

Высокомолекулярные соединения (серия А), 2021, T. 63, № 5, стр. 334-343
ВЛИЯНИЕ КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ КОМПОНЕНТОВ И ГРАНИЧНОГО СЛОЯ МЕЖДУ НИМИ НА МЕХАНИЧЕСКИЕ СВОЙСТВА КОМПОЗИЦИЙ ПОЛИПРОПИЛЕН–ПОЛИЭТИЛЕН ВЫСОКОЙ ПЛОТНОСТИ
Б. Ф. Шклярук a, В. А. Герасин a, М. А. Гусева a, *, В. В. Малетина a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: guseva@ips.ac.ru
Поступила в редакцию 09.04.2021
После доработки 29.04.2021
Принята к публикации 10.05.2021
Аннотация
Методом экструзии в расплаве приготовлены смеси изотактического ПП и ПЭВП различного состава при двух режимах кристаллизации. Первая серия композиций получена быстрым охлаждением расплава (≈8000 град/мин). Методами РСА и микроскопии установлено, что чистый ПП в данных условиях образует смектическую мезофазу. В отличие от него ПП-компонент в смесях кристаллизуется в виде моноклинных кристаллов α-формы, объединенных в сферолиты диаметром ~4–5 мкм. ПЭ-компонент композиций имеет меньшую энтальпию плавления, чем у чистого ПЭ и уменьшается с ростом содержания ПП в системе. Это может быть обусловлено образованием межфазного слоя, где молекулы ПЭ и ПП перемешаны на молекулярном уровне. Оценка по теории Гелфанда показала, что толщина граничного слоя в композициях первой серии составляет около 30 нм. Вторая серия композиций получена медленным охлаждением расплава (≈2 град/мин). Все образцы данной серии содержат крупные (~80–120 мкм) радиальные сферолиты, но в смесях их размер несколько меньше, чем в чистом ПП. Энтальпия плавления ПЭ- и ПП-компонентов в смесях второй серии не зависит от состава и равна соответствующим значениям для чистых ПЭ и ПП. По-видимому, при медленном охлаждении расплава двух полиолефинов молекулы из межфазного слоя успевают переместиться в соответствующие домены и образуется обычная двухфазная система без “переходной” зоны. Благодаря развитой межфазной зоне в композициях первой серии фазы деформируются совместно, и эти материалы обладают хорошими деформационными свойствами. В композитах второй серии из-за отсутствия протяженного граничного слоя все механические показатели хуже, чем у чистых полимеров и смесей первой серии (“антагонистический эффект”).
ВВЕДЕНИЕ
Цель создания смесей на основе ПЭ и ПП – совмещение положительных качеств ПП (высокая прочность, теплостойкость и формоустойчивость) и ПЭ (высокая гибкость, ударная вязкость) в одном материале. Композициям, состоящим из ПЭ и ПП разного типа, посвящено множество статей. На наш взгляд, важнейшими из них являются два обзора [1, 2], охватывающие широкий круг публикаций по смесям ПЭ–ПП с 1959 по 2019 гг. Несмотря на значительное число работ, некоторые экспериментальные факты, относящиеся к системам ПЭ–ПП, до сих пор не получили четкого объяснения и остаются дискуссионными.
Так, некоторые авторы сообщают о “синергетическом эффекте” в композициях ПЭ–ПП, который проявляется в увеличении модуля упругости или других механических характеристик на величину большую, чем предсказывает правило аддитивности [1–4]. Но для других пар ПЭ–ПП синергизма не обнаружили и даже наблюдали “антагонизм” (существенное уменьшение механических показателей смесей по сравнению с их значениями у исходных полимеров). Морфология и механические свойства композиций ПЭ–ПП зависят как от микроструктуры смешиваемых полимеров (разветвленность ПЭ, изотактичность ПП, длина цепей и т.п.), так и от методов получения смесей. В настоящее время остается неясным, при каких условиях в системах ПЭ–ПП будет происходить увеличение механических характеристик.
В твердом состоянии смеси на основе ПЭ и ПП относятся к термодинамически несовместимым, так как при их совмещении любым способом (смешение через раствор, через расплав, полимеризация in situ) общая кристаллическая решетка не образуется. Поскольку ПЭ и ПП близки по химической природе, расплавы данных полимеров частично совместимы, и на границе раздела областей ПЭ и ПП формируется межфазный слой со смешением, близким к молекулярному. [5]. Следует ожидать, что если компоненты смеси кристаллизуются при одинаковой или близких температурах, при охлаждении расплава фазы не успеют разделиться полностью, что позволит улучшить контакт на межфазной границе и структурную однородность таких систем. В связи с этим для исследования были выбраны ПЭ и ПП, имеющие при скорости охлаждения 8 град/мин температуру кристаллизации 114 и 112°С соответственно. Условия переработки и охлаждения (соотношение между скоростью охлаждения и скоростью диффузии макромолекул) оказывают сильное влияние на морфологию и свойства композиций ПЭ–ПП. Данный аспект пока изучен недостаточно, и его изучение составляло одну из задач настоящей работы.
В изотропном состоянии морфология смесей ПЭ–ПП может быть двух типов в зависимости от соотношения компонентов [6]: при содержании второго полимера до φ ≈ 30–40% – обычная дисперсия (домены округлой формы, имеющие размер несколько микрометров, равномерно распределены в изотропной матрице); 2) при φ ≈ 40–60% – система из двух непрерывных матриц. Используя особые приемы экструзии или ориентационной вытяжки, “капли” дисперсной фазы можно растянуть в несколько раз и превратить в волокна нанометрового диаметра [7]. Структурно-механические модели полимерных композитных материалов предсказывают, что волокна с высоким фактором формы обладают гораздо большим усиливающим эффектом, чем изодиаметрические включения [8, 9].
Конечная цель данной работы – установить корреляцию между структурными параметрами и механическими свойствами ориентированных композиций ПЭ–ПП. Метод экструзии в расплаве позволяет получить смеси с любым соотношением компонентов, но некоторые из них разрушаются хрупко и не подходят для ориентационной вытяжки. В связи с этим исследования были начаты с приготовления смесей, имеющих структуру обычной дисперсии. В настоящей статье представлены результаты, полученные для изотропных композиций ПЭ–ПП.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для приготовления смесей использовали ПЭ высокой плотности и изотактический ПП, основные характеристики которых представлены в табл. 1.
Изучение механизма смешения высоковязких жидкостей показывает, что компоненты наиболее равномерно распределяются по объему смеси, если отношение вязкостей диспергируемой фазы и дисперсионной среды не превышает трех [10–12]. Для исследуемой нами пары полимеров ηПЭ/ηПП = 2.2. С ростом температуры смешения увеличивается подвижность макромолекул, что способствует взаимной диффузии компонентов и повышению структурной однородности системы. Кроме того, при высокой температуре начинается термическое окисление полимеров. Увеличение скорости вращения шнеков и времени смешения может вызвать снижение молекулярной массы смешиваемых полимеров.
В связи с этим приготовление композиций было начато с разработки способа экструзии, обеспечивающего качественное смешение при минимальной термо- и механодеструкции ПЭВП и ПП. Были выбраны следующие параметры смешения: температура смешения 190°С, скорость вращения шнеков 80 об/мин, время смешения 10 мин. В микроэкструдер “MiniLab НААКЕ Rheomex CTW5” сначала загружали более вязкий ПЭВП, постепенно добавляя к нему гранулы ПП. Экструзию проводили в атмосфере азота.
Из экструдатов методом горячего прессования при 190°С получали пленки диаметром 60 мм и толщиной 0.35 мм. Первую серию смесей готовили быстрой закалкой в ледяной воде (скорость охлаждения q ~ 8000 град/мин), вторую – постепенным охлаждением в прессе со скоростью q = = 2 град/мин.
С помощью штампа из пленок вырубали двусторонние “лопатки” с размером рабочей части 10 × 3 × 0.35 мм. Приготовленные “лопатки” растягивали на разрывной машине “TIRAtest-2000” (ГДР) со скоростью 20 мм/мин до разрыва при комнатной температуре. В каждой серии испытывали 7–10 образцов и определяли усредненные механические характеристики: модуль упругости Е, предел текучести σт, удлинение в пределе текучести εт, прочность на разрыв σр, разрывное удлинение εр.
Термодинамические параметры плавления и кристаллизации образцов с разной термической предысторией определяли на калориметре DTAS-30. Термограммы первого нагревания смесей и исходных полимеров с навеской 15–20 мг регистрировали в температурном диапазоне от 20 до 190о С со скоростью нагревания 8 град/мин. Погрешность при определении температуры и энтальпии составляет ±1° и ±5% соответственно.
Рентгенограммы в больших углах дифракции получали на дифрактометре DRON-3 (излучение CuKα с длиной волны λ = 0.154 нм). Микрофотографии прессованных пленок и деформированных “лопаток” получали в режиме “на прохождение” на оптическом микроскопе “Bresser Micro 5MP”.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Кристаллическая структура смесей ПЭВП–ПП
В публикации [13] было показано, что если расплав изотактического ПП в пресс-форме толщиной 100 мкм охлаждать со скоростью большей, чем 100 град/с, в нем образуется смектическая мезофаза. Два широких “размытых” максимума в области углов 2θ ≈ 15° и 21° на рентгенограмме (рис. 1) и отсутствие каких-либо кристаллических образований на микрофотографиях (рис. 2а) подтверждают, что чистый ПП серии 1 находится в мезоморфном состоянии. Изначально ожидали, что ПП-компонент в смесях серии 1 будет иметь такую же структуру. Тем не менее, на рентгенограммах композиций наблюдаются четко выраженные брэгговские пики при 2θ = 14.1°, 16.9°, 18.8° и 21.8°, соответствующие рефлексам 110, 040, 130 и 111/041 α-модификации ПП. На микрофотографиях видны небольшие сферолиты ПП диаметром до 4–5 мкм (рис. 2б).
Рис. 1.
Дифрактограммы образцов серии 1: 1 – ПП; 2, 3 – композиция ПП–ПЭВП состава 95 : 5 (2) и 70 : 30 (3).
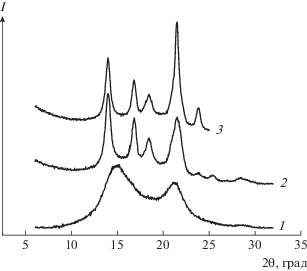

На рентгенограммах смесей серии 1 также имеются кристаллические рефлексы при 2θ = 21.5° и 23.9°, отвечающие рассеянию от орторомбических кристаллов ПЭВП. Интенсивность этих максимумов возрастает с повышением содержания ПЭВП в смеси.
Данные РСА и микроскопии показывают, что при охлаждении со скоростью 2 град/мин ПП успевает закристаллизоваться в виде крупных (80–120 мкм) сферолитов, состоящих из моноклинных кристаллов α-формы (рис. 3, 4). Добавление ПЭВП приводит к тому, что размер сферолитов ПП несколько уменьшается, а их границы из-за контакта с “каплями” ПЭ становятся менее ровными (рис. 4б).
Рис. 3.
Дифрактограммы образцов серии 2: 1 – ПП; 2, 3 – композиция ПП–ПЭВП состава 70 : 30, полученная экспериментально (2) и суммированием кривых (3).
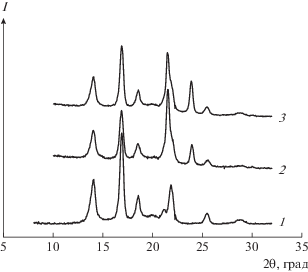

Было установлено, что рентгенограммы смесей серии 2, полученные экспериментально, практически совпадают с рентгенограммами, полученными алгебраическим сложением кривых рассеяния для чистых ПЭВП и ПП с учетом соответствующих объемных долей (рис. 3). Это свидетельствует о том, что композиции серии 2 представляют собой простые двухфазные дисперсии, а переходный слой между компонентами имеет очень маленькую толщину.
Термодинамические параметры плавления и кристаллизации смесей
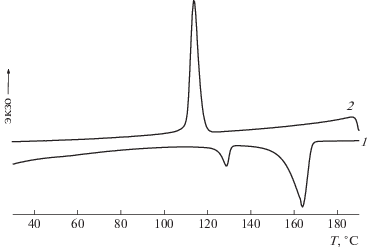
На кривых нагревания (рис. 5) композиций всех составов обеих серий наблюдаются два пика, отвечающих плавлению орторомбических кристаллов ПЭ при 132°С и моноклинных кристаллов ПП при 165°С. Одинарный экзотермический пик при 112°С на кривых охлаждения показывает, что интервалы кристаллизации ПП и ПЭВП перекрываются.
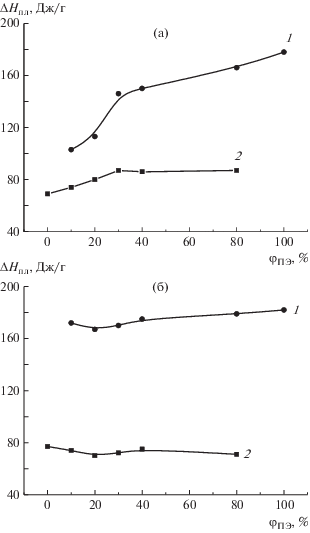
Для серии 1 было обнаружено, что энтальпия плавления ПЭВП в композиции с ПП значительно меньше, чем у чистого ПЭВП (рис. 6а). Особенно низкие значения энтальпии ПЭВП были зарегистрированы для смесей с его малым содержанием (φПЭ < 20%). Наоборот, энтальпия плавления ПП увеличивается с повышением концентрации ПЭВП в смеси (рис. 6). В композициях серии 2 энтальпии плавления обеих компонент практически не зависят от состава и равны соответствующим значениям для чистых полимеров (рис. 6б).
Рис. 6.
Зависимости энтальпии плавления ПЭВП (1) и ПП (2) от содержания ПЭВП в смесях серий 1 (а) и 2 (б).
Согласно теории кристаллизации [14–16], она всегда начинается с образования первичных центров кристаллизации, которые могут быть гомогенными или гетерогенными. Чистый ПП без добавок имеет небольшое число собственных центров кристаллизации, но очень легко поддается воздействию различных нуклеирующих агентов [17–19]. В качестве нуклеатоторов ПП могут выступать как неорганические наполнители (кварц, каолин, тальк, монтмориллонит), так и органические вещества (соли бензойной и стеариновой кислот, терефталевая кислота, производные сорбитола и т.д.). Как правило, введение нуклеаторов в изотактический ПП вызывает увеличение его температуры кристаллизации на 15–25°С, существенное сокращение размеров сферолитов и повышение общей степени кристалличности. Кроме того, нуклеаторы могут стимулировать кристаллизацию ПП в β-форме [20, 21].
В исследованных нами композициях ПП–ПЭВП температура кристаллизации ПП не повышалась, а в смесях с высоким содержанием ПЭНП наблюдали даже снижение Ткр ПП со 119° до 97°С [4]. По-видимому, в смесях с ПЭ любого вида кристаллизация ПП становится возможной после образования первых кристаллитов ПЭ, которые играют роль гетерогенных зародышей для макромолекул ПП. В ПП, содержащем часто используемые органические нуклеаторы, сферолиты имеют диаметр ~1 мкм [17–19]. В композициях серии 1 средний размер сферолитов ПП несколько больше (3–5 мкм), а в смесях серии 2 – значительно больше (60–90 мкм). Суммируя все данные, можно заключить, что ПЭВП является не очень эффективным агентом нуклеации ПП и способствует его кристаллизации в α-форме.
ПЭ высокой плотности, обладающий линейной структурой, содержит большое количество гомогенных центров кристаллизации и имеет очень высокую скорость кристаллизации [14–16]. В связи с этим многочисленные минеральные наполнители и органические вещества, используемые в качестве эффективных нуклеаторов для других полимеров, практически не влияют на процесс кристаллизации ПЭВП и структуру его кристаллов [22]. Из-за наличия некоторого количества боковых ответвлений кристаллизуется с меньшей скоростью, чем ПЭВП, что делает его более восприимчивым к воздействию разных нуклеирующих агентов [23].
Уменьшение энтальпии плавления ПЭВП в композитах серии 1 может быть вызвано образованием протяженного межфазного слоя на границе с ПП.
Согласно теории Helfand [24, 25], толщина межфазного слоя, возникающего при контакте расплавов двух полимеров, определяется выражением
(1)
$~~{{d}_{{{\text{AB}}}}} = \;~2{{\left( {\frac{{{{b}_{{\text{A}}}}{{b}_{{\text{B}}}}}}{{6\chi }}} \right)}^{{1/2}}},~$(2)
$~{{{{\gamma }}}_{{{\text{AB}}}}} = {{\left( {\frac{1}{6}\chi {{\rho }_{{\text{A}}}}{{\rho }_{B}}{{b}_{A}}{{b}_{B}}} \right)}^{{\frac{1}{2}}}}RT~$Здесь γAB – межфазное натяжение, ρА, ρВ – молярная плотность полимеров А и В соответственно, R – универсальная газовая постоянная, Т – температура.
Для расчетов по формулам (1) и (2) были использованы следующие значения: при Т = 190°С параметр γAB = 2.4 мН/м [26], ρПЭ = 3.14 × 104 моль/м3, ρПП = 2.09 × 104 моль/м3 [27], bПЭ = 1.4 нм ≈ bПП [28]. В результате расчетов было установлено, что толщина межфазного слоя в смеси расплавов ПЭ и ПП при температуре 190°С составляет ~ 27 нм, что по порядку величины соответствует размеру граничного слоя для других пар полимеров [25]. Следует отметить, что теория Helfand была разработана для невозмущенных расплавов полимеров. В неравновесных условиях под действием механических напряжений в процессе экструзии и формования толщина межфазного слоя может существенно увеличиваться [29].
При охлаждении и кристаллизации расплава смеси полимеров молекулы в граничном слое разделяются и стремятся диффундировать к молекулам своего типа в соответствующие домены. Необходимое для этого время можно оценить на основе теории рептаций [28, 30]. Чтобы воспользоваться диффузионным соотношением, нужно знать коэффициент диффузии в межфазном слое DAB. Если предположить, что он незначительно отличается от коэффициента самодиффузии полиэтилена Ds и имеет такой же порядок величины 10–16 м2/с [28], получим t ~ (dAB)2/Ds ~ 7 c.
При охлаждении расплава со скоростью q = 8000 град/мин понижение температуры от 190 до 120°С (температура начала кристаллизации ПЭВП и ПП) происходит приблизительно за 0.5 с, а при q = 2 град/мин – за 35 мин. Естественно ожидать, что в первом случае в значительной мере удается зафиксировать структуру с широким межфазным слоем, существовавшую в расплаве. Во втором случае макромолекулы ПЭВП и ПП из межфазного слоя успевают диффундировать в соответствующие домены, число сегментов в межфазной области существенно сокращается, и кристаллизация ПЭ- и ПП-компонентов протекает практически независимо друг от друга.
Механические свойства
Деформационные кривые чистых ПП и ПЭВП серии 1 (рис. 7, 8) имеют вид, типичный для кристаллизующихся полимеров и содержат четыре этапа: область упругих деформаций, стадию формирования шейки, стадию распространения шейки по образцу и деформационное упрочнение. У композиции ПП–ПЭВП состава 90 : 10 предельное удлинение немного больше, а у смеси состава 80 : 20 и 70 : 30 немного меньше, чем у чистого ПП. Все эти образцы деформируются с образованием шейки, но композиции ПП–ПЭВП состава 80 : 20 и 70 : 30 имеют более короткую зону деформационного упрочнения, чем чистый ПП и образец 90 : 10. Композиции ПП–ПЭВП состава 60 : 40 и 20 : 80 разрушаются хрупко сразу после достижения верхнего предела текучести.
Рис. 7.
Диаграммы растяжения образцов серии 1 с матрицей ПП: 1 – ПП; 2–5 – композиция ПП–ПЭВП состава 90 : 10 (2), 80 : 20 (3), 70 : 30 (4) и 60 : 40 (5).
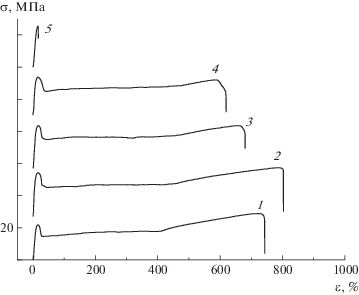
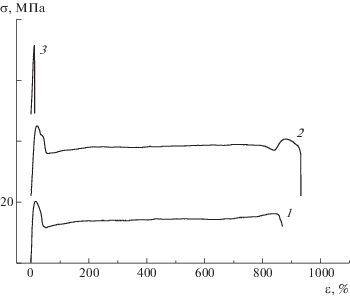
Рис. 8.
Диаграммы растяжения образцов серии 1 с матрицей ПЭВП: 1 – ПЭВП (1); 2, 3 – композиция ПП–ПЭВП состава 10 : 90 (2) и 20 : 80 (3).
Определенные из диаграмм растяжения механические характеристики представлены в табл. 2. Модуль упругости исходных ПЭВП и ПП имеет близкие значения, поэтому модуль приготовленных из них композиций практически не зависит от состава и попадает в тот же диапазон значений (Е ≈ 300–350 МПа). Параметрами, наиболее чувствительными к составу, оказались предел текучести σт (рис. 9а) и разрывное удлинение εр (рис. 9б). Видно, что экспериментальная зависимость предела текучести от концентрации ПЭВП для композиций, где ПП является матрицей, проходит выше, чем прямая, рассчитанная по правилу аддитивности (σ = φППσПП + φПЭσПЭ), и достигает максимального значения при φПЭ ≈ 10–20%.
Таблица 2.
Механические свойства композиций серии 1
| Состав композиций ПП–ПЭВП | Е, МПа | σт, МПа | εт, % | σр, МПа | εр, % |
|---|---|---|---|---|---|
| 100 : 0 | 319 (±73) | 21.3 (±1.0) | 16 (±2) | 27.2 (±3.9) | 679 (±134) |
| 90 : 10 | 363 (±63) | 26.8 (±0.5) | 16 (±1.3) | 29.5 (±1.1) | 743 (±49) |
| 80 : 20 | 330 (±53) | 26.2 (±1.4) | 16 (±2) | 22.8 (±4.4) | 588 (±89) |
| 70 : 30 | 302 (±51) | 24.2 (±1.3) | 17 (±1) | 22.1 (±0.3) | 596 (±42) |
| 60 : 40 | 288 (±102) | 24.6 (±2.2) | 14 (±3) | 24.6 (±2.2) | 14 (±3) |
| 20 : 80 | 259 (±26) | 21.9 (±1.0) | 13 (±1) | 21.9 (±1.0) | 13 (±1) |
| 10 : 90 | 277 (±23) | 21.7 (±0.9) | 21 (±2) | 18.7 (±1.3) | 845 (±173) |
| 0 : 100 | 312 (±47) | 21.5 (±0.9) | 17 (±1.2) | 18.2 (±2.6) | 886 (±176) |
Рис. 9.
Зависимости предела текучести (а) и разрывного удлинения (б) от содержания ПЭВП в композициях серии 1.
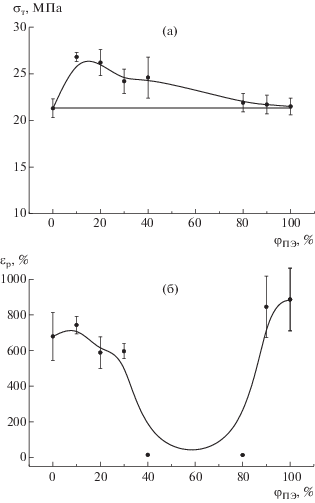
На первый взгляд, такой характер изменения σт(φ) может свидетельствовать о “синергетическом эффекте”. Однако необходимо учесть, что все известные структурно-механические модели [8, 9] предполагают, что структура компонентов в смеси полностью идентична структуре исходных гомополимеров, и это условие не выполняется для композиций с ПП-матрицей. Как уже отмечалось выше, при охлаждении со скоростью 8000 град/мин чистый ПП имеет мезоморфную структуру, тогда как ПП-компонент в смесях с ПЭВП – α-моноклинную. Для пластической деформации твердой кристаллической структуры необходимы большие нагрузки, чем для деформации менее упорядоченной мезофазы, что приводит к повышению предела текучести композиций.
Кристаллическая структура ПЭ-компонента в смесях с ПП практически такая же, как у чистого ПЭВП, поэтому предел текучести композиций соответствуют аддитивным значениям.
Предельная деформируемость образцов ПП–ПЭВП состава 90 : 10, 80 : 20, 70 : 30 и 10 : 90 почти такая же, как у чистых ПП и ПЭ (с учетом разброса экспериментальных данных). Композиции ПП–ПЭВП состава 60 : 40 и 20 : 80 разрушаются хрупко при удлинении 12–14% до начала процесса ориентации. По всей вероятности, изменение характера разрушения в этих образцах обусловлено началом инверсии фаз [6]. Когда смесь имеет структуру обычной дисперсии (“капли в матрице”) и жесткость дисперсной фазы меньше или примерно равна жесткости дисперсионной среды (ЕПЭ ≈ ЕПП), матрица передает напряжение “каплям”, и они растягиваются совместно с ней, превращаясь в вытянутые эллипсоиды. Предельная деформируемость таких смесей остается на уровне матричного полимера и может даже превышать ее, если второй полимер имеет очень большую разрывную деформацию.
При увеличении концентрации второго полимера он начинает образовывать более крупные кластеры, которые сосуществуют наряду с мелкими “островками”. При дальнейшем повышении концентрации число дискретных доменов уменьшается и образуется единый непрерывный объем сложной конфигурации. Известно, что большую вероятность образовать непрерывную фазу имеет менее вязкий полимер (ПП), поэтому в композитах с ПЭ-матрицей обращение фаз начинается при меньшем содержании дисперсной фазы (φПП ≈ 20%), чем в композитах с ПП-матрицей (φПЭ ≈ 40%). В системе из двух непрерывных взаимопереплетенных матриц фазы препятствуют деформированию друг друга, и разрушение происходит по межфазной границе при малых удлинениях.
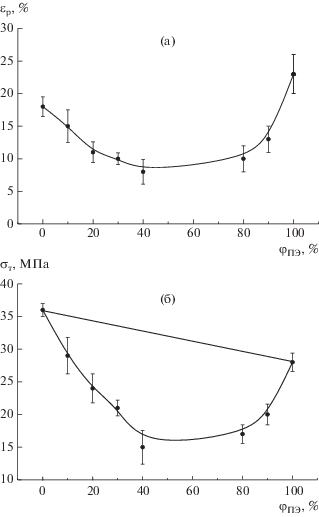
Как уже отмечалось, пленки ПП, охлажденного со скоростью 2 град/мин, состоят из крупных сферолитов диаметром до 120 мкм. Такие большие и жесткие кристаллические образования практически не способны деформироваться пластически, и образцы ПП с такой морфологией разрушаются хрупко, при малых удлинениях [13]. Несмотря на то, что в смесях ПП–ПЭВП размеры сферолитов уменьшаются по сравнению с чистым ПП (рис. 4), это не приводит к улучшению пластических свойств. Предельная деформируемость композиций серии 2 снижается с повышением содержания второго полимера (т.е. с увеличением протяженности межфазной границы) (рис. 10а). По всей вероятности, основная причина преждевременного разрушения образцов данной серии – отсутствие протяженного граничного слоя, соединяющего компоненты и передающего нагрузку между ними. Смеси серии 2 разрушаются до того, как начинается процесс ориентации, и предел текучести в таком случае совпадает с разрывной прочностью. Зависимость предела текучести от состава во всем диапазоне значений проходит ниже, чем прямая, рассчитанная по правилу аддитивности (так называемый “антагонистический эффект”) (рис. 10б).
Рис. 10.
Зависимости разрывного удлинения (а) и предела текучести (б) от содержания ПЭВП в композициях серии 2.
Поскольку в дальнейшем планируется получать ориентированные пленки и волокна, целесообразно для этого использовать смеси серии 1 с содержанием ПЭВП 10–30% и ~90%, сохраняющие хорошие пластические свойства.
ЗАКЛЮЧЕНИЕ
Методом экструзии в расплаве приготовлены две серии композиций ПП–ПЭВП, отличающиеся скоростью охлаждения: 8000 град/мин – серия 1 и 2 град /мин – серия 2.
Обнаружено, что ПЭВП влияет на нуклеацию ПП и способствует его кристаллизации в α-форме. Если ПП имеет сферолитную структуру, добавление ПЭВП приводит к уменьшению размера сферолитов и искажению их формы.
В первой серии образцов энтальпия плавления ПЭВП и ПП увеличивается с ростом содержания ПЭВП в смеси. Во второй серии значения энтальпии плавления ПЭВП и ПП не зависят от состава и равны соответствующим значениям для чистых полимеров. Это обусловлено тем, что при быстром охлаждении расплава большое число макромолекул ПЭВП и ПП остаются в граничном слое, тогда как при медленном охлаждении компоненты разделяются и образуется двухфазная система.
Благодаря развитой межфазной зоне в композициях серии 1 фазы деформируются совместно, и эти материалы обладают хорошими пластическими свойствами. В композитах серии 2 из-за отсутствия граничного слоя все механические показатели хуже, чем у чистых полимеров и смесей серии 1 (“антагонистический эффект”).
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 20-03-00168-а).
Список литературы
Teh J.W., Rudin A., Keung J.C. // Adv. Polym. Technol. 1994. V. 13. № 1. P. 1.
Graziano A., Jaffer S., Sain M. // J. Elastomers Plastics. 2019. V. 51. № 4. P. 291.
Erina N.A., Prut E.V. // Polymer Science A. 2000. V. 42. № 2. P. 183.
Mofokeng T.G., Ojijo V., Ray S.S. // Macromol. Mater. Eng. 2016. V. 301. № 10. P. 1191.
Кришевски М., Пакула Т., Грембович Я. // Высокомолек. соед. А. 1974. Т. 26. № 7. С. 1569.
Jose S., Aprem A.S., Francis B., Chandy M.C., Werner P., Alstaedt V., Thomas S. // Eur. Polym. J. 2004. V. 40. P. 2105.
Krajenta J., Pawlak A., Gałęski A. // Polimery. 2015. V. 60. № 10. P. 664.
Nielsen L., Lawrence E. Mechanical Properties of Polymers and Composites. New York: Marcel Dekker, 1974.
Manson J., Sperling L. Polymer Blends and Composites. New York: Acad. Press, 1976.
Wu S. // Polym. Eng. Sci. 1987. V. 27. № 5. P. 335.
Мийченко И.П. Технология полуфабрикатов полимерных материалов. Уч. пособие. СПб.: Научные основы и технологии, 2012.
Кулезнев В.Н. Смеси и сплавы полимеров. СПб.: Научные основы и технологии, 2013.
Zia Q., Radusch H.-J., Androsch R. // Polym. Bull. 2009. V. 63. P. 755.
Van Krevelen D.W. Properties of Polymers Correlations with Chemical Structure. Amsterdam; London; New York: Elsevier, 1972.
Mandelkern L. Crystallization of Polymers. New York; San-Francisco; Toronto; London: McGraw-Hill, 1964.
Hoffman J.D., Davis G.T., Lauritzen J.I. // Treatise on Solid State Chemistry. Boston: Springer, 1976. V. 3. P. 497.
Libster D., Aserin A., Garti N. // Polym. Adv. Technol. 2007. V. 18. № 9. P. 685.
Marco C., Gómez M.A., Ellis G., Arribas J.M. // J. Appl. Polym. Sci. 2002. V. 84. № 9. P. 1669.
Гликштерн М.В. // Полим. матер. 2002. № 6. С. 13.
Marco C., Gomez M.A., Ellis G., Arribas J.M. // J. Appl. Polym. Sci. 2002. V. 86. № 3. P. 531.
Li X., Hu K., Ji M., Huang Y., Zhou G. // J. Appl. Polym. Sci. 2002. V. 86. № 3. P. 633.
Hoffmann K., Huber G., Mäder D. // Macromol. Symp. 2001. V. 176. № 1. P. 83.
Li J., Xu J., Mannion M.J. Pat. № 7781511 USA, 2010.
Helfand E., Tagami Y. // J. Polym. Sci., Polym. Phys. 1971. V. 9. № 10. P. 741.
Helfand E., Tagami Y. // J. Chem. Phys. 1972. V. 56. № 7. P. 3592.
Souza A.M.C., Demarquette N.R. // Polymer. 2002. V. 43. № 4. P. 1313.
Антипов Е.М. Дис. … д-ра хим. наук. М.: ИНХС РАН, 1990.
Rubinstein M. and Colby R. Polymer Physics. Oxford: Oxford Univ. Press, 2003.
Липатов Ю.С. Межфазные явления в полимерах. Киев: Наукова думка, 1980.
Гросберг А.Ю., Хохлов А.Р. Физика в мире полимеров. М.: Наука, 1989.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)