

Высокомолекулярные соединения (серия А), 2022, T. 64, № 1, стр. 72-80
О ПРИМЕНИМОСТИ КОНЦЕПЦИИ КРИТИЧЕСКОЙ ХРОМАТОГРАФИИ ДЛЯ ИССЛЕДОВАНИЯ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕОТИДОВ В ОДНОНИТЕВЫХ ДНК
А. В. Горшков a, *, И. Г. Роздина a, В. В. Евреинов a
a Федеральный исследовательский центр химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: alex.gorshkov@gmail.com
Поступила в редакцию 13.04.2021
После доработки 15.09.2021
Принята к публикации 25.10.2021
- EDN: UYLOLZ
- DOI: 10.31857/S2308112022010035
Аннотация
Решеточная модель адсорбции макромолекул применена для описания разделения однонитевых ДНК в изократическом и градиентном режимах на обращенной фазе в смеси воды и ацетонитрила. Показано, что данная модель, дополненная корреляционными соотношениями зависимости эффективной энергии звена от состава растворителя, дает возможность с хорошей точностью описать закономерности хроматографического разделения как гомополимеров нуклеотидов, так и их гетерополимеров. Для гетерополимеров корреляция между расчетом и экспериментальными данными имеет меньшую точность, чем для гомополимеров, что связано с малым вкладом энтропийной составляющей адсорбционного взаимодействия при вариации последовательности. Обсуждаются варианты уточнения модели для учета эффекта соседних звеньев.
ВВЕДЕНИЕ
ДНК и РНК – высокомолекулярные гетерополимеры, играющие главную роль в передаче наследственных признаков и в управлении процессами биосинтеза белков. Они состоят из последовательности связанных в цепь нуклеотидов четырех типов, три из которых (A, G и C) типичны для ДНК и РНК, а два других T и U входят в состав только ДНК и РНК соответственно. Благодаря свойству комплементарности нуклеотиды попарно взаимодействуют как в одной цепи, так и между соседними цепями, образуя сложные пространственные структуры, определяемые последовательностью, иначе первичной структурой ДНК или РНК.
Для проявления текста последовательности ДНК, например, при адсорбционном взаимодействии с поверхностью в хроматографическом эксперименте, структура двойной спирали должна быть разрушена, переведена в однонитевое состояние. В таком состоянии адсорбционные свойства однонитевых ДНК и РНК оказываются весьма схожими. Олигонуклеотиды однонитиевых ДНК с относительно небольшой степенью полимеризации n = 10–60 применяются в разнообразных биологических исследованиях. Для чтения текстов нуклеотидных последовательностей и отбора особых, важных для биологии олигонуклеотидов, используются методы жидкостной хроматографии с масс-спектрометрическим детектированием.
Вопросам хроматографического исследования последовательности олигонуклеотидов посвящено большое число работ. Основным итогом их явилось создание простой корреляционной теории, связывающей объем удерживания с составом цепи олигонуклеотида [1]. Такая теория основывается на аддитивном вкладе нуклеотида в объем удерживания, дополненном логарифмической поправкой на длину цепи n, и по этой причине связана в первую очередь со средним нуклеотидным составом цепи. Идейно данная теория весьма близка к различным вариантам аддитивных моделей, разработанных для предсказания объемов удерживания пептидов и полимеров, состоящих из аминокислот [2–5]. Как было показано нами в работе [6], эмпирические поправки, вводимые в аддитивные корреляционные теории для их уточнения, автоматически вытекают из физических моделей адсорбции, представляющих макромолекулы в виде жестких стержней или в виде цепи случайных блужданий, адсорбирующихся в ограниченных объемах.
Подобно любым макромолекулам, цепочечная структура онДНК приводит к фазовому характеру адсорбции, происходящей в узком диапазоне состава растворителя. Как и в любых цепочечных макромолекулах, объем удерживания однонитевых ДНК может определяться не только средним составом, но и характером чередования звеньев с различной энергией адсорбции. В частности, макромолекулы, имеющие один и тот же набор нуклеотидов, но различающиеся последовательностью их расположения в цепи, могут заметно отличаться по своим адсорбционным (и не только) свойствам.
Зависимость адсорбционных свойств от последовательности вытекает из физической сущности связывания звеньев в цепь и включения во взаимодействие с поверхностью энтропийной составляющей. В наиболее простом виде адсорбционное взаимодействие описывается моделью решеточной цепи [7], лежащей в основе модели BioLCCC [8]. В отличие от корреляционных теорий, в которых влияние вариации текста присутствует в виде поправок, в модели BioLCCC зависимость адсорбционных свойств от последовательности звеньев возникает как результат некоммутативного произведения переходных матриц [7, 8].
Хотя для практического применения корреляционные теории в большинстве случаев достаточны, с идейной точки зрения представляет интерес возможность описания адсорбционных свойств однонитевых ДНК в рамках модели адсорбции решеточной цепи или жесткого стержня, что делается в настоящей работе. Такая модель дает глубокое понимание физики процесса адсорбции и его связи с общей теорией критических явлений. Взаимосвязь параметров, описывающих макромолекулу, адсорбент и растворитель, открывает широкие возможности для оптимизации разделения по тем или иным признакам строения цепи.
МОДЕЛЬ АДСОРБЦИОННОГО ВЗАИМОДЕЙСТВИЯ
В отличие от пептидов макромолекулы однонитевых ДНК являются полиэлектролитами, в которых каждое звено может быть заряжено. Для жидкостной хроматографии, обычно проводимой в водных средах на обращенной фазе, используется растворитель сложного состава, содержащий, кроме воды и ацетонитрила, добавки ацетата триэтиламмония при pH 7.0, что позволяет нейтрализовать заряженность звена и включить его адсорбционное взаимодействие. В таком растворителе взаимодействие звена с поверхностью на молекулярном уровне имеет сложный характер и не сводится к чисто гидрофобному. Однако для рассматриваемых ниже моделей адсорбции решеточной цепи или стержня точный механизм взаимодействия в масштабе звена не важен. Важно лишь, чтобы это взаимодействие было локальным, т.е. действие адсорбционных сил проявлялось на расстояниях порядка размера звена, что будет предполагаться ниже.
Значительным упрощением для построения модели адсорбционного взаимодействия онДНК является тот факт, что по крайней мере для трех оснований T, C и A возможен синтез гомополимеров, представляющих не что иное, как обычные макромолекулы олигомеров (здесь и далее используются стандартные буквенные обозначения нуклеотидов). Закономерности хроматографического разделения олигомеров с функциональными группами, в частности переход через критическую точку, смена режима разделения с эксклюзионного на адсорбционный, исчезновение молекулярно-массового распределения, хорошо известны [9]. Как и для полимеров, для гомополимеров однонитевых ДНК переход в адсорбированное состояние осуществляется в узком диапазоне состава растворителя и имеет черты фазового перехода. Это упрощает нахождение основного феноменологического параметра модели – эффективной энергии адсорбции звена ε. Подробное экспериментальное исследование разделения онДНК приведено в работе [1], данные которой будут сопоставлены с результатами расчета.
Эксперименты в работе [1] проводили на колонке Xterra MS C18 размером 30 × 4.6 мм (или 50 × 4.6 мм для разделения в градиенте), диаметр пор адсорбента 125 Å. Для расчетов примем размер звена нуклеотида 5 Å, тогда размер пор, выраженный в звеньях, D = 25. Отметим, что точный выбор размера звена не очень важен для применения модели, однако от него будет зависеть величина эффективной энергии.
Чтобы связать рассчитываемый в теории [6, 7] коэффициент распределения Kd (численно равный статистической сумме макромолекулы в поре) с экспериментально измеряемым коэффициентом емкости k примем, что объем жидкой фазы V 0 = 0.9 от общего объема колонки, а объем пор Vp = 0.4 V 0. Тогда коэффициент емкости можно записать как
объем удерживания VR макромолекул а время удерживания tRгде u – объемная скорость подачи растворителя.Эффективную энергию адсорбции звеньев ε всюду в дальнейшем будем выражать в единицах kT.
Первый вопрос, на который необходимо ответить, анализируя данные по разделению гомополимеров [1], какую модель макромолекулы, жесткого стержня [6] или решеточной цепи блужданий [7], взять за основу? Качественно эти модели в доступных экспериментально режимах разделения, например пептидов, близки и обе предсказывают существование критической точки с резким изменением адсорбционных свойств. Тем не менее, они имеют заметные различия в том, что касается зависимости адсорбционного взаимодействия от длины цепи n.
Принято считать, что цепи нуклеотидов достаточно жесткие. Кроме того, учитывая полиэлектролитный характер цепи, казалось бы, логично предположить, что ей ближе соответствует модель стержня. Однако анализ экспериментальных данных в изократическом режиме для гомополимеров типа (dT)n показал, что для таких гомополимеров зависимость k от степени полимеризации n точнее соответствует модели цепи случайных блужданий [7], а не модели жесткого стержня (рис. 1). Модель стержня [6] предсказывает более резкую зависимость, заметно отличающуюся от экспериментальной.
Рис. 1.
Зависимость lnk от степени полимеризации n гомополимера (dT)n в изократических условиях при объемной доле ацетонитрила Vb = 0.08. 1 – эксперимент, 2 – расчет по модели клубка, 3 – расчет по модели стержня.
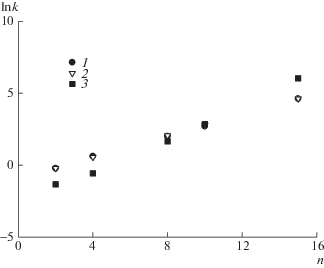
Возможным объяснением того, что решеточная цепь точнее соответствует эксперименту, может быть то, что состав растворителя в области, близкой к критической точке, нейтрализует заряженность звеньев цепи, и ее конформация становится близкой к клубку. Нельзя исключить, однако, что совпадение расчета в рамках модели BioLCCC и эксперимента обусловлено случайными причинами, которые рассматриваемая упрощенная модель не учитывает.
При каждом значении объемной доли Vb ацетонитрила в смеси молекулярно-массовые зависимости рис. 2 следуют модели решеточной цепи при одном и том же значении эффективной энергии адсорбции звена ε. Эффективные энергии звеньев, соответствующие экспериментальным точкам при разных Vb, приведены ниже.
Рис. 2.
Молекулярно-массовые зависимости коэффициента емкости k гомополимера T'(dT)n –2T' для изократического состава Vb = 0.08 (1, 2), 0.09 (3, 4), 0.1 (5, 6), 0.105 (7, 8). 1, 3, 5, 7 – эксперимент; 2, 4, 6, 8 – расчет.
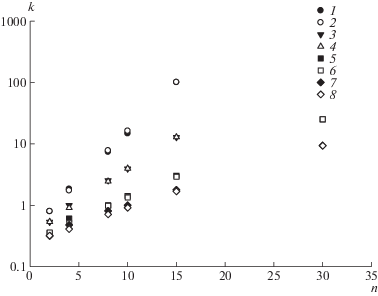
| ε | 1.60 | 1.17 | 0.72 | 0.56 | 0.45 | 0.41 | 0.37 | 0.31 | 0.26 |
| Vb | 0.04 | 0.06 | 0.08 | 0.09 | 0.10 | 0.105 | 0.11 | 0.12 | 0.13 |
| Nb | 0.014 | 0.021 | 0.029 | 0.033 | 0.037 | 0.039 | 0.041 | 0.045 | 0.049 |
(Nb – мольная доля ацетонитрила).
Для точного совпадения экспериментальных и теоретических величин k в модель дополнительно была введена энергия адсорбции концевых звеньев ε'(T '), отличающаяся на постоянную величину от энергии срединных звеньев цепи ε(T). Такое различие имеет определенное обоснование: химические структуры звена цепи T и концевого звена T ' различаются. Разница энергий ε'(T ') – ε(T) ≈ 1.0, постоянная и не зависящая от состава растворителя, позволяет получить точное соответствие расчетных и экспериментальных величин (рис. 2).
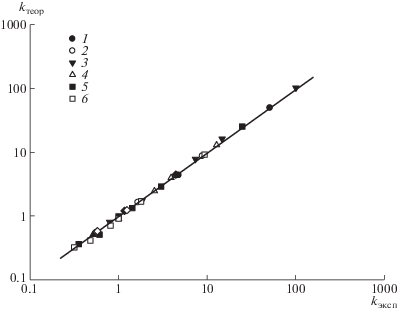
В целом решеточная цепь весьма хорошо описывает изократический эксперимент для гомополимера T '(dT)n –2T ' (рис. 3). По этой причине далее в работе для однонитевых ДНК разного состава будет применяться решеточная модель, аналогичная модели BioLCCC [8], ранее использованной авторами для описания разделения пептидов и белков.
Рис. 3.
Сравнение расчетных kтеор и экспериментальных kэксп коэффициентов емкости в изократических режимах для гомополимера T'(dT)n –2T' с n = 2 (1), 4 (2), 8 (3), 10 (4), 15 (5) и 30 (6) в диапазоне составов растворителя с объемной долей ацетонитрила Vb = 0.04–0.13.
Кратко содержание модели BioLCCC сводится к следующему. Поровое пространство адсорбента представляется в виде одномерной щели, размер которой D соответствует реальному размеру пор, выраженному в единицах размера звена. Макромолекула, адсорбирующаяся в такой поре, описывается цепью случайных блужданий длиной n на кубической решетке, размер ячеек которой совпадает с размером звена. Все возможные конфигурации цепи задаются произведением n переходных матриц W(εi), элементы которых зависят от энергии адсорбции εi соответствующего звена i последовательности, а размерность совпадает с размером поры D:
Коэффициент распределения Kd, численно совпадающий со статистической суммой макромолекулы, получается суммированием всех конфигураций цепи с помощью единичного вектора U, компоненты которого равны единице:
(2)
${{K}_{d}} = \frac{1}{D}{{U}^{T}} \times \left[ {\prod\limits_i {W({{\varepsilon }_{i}})} } \right] \times U$Порядок произведения матриц соответствует последовательности разных нуклеотидов i в цепи. Произведение матриц некоммутативно, следовательно, Kd зависит от последовательности нуклеотидов.
ОДНОНИТЕВЫЕ ДНК В ГРАДИЕНТЕ РАСТВОРИТЕЛЯ
Для описания зависимости VR от градиентного профиля необходимо связать эффективную энергию адсорбции звена ε с составом растворителя – объемной Vb или мольной долей Nb компонента b (ацетонитрила в данном случае). В модели BioLCCС используется приближение Ленгмюра, развитое для адсорбционной хроматографии в работах L.R. Snyder [10]. Хотя данное приближение для разделения на обращенной фазе плохо обосновано, как показывает опыт [7], оно оказывается вполне удовлетворительным для моделирования разделения пептидов и белков. Причина заключается в том, что для макромолекул даже не очень большой молекулярной массы существенное изменение адсорбционных свойств происходит в узком диапазоне состава растворителя вблизи критической точки адсорбции ${{\varepsilon }_{c}}({{N}_{c}})$, и детальный вид зависимости энергии адсорбции звена $\varepsilon ({{N}_{b}})$ в этом узком диапазоне оказывается несущественным. Без потери общности для бинарного растворителя с компонентами a и b можно принять энергию адсорбции “слабого компонента” a (воды) на обращенной фазе ${{\varepsilon }_{a}} = 0$, тогда зависимость ${{\varepsilon }_{{ab}}}({{N}_{b}})$ смеси от концентрации “сильного компонента” b (ацетонитрила) записывается следующим образом [7]:
где ${{\varepsilon }_{b}}$ – энергия адсорбции компонента b. Для энергии звена $\varepsilon ({{N}_{b}})$ имеемЗдесь ${{\varepsilon }^{0}}$ – энергия звена в “чистом” компоненте a, ${{\varepsilon }_{c}}$ – энергия звена, соответствующая критической точке (равная $\ln (6{\text{/}}5)$ для рассматриваемой модели), которой соответствует состав Nc ≈ 0.05 для гомополимера (dT)n. В последнем выражении учтено, что обычно энергия адсорбции сильного компонента (ацетонитрила) εb ≫ 1.
На рис. 4а показано соответствие экспериментально найденных эффективных энергий звеньев для растворителя разного состава (см. выше) и приведенного выше приближения Ленгмюра. Как видно, данное приближение неплохо соответствует экспериментальным точкам и могло бы быть взято для описания градиентного разделения.
Рис. 4.
Зависимость эффективной энергии ε звена от мольной доли ацетонитрила Nb в бинарной смеси для клубка (1) и приближения Ленгмюра (2) (а), а также зависимость ln(ε) от объемной доли ацетонитрила Vb для гомополимера (dT)n (б).
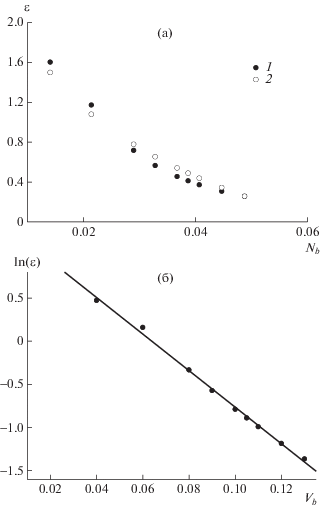
Однако в настоящей работе мы не будем применять приближение Ленгмюра. Для общности мы покажем, что в модели BioLCCC в масштабе звена можно использовать любое корреляционное или эмпирическое выражение, описывающее зависимость его эффективной энергии от состава растворителя. Например, как следует из рис. 4б, для звеньев T имеет место практически линейная зависимость $\ln \varepsilon $ от объемной доли ацетонитрила Vb:
Воспользуемся далее этой простой эмпирической зависимостью для описания градиентного разделения. Наблюдаемая линейная зависимость похожа на линейное приближение lnk от Vb [11]. Параметр ln(ε0) соответствует в данном случае энергии в некотором “стандартном” растворителе, например, в составе, реализующем критическую точку адсорбции, или в “чистом” компоненте a растворителя при Vb = 0.
В градиенте растворителя Kd и связанный с ним в соответствии с уравнением (1) коэффициент k зависят от состава растворителя Vb(V), изменяющегося при изменении времени или объема V, прокачанного через колонку. Объем удерживания VR (или время удерживания tR) определяется известным уравнением
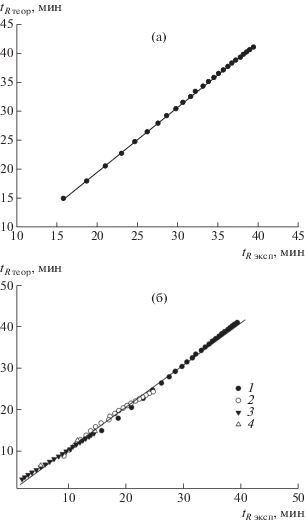
Результат расчета разделения гомополимера (dT)n для градиента с начальной точкой 5% и наклоном 0.15% ацетонитрила/мин [1] показан на рис. 5a. Как и следовало ожидать, из точного описания изократического режима следует весьма хорошее соответствие расчета и эксперимента для градиента.
Рис. 5.
Корреляция между расчетными tR теор и экспериментальными tR эксп временами удерживания при градиентном разделении для гомополимера (dT)n (а) и общая корреляция для гомополимеров (dT)n (1), (dA)n (2), (dC)n (3) и (CCG)n (4) при градиентном разделении [1] (б).
Для других гомополимеров, для которых в работе [1] не приводятся результаты разделения в изократическом режиме, примем, что различие заключается только в разнице стандартных энергий адсорбции звеньев, в то время как наклон S для них одинаков. Из представленных в работе [1] экспериментальных данных по градиентному разделению гомополимеров можно найти энергии звеньев A и C. Для звена G энергию можно определить из данных по градиентному разделению макромолекул типа (CCG)n. Соответствующие энергии нуклеотидов приведены в табл. 1.
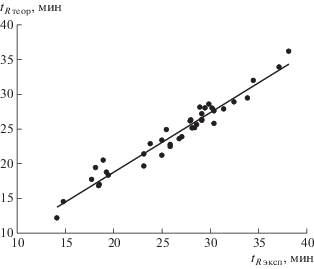
С использованием найденных таким образом энергий адсорбции звеньев T, A, C и G были рассчитаны времена удерживания гетерополимеров однонитевых ДНК. Последовательность букв в структуре олигонуклеотида задает порядок произведения переходных матриц в выражении (2) с различной энергией звена, соответствующей букве. Результаты сопоставлены с экспериментальными данными в табл. 2 и на рис. 6. В работе [1] удерживание макромолекул олигонуклеотидов приведено к объемному содержанию ацетонитрила в растворителе, при котором макромолекула выходит из колонки. Эти величины были пересчитаны нами в tR с помощью параметров градиентного профиля (начальной точки 5% и скорости изменения 0.15% ацетонитрила/мин).
Таблица 2.
Последовательности однонитевых ДНК и их экспериментальные и расчетные времена выхода tR
| n | Последовательность | %B* [1] | tR эксп. мин | tR теор. мин |
|---|---|---|---|---|
| 16 | GTAGCAGCAGCCAGAC | 7.12 | 14.13 | 12.15 |
| 16 | GTCTGGCTGCTGCTAC | 7.66 | 17.73 | 17.71 |
| 16 | GACATGGGCTCCGCTC | 7.22 | 14.80 | 14.49 |
| 18 | GAGATCTAGGTGATGTGC | 7.84 | 18.93 | 20.48 |
| 18 | ACAGTATTGCTGGCGGAG | 7.92 | 19.47 | 18.32 |
| 19 | GAAGAATTGGTAGGAGTCC | 7.72 | 18.13 | 19.41 |
| 20 | ACTGGGAACGAGGGCGACGT | 7.77 | 18.47 | 16.79 |
| 20 | TCAGACGAGAAGGACCTGAC | 7.78 | 18.53 | 16.98 |
| 20 | TTCGCCAGCAACAATGTGAC | 8.47 | 23.13 | 19.64 |
| 22 | GTCCTTTGAATGCAGAAATGGC | 8.57 | 23.80 | 22.84 |
| 22 | ATGGTTCCTGTGCAACTGTGAC | 8.75 | 25.00 | 23.36 |
| 23 | CAGCCCCTTCCCTCTGCCGCCAG | 7.89 | 19.27 | 18.75 |
| 25 | ACCTCTGCACCCATCTCTCTCCTCA | 9.02 | 26.80 | 23.56 |
| 26 | GGGGTACCAGTATCGAAAGCACCTCC | 8.47 | 23.13 | 21.36 |
| 26 | GCGAATTCCAATACCGATATTTCGGA | 9.26 | 28.40 | 25.14 |
| 26 | GCGAATTCTTATTTAGGATTGATAGC | 9.42 | 29.47 | 28.00 |
| 27 | TGGGAATTCAGTGCTTGGTCACTTCCG | 9.2 | 28.00 | 26.28 |
| 30 | GGGCTTCATGATGTCCCCATAATTTTTGGC | 9.48 | 29.87 | 28.55 |
| 30 | AGCGTTGGCTACCCGTGATATTGCTGAAGA | 9.37 | 29.13 | 26.33 |
| 32 | CTTACCCAACAAGTGCGGCCGATCGAATTGAG | 9.22 | 28.13 | 25.12 |
| 32 | CTCAATTCGATCGGCCGCACTTGTTGGGTAAG | 9.37 | 29.13 | 27.16 |
| 32 | GCCAGGTGCCAGGCTGGGTTGCCATTGATGGC | 8.82 | 25.47 | 24.88 |
| 32 | CCGCTCGAGGGTTCTCTGGATAACATCTCAGC | 9.19 | 27.93 | 26.14 |
| 33 | CTCCAACGGGAACCTGGCCCCCTGGGCAGGGGG | 8.75 | 25.00 | 21.18 |
| 33 | CCCCCTGCCCAGGGGGCCAGGTTCCCGTTGGAG | 8.88 | 25.87 | 22.74 |
| 34 | CCTGCGCATCATTTAATCAGAATGGCATTCACCG | 9.71 | 31.40 | 27.86 |
| 34 | CGGTGAATGCCATTCTGATTAAATGATGCGCAGG | 9.53 | 30.20 | 27.98 |
| 37 | AATGGATCCGGAGGAGGAGGGGACACCTTCATCCGTC | 9.56 | 30.40 | 25.76 |
| 39 | CCCAAGCTTATGGGAAACGCGGCGGCGGTGGGCCAGGCG | 9.06 | 27.07 | 23.85 |
| 39 | CCATCCAGGGGGGAGCGGCCGCCCCTCGACCTCAGAGCC | 8.88 | 25.87 | 22.44 |
| 39 | CATCCTCAGGTAGGCACTGAGCATCCCCCCATACGCACC | 9.37 | 29.13 | 26.21 |
| 39 | CCTCAGGTAGGCACTGAGCATCCCCCCATAAGCTCCACC | 9.29 | 28.60 | 25.59 |
| 41 | CATCCTCAGGTAGGCACTGAGCATCCCCCCATAACTTCCTC | 9.34 | 28.93 | 28.11 |
| 48 | GGAGCCACTGGCCACGCCGCCGCTTTGGATGCGCGGGAGGTGATTCCC | 9.56 | 30.40 | 27.57 |
| 54 | TATAAGCTTGAAGGGGAACGAAAGATGGCGGCGGAAACACTGCTGTCCAGTTTG | 10.08 | 33.87 | 29.43 |
| 56 | AGAGAATTCCCACCATGGACTACAAGGACGACGATGACAAGTCTCAGAGCAACCGG | 9.87 | 32.47 | 28.88 |
| 57 | ATTTCTAGATTACTATTTGTCATCGTCCTTATAGTCAAAGTCAAATTCGTCCAGGTC | 10.72 | 38.13 | 36.17 |
| 57 | CAATATTTTACATGAACTGGAGGTCCGTCAATGACAGTGTAGGCTGGAGCTGCTTCG | 10.17 | 34.47 | 31.97 |
| 60 | GACTTAGACTTAGACTTAGACTTAGACTTAGACTTAGACTTAGACTTAGACTTAGACTTA | 10.57 | 37.13 | 33.89 |
Рис. 6.
Корреляция между расчетными tR теор и экспериментальными tR эксп временами удерживания для гетерополимеров онДНК со степенью полимеризации n = 16–60 при градиентном разделении [1].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Как следует из приведенных выше результатов, модель решеточной цепи, дополненная корреляционной зависимостью ε(Vb), правильно схватывает основные черты разделения однонитевых ДНК. Необходимо, однако, отметить следующее.
Хотя общий коэффициент корреляции R2 = 0.99 для гомополимеров близок к единице, на рис. 5б можно заметить, что для взятого отдельно гомополимера (dA)n корреляция между расчетом и экспериментом не столь хороша. Варьируя параметры ln(ε0) и S, вероятно, можно улучшить корреляцию и для (dA)n. Как уже отмечалось, для модели совершенно неважно, каким образом описывать зависимость эффективной энергии от состава растворителя, в частности, можно использовать для этого и нелинейную аппроксимацию. В настоящей работе мы не стали прибегать к нелинейной аппроксимации, поскольку она вряд ли улучшит описание для гетероцепей однонитевых ДНК, а модель при такой аппроксимации потеряет простоту. Тем не менее, необходимо сказать, что исследование зависимости lnk гомополимера (dA)n от состава растворителя в изократическом режиме, подобное сделанному для (dT)n, может прояснить особенности адсорбционного взаимодействия звеньев A c поверхностью и уточнить описание его градиентного разделения.
Также отметим, что принятая в работе корреляционная зависимость энергии от состава в отличие от приближения Ленгмюра нарушает аддитивность вкладов звена и растворителя, что неявно подразумевалось в модели BioLCCC и в ее приложении для пептидов. Разница энергий взаимодействия нуклеотидов T, A, C, G не абсолютна, а зависит от состава растворителя.
Наконец, коэффициент корреляции R2 = 0.95 между расчетом и экспериментом для гетерополимеров (рис. 6) оказывается заметно хуже, чем для гомополимеров. Другими словами, предсказание именно последовательности гетерополимера, а не только среднего состава, из измеренного времени удерживания tR получается недостаточно точным. Рассмотрим данный вопрос более подробно.
Зависимость объема или времени удерживания от последовательности имеет две составляющие – физическую и химическую. В рассматриваемой модели учитывается только физическая составляющая зависимости. Предполагается, что вариация последовательности, составленной из одного и того же набора “букв” – нуклеотидов, не меняет их энергию, т.е. словами, при вариации последовательности средняя энергия звена сохраняется. В этом случае зависимость tR от последовательности вблизи критической точки имеет в основном энтропийную природу (заметим, что в градиенте макромолекулы перемещаются в колонке в составе растворителя, близком к критическому). Чтобы вариация последовательности (например, перестановка звеньев) была значимой, согласно размерным оценкам работы [12], длина цепи n должна соответствовать условию
где Δε – дисперсия энергии взаимодействия звеньев. Другими словами, для проявления последовательности при небольшой дисперсии цепь должна быть достаточно длинной. Оценка дисперсии взаимодействия звеньев гипотетической гетероцепи однонитевых ДНК с равным содержанием нуклеотидов A, T, G, C в составе растворителя, при котором она выходит из колонки (табл. 1), дает Δε ≈ 0.04, так что для цепей с длиной n = 40–60 физическая составляющая, по-видимому, малозначима. Объем удерживания определяется длиной цепи и средним нуклеотидным составом, тем самым подтверждая аддитивный подход [1]. Макромолекулу гетерополимера при этом можно рассматривать как гомополимер с некоторой средней энергией взаимодействия звена с поверхностью. Конечно, возможно существование каких-то уникальных последовательностей, значительно отклоняющихся по времени удерживания от средней величины, но для большинства макромолекул однонитевых ДНК вариация последовательности приводит к отклонению времени удерживания порядка ширины пика.Вместе с тем существует и химический фактор в зависимости адсорбционных свойств от последовательности нуклеотидов. При перестановке нуклеотид X, имеющий изначально соседями Y1 и Z1, оказывается в локальном окружении других звеньев – соседей Y2, Z2:
В соответствии с общими химическими законами, за счет эффектов соседа взаимодействие X с поверхностью может измениться. Тем самым, средняя энергия звена меняется, и вариация последовательности оказывается заметной уже за счет энергетической составляющей. Вероятно, для коротких цепей однонитевых ДНК химический фактор преобладает, что и вызывает расхождение расчета и эксперимента для гетерополимеров. Макромолекулы, составленные из одного и того же набора нуклеотидов, но различающиеся последовательностью, можно рассматривать как гомополимеры с различной средней энергией звена и, следовательно, с разной критической точкой и временем выхода.
В рамках рассматриваемой модели химический фактор можно учесть. Для этого необходимо синтезировать порядка 30 модельных однонитевых ДНК, имеющих в середине цепи структуру типа
в которой X, Y, Z последовательно замещаются на четыре типа возможных нуклеотидов. Исследование разделения таких макромолекул в изократическом режиме позволит определить поправки к энергии звеньев X, зависящие от соседей, и внести их в схему расчета.Наконец, не стоит забывать, что однонитевая ДНК может замыкаться сама на себя за счет комплементарности составляющих ее нуклеотидов. Если замыкание происходит, то образующиеся вторичные структуры меняют адсорбционные свойства макромолекулы и, в какой-то степени, маскируют ее последовательность. Скорее всего в рассматриваемом сложном растворителе при температуре 60°С вероятность такого замыкания небольшая. В работе [13] показано, каким образом в модель BioLCCC можно включить различные вторичные структуры – складки, циклы, листы. В соответствии с расчетами для относительно коротких пептидов влияние таких структур малозаметно, что приближает их адсорбционные свойства к соответствующим принципу аддитивности.
ЗАКЛЮЧЕНИЕ
Подводя итог можно сказать, что модель адсорбции решеточной цепи, дополненная корреляционной зависимостью эффективной энергии звена от состава растворителя, правильно схватывает основные закономерности разделения олигонуклеотидов на обращенной фазе. Она может использоваться как для предсказания времен удерживания олигонуклеотидов с известной последовательностью для их отбора из сложной смеси, так и для оптимизации условий разделения макромолекул с различными последовательностями. В силу малой дисперсии энергий адсорбции разных нуклеотидов на обращенной фазе разделение в основном чувствительно к составу нуклеотидов в цепи, а не к тексту последовательности. По этой причине предсказательные возможности модели аналогичны аддитивной модели, предложенной в работе [1].
Модель оказывается весьма содержательной и единым образом описывает адсорбционные свойства широкого класса макромолекул, имеющих цепочечное строение – синтетических полимеров, пептидов, белков и однонитевых ДНК. Отметим также что данная модель перекидывает мост между макромолекулами и низкомолекулярными соединениями: она содержит в себе также и возможность описания хроматографии низкомолекулярных соединений, которым соответствует длина цепи n = 1. Это дает возможность рассматривать хроматографию единым образом, не выделяя отдельно хроматографию низкомолекулярных веществ и макромолекул. Наконец, модель имеет широкие возможности для более точного описания структуры макромолекулы и может быть основой для рассмотрения не только адсорбции на поверхности, но и других взаимодействий с участием макромолекул.
Современные методы компьютерного моделирования дают возможность детального описания структуры макромолекул. Однако надо иметь в виду, что для моделирования необходимо точное знание множества потенциалов взаимодействия внутри цепи, а также их зависимость от внешних условий. Определение потенциалов представляет собой сложную экспериментальную задачу. Детализация структуры макромолекулы требует столь же подробной детализации поверхности. Эти вопросы значительно усложняют как моделирование взаимодействия макромолекулы с поверхностью, так и интерпретацию результатов. Хотя такая программа исследования представляется перспективной, на наш взгляд, упрощенные модели, подобные рассмотренной в настоящей работе, правильно дополненные небольшим числом эмпирических параметров, не потеряли своей актуальности.
Список литературы
Gilar M., Fountain K.J., Budman Y., Neue U.D., Yardley K.R., Rainville P.D., Russel II R.J., Gebler J.C. // J. Chromatogr. A. 2002. V. 958. P. 167.
Guo D., Mant C.T., Taneja A.K., Parker J.M.R., Hodges R.S. // J. Chromatogr. 1986. V. 359. P. 499.
Krokhin O.V. // Anal. Chem. 2006. V. 78. I. 22. P. 7785.
Reimer J., Spicer V., Krokhin O.V. // J. Chromatogr. A. 2012. V. 1256. P. 160.
Gilar M., Hie H.W., Jaworski A. // Anal Chem. 2010. V. 82. P. 265.
Gorshkov A.B., Goloborod’ko A.A., Pridatchenko M.L., Tarasova I.A., Rozdina I.G., Evreinov V.V., Gorshkov M.V. // Polymer Science A. 2017. V. 59. № 3. P. 354.
Gorshkov A.V., Evreinov V.V., Tarasova I.A., Gorshkov M.V. // Polymer Science B. 2007. V. 49. № 3–4. P. 93.
Tarasova I.A., Gorshkov A.V., Evreinov V.V., Adams K., Zubarev R.A., Gorshkov M.V. // Polymer Science A. 2008. V. 50. № 3. P. 309.
Entelis S.G., Evreinov V.V., Gorshkov A.V. // Adv. Polym. Sci. 1986. V. 76. P. 129.
Snyder L.R. Principles of Adsorption Chromatography. New York: Marcel Dekker, 1968.
Snyder L.R., Dolan J.W., Gant J.R. // J. Chromatography B. 1979. V. 165. P. 3.
Обухов С.П. // Журн. эксперим. и теорет. физики. 1987. T. 93. № 6. С. 1973.
Gorshkov A.V., Rozdina I.G., Pridatchenko M.L., Evreinov V.V. // Polymer Science A. 2020. V. 62. № 4. P. 437.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)