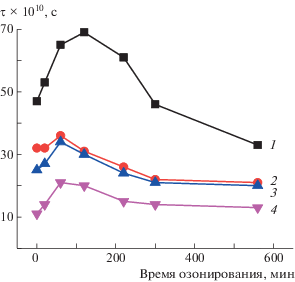
Таким образом, воздействие озона обусловливает рост времени корреляции зонда в волокнах, а значит, уменьшение молекулярной подвижности на ранних стадиях окисления, что вызвано как межмолекулярной физической сшивкой макромолекул ПГБ, так и процессами доориентации цепей. Сшивка на начальной стадии окисления преобладает над процессами деструкции цепей, при этом нарастает жесткость полимерных молекул в результате формирования сетки водородных связей, которая препятствует свободному движению сегментов ПГБ. При более глубоких степенях окисления преобладают процессы деструкции цепей, что приводит к росту молекулярной подвижности в аморфной фазе волокон. Наличие мелкодисперсной добавки SnCl2–ТФП, разрыхляющей аморфную структуру металлокомпозита, ослабляет эффект замедления молекулярной подвижности на начальном этапе озонолиза. В отличие от этого на начальной стадии озонолиза композиции ПГБ–FeCl–ТФП происходит все более резкий рост времени корреляции с ростом содержания FeCl–ТФП [14]. Наблюдаемые различия можно объяснить разницей в размере частиц, внедренных в структуру ПГБ.
ЗАКЛЮЧЕНИЕ
В работе показано, что добавление комплекса SnCl2–ТФП в ПГБ обусловливает резкий рост энтальпии плавления ультратонких волокон. Экспозиция волокон ПГБ–SnCl2–ТФП в водной среде в течение 30 мин при 70°С приводит к росту энтальпии плавления в образцах, содержащих 0, 1 и 3% добавки, и к ее значительному снижению в волокнах с концентрацией металлокомплекса 5%. С ростом времени отжига энтальпия плавления всех волокон снижается, особенно значительно в полимере с содержанием добавки 5%. Время корреляции уменьшается с увеличением времени экспозиции в водной среде, что вызвано образованием гидратированных комплексов, разрыхляющих аморфные и кристаллические области.
Отжиг волокон при 140°С приводит к резкому увеличению доли кристаллической фазы и снижению концентрации радикала в аморфных областях во всех изучаемых волокнах. Время корреляции уменьшается. Изотермы сорбции паров воды нетканых волокнистых материалов показали, что с ростом концентрации добавки увеличивается сорбционная емкость полимера.
Озонное воздействие на волокна изучаемой смеси на начальном этапе приводит к росту времени корреляции, что объясняется увеличением доли выпрямленных цепей и усилением межмолекулярного взаимодействия вследствие накопления кислородсодержащих групп. При более глубоком окислении подвижность радикала возрастает, что объясняется интенсивной деструкцией цепей полимера.
Взаимное влияние кристаллических и аморфных областей в биоразлагаемых высококристаллических полимерах и их композициях остается сложной, мало изученной проблемой современного полимерного материаловедения. Исследования состояния полимерной матрицы и роли добавки впервые позволили интерпретировать на молекулярном уровне воздействие ряда агрессивных факторов (таких как температура и озон) на структурно-динамические характеристики волокон смесей ПГБ–SnCl2–ТФП.
В данной работе измерения кристалличности проводили методом ДСК на приборе “DSC 204 F1” фирмы “Netzsch”, Германия (Центр Коллективного пользования Института биохимической физики РАН “Новые материалы и технологии”).
Авторы выражают благодарность А.В. Лобанову (Институт химической физики им. Н.Н. Семенова РАН) за предоставленный металлокомплекс SnCl2–ТФП.
Список литературы
Jana S., Maiti S., Jana S. Biopolymer-Based Composites: Drug Delivery and Biomedical Application / Eds by S. Jana, S. Maiti, S. Jana. Kidlington: Woodhead Publ. Elsevier Ltd., 2017. ISBN 978-0-08-101914-6.
Zykova A.K., Pantyukhov P.V., Popov A.A., Kolesnikova N.N., Monakhova T.V. // J. Polym. Environment. 2018. V. 26. № 4. P. 1343.
Sangroniz A., Zhu J.-B., Tang X., Etxeberria A., Chen E.Y.-X., Sardon H. Packaging Materials with Desired Mechanical and Barrier Properties and Full Chemical Recyclability // Nat. Commun. 2019. V. 10. 3559.
Rosa A., Ricciardi G., Baerends E.J. // Inorg. Chem. 2005. V. 44. № 19. P. 6609.
Ray P.C., Leszczynski J. // Chem. Phys. Lett. 2006. V. 419. № 4. P. 578.
Li C., Ly J., Lei B. // J. Phys. Chem. B. 2004. V. 108. № 28. P. 9646.
Balzani V., Credi A., Venturi M. // Chem. Eur. J. 2008. V. 14. № 1. P. 26.
Bychkova A.V., Iordanskii A.L., Kosenko R.Y. // Nanotechnol. Russia. 2015. V. 10. № 3–4. P. 325.
Karpova S.G., Ol’khov A.A., Shilkina N.G., Tyubaeva P.M., Popov A.A., Iordanskii A.L. // Polymer Science A. 2017. V. 59. № 3. P. 342.
Karpova S.G., Ol’khov A.A., Shilkina N.G., Popov A.A., Filatova A.G., Kucherenko E.L., Iordanskii A.L. // Polymer Science A. 2017. V. 59. № 1. P. 58.
Iordanskii A.L., Ol’khov A.A., Karpova S.G., Kucherenko E.L., Kosenko R.Y., Rogovina S.Z., Chalykh A.E., Berlin A.A. // Polymer Science A. 2017. V. 59. № 3. P. 352.
Karpova S.G., Ol’khov A.A., Iordanskii A.L., Lomakin S.M., Shilkina N.S., Popov A.A., Gumargalieva K.Z., Berlin A.A. // Polymer Science A. 2016. V. 58. № 1. P. 76.
Olkhov A.A., Staroverova O.V., Bonartsev A.P., Zharkova I.I., Sklyanchuk E.D., Iordanskii A.L., Rogovina S.Z., Berlin A.A., Ishchenko A.A. // Polymer Science D. 2015. V. 8. № 2. P. 100.
Karpova S.G., Olkhov A.A., Bakirov A.V., Shilkina N.G., Popov A.A. // Russ. J. Phys. Chem. B. 2018. V. 12. № 1. P. 142.
Karpova S.G., Ol’khov A.A., Krivandin A.V., Shatalova O.V., Popov A.A., Lobanov A.V., Iordanskii A.L. // Polymer Science A. 2019. V. 61. № 1. P. 70.
Karpova S.G., Ol’khov A.A., Lobanov A.B., Popov A.A., Iordanskii A.L. // Nanotechnol. Russia. 2019. V. 14. № 3–4. P. 132.
Глазкова М.Е., Агеева Т.А., Николаева О.И., Румянцева Ю.В., Койфман О.И. // Изв. вуз. Сер. Химия и хим. технология. 2011. № 3. С. 104.
Филимонов Д.А., Самолетов О.В., Базанов М.И., Семейкин А.С., Петров А.В. // Электрохим. энергетика. 2009. Т. 9. № 2. С. 91.
Ольхов А.А., Староверова О.В., Гольдштрах М.А., Хватов А.В., Гумаргалиева К.З., Иорданский А.Л. // Хим. физика. 2016. Т. 35. № 10. С. 53.
Amati A., Cavigli P., Demitri N., Natali M. // Inorg. Chem. 2019. V. 58. № 7. P. 4399.
Kim H., Kim W., Mackeyev Y., Lee G.-S., Kim H.-J., Tachikawa T. // Environmental Sci. Technol. 2012. V. 46. № 17. P. 9606.
Huang H., Chauhan S., Geng J., Qin Y., Watson D.F. // Biomacromolecules. 2017. V. 18. № 2. P. 562.
Mintrop L., Windisch J., Gotzmann C., Alberto R., Probst B. // J. Phys. Chem. B. 2015. V. 119. № 43. P. 13698.
Tran C.D., Duri S., Harkins A.L. // J. Biomed. Mater. Res. A. 2013. № 8. P. 2248.
Филатов Ю.Н. Электроформование волокнистых материалов (ЭФВ-процесс). М.: Нефть и газ, 1997.
Liang Z., Freed J.H. // J. Phys. Chem. B. 1999. № 10. P. 6384.
Тимофеев В.П., Мишарин А.Ю., Ткачев Я.В. // Биофизика. 2011. Т. 56. С. 420.
Вассерман A.M., Бучаченко A.Л., Коварский A.Л., Нейман M.Б. // Высокомолек. соед. А. 1968. Т. 10. № 8. С. 1930.
Stephen Z.D. Handbook of Thermal Analysis and Calorimetry, Applications to Polymers and Plastics. Amsterdam; Boston; London: Elsevier, 2002. V. 3.
Dobrovol′skaya I.P., Lebedeva I.O., Yudin V.E., Popryaduhin P.V., Ivan′kova E.M., Elohovskiy V.Yu. // Polymer Science A. 2016. V. 58. № 2. P. 246.
Tertyshnaya Y.V., Shibryaeva L.S. // Polymer Science B. 2013. V. 55. № 3–4. P. 164.
Камаев П.П. Дис. … канд. хим. наук. М.: ИХФ РАН им. Н.Н. Семенова, 2001.
Иванцова Е.Л., Иорданский А.Л., Косенко Р.Ю., Роговина С.З. // Докл. РАН. Сер. Физическая химия. 2010. Т. 431. С. 500.
Karpova S.G., Ol’khova A.A., Chvalund S.N., Tyubaevaa P.M., Popova A.A., Iordanskii A.L. // Nanotechnol. Russia. 2019. V. 14. № 7–8. P. 367.
Озерин А.Н. Дис. … канд. хим. наук. М.: Научно-исследовательский физико-химический ин-т им. Л.Я. Карпова, 1977.
Карпова С.Г., Попов А.А., Заиков Г.Е. // Высокомолек. соед. 1991. Т. 33. № 5. С. 931.
Karpova S.G., Iordanskii A.L., Popov A.A., Chvalun S.N., Berlin A.A. // Russ. J. Phys. Chem. B. 2012. V. 6. № 1. P. 72.
Karpova S.G., Ol’khov A.A., Tyubaeva P.M., Shilkina N.G., Popov A.A., Iordanskii A.L. // Rus. J. Phys. Chem. B. 2019. V. 13. № 2. P. 313.
Karpova S.G., Ol’khov A.A., Iordanskii A.L., Lomakin S.M., Shilkina N.S., Popov A.A., and Berlin A.A. // Polym. Science-Series A. Focus on Physics. 2016. V. 58. № 1. P. 76.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)