

Высокомолекулярные соединения (серия А), 2022, T. 64, № 4, стр. 260-266
ПЛАСТИФИКАЦИЯ СОПОЛИМЕРА БУТИЛМЕТАКРИЛАТА С МЕТАКРИЛОВОЙ КИСЛОТОЙ ДИМЕТИЛФТАЛАТОМ ДЛЯ КОНСТРУИРОВАНИЯ ТРАНСДЕРМАЛЬНЫХ ТЕРАПЕВТИЧЕСКИХ СИСТЕМ
В. Ф. Урьяш a, b, *, С. В. Чупрова a, Н. Ю. Кокурина b
a Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
b Научно-исследовательский институт химии
603022 Нижний Новгород, пр. Гагарина, 23/5, Россия
* E-mail: su2101@rambler.ru
Поступила в редакцию 15.11.2021
После доработки 17.02.2022
Принята к публикации 04.03.2022
- EDN: WWMUYF
- DOI: 10.31857/S2308112022200032
Аннотация
Методом дифференциального термического анализа в области 80–450 К изучено влияние кристаллизующегося пластификатора диметилфталата на температуру стеклования сополимера бутилметакрилата с метакриловой кислотой (7.6 мол. %). Калориметрическим методом определена растворимость диметилфталата в сополимере при температуре плавления пластификатора. По полученным данным построена и проанализирована диаграмма физических состояний системы акриловый сополимер–диметилфталат. Сопоставлены пластифицирующие характеристики двух пластификаторов – диметилфталата и ди-2-этилгексилфталата.
ВВЕДЕНИЕ
Пластификация является распространенным методом модификации физико-химических, механических и других свойств синтетических полимеров и сополимеров [1–3]. В последнее время системы полимер–пластификатор широко применяются в медицине, позволяя значительно повысить ее эффективность и расширить возможности в области диагностики и лечения заболеваний [4–10]. Необходимым условием, определяющим возможность использования полимерного материала в любой сфере медицины, является наличие у него таких свойств, как нетоксичность, стойкость по отношению к биологическим средам, способность выдерживать стерилизацию без существенного изменения свойств. Более того, он не должен обладать сенсибилизирующим, канцерогенным, мутагенным и тератогенным действием. Одной из перспективных областей применения полимеров в медицине можно назвать конструирование трансдермальных терапевтических систем (ТТС), способных обеспечить доставку лекарственных веществ в определенной дозе к заданному органу – мишени в строго регламентированных временных интервалах [5, 6, 11–16]. Такие терапевтические системы нового поколения являются пролонгированными, не вызывают болевого шока, а лекарственное вещество не подвергается разрушающему действию в желудочно-кишечном тракте. При создании ТТС широкое применение находят акриловые полимеры и сополимеры [8, 13, 17, 18].
Ранее в работах [18, 19] была изучена температура стеклования в ряде сополимеров бутилметакрилата (БМА) с метакриловой кислотой (МАК) с содержанием последней от 7.6 до 37.3 мол. %, измерена их теплоемкость в области 6–330 К и рассчитаны термодинамические характеристики (энтальпия, энтропия и функция Гиббса) от Т →0 до 330 К. Построены изотермы концентрационных зависимостей указанных характеристик сополимера БМА–МАК. Также рассмотрено влияние некристаллизующегося пластификатора ди-2-этилгексилфталата (диоктилфталата, ДОФ) на температуру стеклования сополимеров БМА–МАК (7.6 и 25.1 мол. %) и построены диаграммы физических состояний указанных систем сополимер–пластификатор [18].
На основании изложенного выше, с научной и практической точки зрения необходимо было изучить влияние кристаллизующегося пластификатора диметилфталата (ДМФ) на температуру стеклования сополимера БМА–МАК, определить растворимость ДМФ в сополимере и сопоставить полученные результаты с ранее изученной системой БМА–МАК (7.6 мол. %) + ДОФ. Был использован дифференциальный термический анализ в области 80–450 К [9, 18] и низкотемпературная адиабатная калориметрия в области 80–320 К [9, 20, 21]. Проведенные исследования позволили выбрать оптимальный качественный и количественный состав связующего для конструирования матричной трансдермальной терапевтической системы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез сополимера БМА–МАК с содержанием последнего 7.6 мол. % осуществляли суспензионной полимеризацией по запатентованной методике [18, 19, 22]. Предложенная методика позволяет синтезировать сополимеры БМА с МАК до глубокой степени конверсии мономеров (99.5–99.8%), свободных от остаточного мономера БМА и содержащих незначительное количество воды (<0.5 мас. %). Благодаря этому получаемый сополимер нетоксичен. Такие сополимеры могут быть использованы в медицине для создания связующего для ТТС.
Синтез проводили путем суспензионной полимеризации мономеров в водной среде при рН 3 в присутствии инициатора – перекиси бензоила в две стадии: в течение 2 ч при температуре 343 К и 2.5 ч при 353 К в присутствии стабилизатора (0.3–0.6 мас. %) – сополимера метакрилата натрия (84 мас. %) с метакриловой кислотой (16 мас. %) при концентрации инициатора 1.5 мас. %. После этого сополимер промывали несколько раз дистиллированной водой и сушили. Состав сополимера определяли методом кислотно-основного неводного титрования карбоксильных групп МАК на высокочастотном титраторе ТВ-6Л1 [23]. Молекулярную массу (ММ) сополимера устанавливали вискозиметрическим методом после метилирования карбоксильных групп, а также методом гель-проникающей хроматографии. Значения ММ, найденные данными методами, хорошо совпали; ММ = 8.8 × 104. Рентгеноструктурный анализ показал, что полученный статистический сополимер БМА–МАК полностью аморфен. Молярную массу повторяющегося звена сополимера (услов. моль) рассчитывали по уравнению:
где Х1 и Х2 – мольные доли БМА и МАК, М1 и М2 – молярные массы повторяющегося звена БМА и МАК соответственно.Мономеры БМА и МАК очищали фракционированием при пониженном давлении. По данным хроматографического анализа содержание основного компонента в отобранных фракциях мономеров составляло 99.9 мас. %. Показатели преломления nD при 293 К для БМА и МАК составляли 1.4240 и 1.4314 соответственно, что совпадало с надежными литературными данными [24].
Пластификатором служил “чистый” ДМФ. Его дополнительно очищали фракционированием при пониженном давлении [25]. Отбирали фракцию, имеющую следующие физико-химические характеристики при 293 К: nD = 1.5158, ρ = = 1.1898 г/см3, η = 13.93 сПз, которые соответствуют надежным литературным данным [26].
Пластифицированные образцы готовили смешением сополимера и пластификатора в присутствии гомогенизатора этанола [3, 9, 18]. Приготовление образцов проводили в соответствии с ТУ 2216–001–25567297–2001. Для этого сначала смешивали при комнатной температуре этиловый спирт и пластификатор в соотношении 1.0 : 1.5 по объему. Затем в смесь жидких компонентов (3.57 объемных частей) вносили сополимер (1 мас. часть). Композицию перемешивали до получения вязкой однородной прозрачной массы. Образцы нагревали при температуре 353 К в течение 6 ч, после чего гомогенизатор удаляли в вакууме. Отсутствие этанола в образцах контролировали хроматографически. В результате получали визуально однородные прозрачные образцы.
ДТА в области 80–450 К осуществляли на установке, конструкция которой и методика работы описаны в работах [3, 9, 18]. Навески образца и эталона составляли 0.3–0.5 г. Эталоном служил кварц. Эталон и образцы загружали в герметизируемые алюминиевые тигли, чтобы предотвратить испарение ДМФ из последних. Эксперимент выполняли в атмосфере гелия. Скорость нагревания в опытах составляла 5 град/мин. Отклонение от линейности не превышало 1%. Для измерения температуры образца и разности температуры между образцом и эталоном использовали хромель-копелевые термопары. Измерительную термопару калибровали по образцовому платиновому термометру сопротивления и реперным веществам во всей области температуры. Погрешность измерения температуры ±0.5 К. Для проверки работы установки ДТА находили температуру плавления Тпл эталонного н-гептана и температуру стеклования Тс очищенного глицерина. Полученные результаты совпали с соответствующими литературными данными для н-гептана с погрешностью 0.2 К [27], для глицерина − 1 К [28]. По рекомендации International Confederation for Thermal Analysis and Calorimetry (ICTAC) [29] за Тс принимали среднюю температуру интервала, в котором происходит переход, так как именно она обладает наилучшей воспроизводимостью.
Для определения предела растворимости ДМФ в сополимере БМА–МАК (7.6 мол. %) калориметрическим методом [1–3, 9, 14, 18] приготовили образец (m = 3.5577 г), содержавший 85.1 мас. % пластификатора. Измерение теплоемкости ($C_{p}^{{^{ \circ }}}$) в области 80–320 К проводили в вакуумном адиабатическом калориметре конструкции Харьковского физико-технического института низких температур АН Украины [9, 20]. Температуру измеряли платиновым термометром сопротивления. Энергетический эквивалент калориметра устанавливали путем измерения теплоемкости калориметрической ампулы, заполненной гелием до давления 8.5 кПа. Он не превышал 40% от $C_{p}^{{^{ \circ }}}$ калориметра с веществом в изученном интервале температуры. Для проверки методики на отсутствие систематической ошибки определяли теплоемкость эталонной бензойной кислоты марки К-1 (Всероссийский научно-исследовательский институт метрологии им. Д.И. Менделеева, Санкт-Петербург). Различие полученных результатов и данных работы [30] составило 0.3% при Т = 80−320 К. Подъем температуры в опытах по измерению теплоемкости составлял 1.0−3.0 К, поэтому поправку на кривизну функции $C_{p}^{{^{ \circ }}}$ = f(T) не вводили. Отклонение экспериментальных данных от усредняющих кривых в пределах 0.3%.
Расчет концентрации ω (мас. %) насыщенного при Тпл ДМФ раствора его в сополимере БМА–МАК выполняли по формуле (2) [1–3, 9, 14, 18]
Здесь q (Дж) − количество энергии в форме теплоты, необходимое для плавления кристаллов фазы “свободного” ДМФ; Δh (87.3 ± 0.2 Дж/г [3, 26]) − удельная энтальпия плавления ДМФ; m (г) − масса образца; С2 (мас. %) − массовая доля ДМФ в образце сополимера. Погрешность определения растворимости этим методом составляла 0.5–1.0% (выражена доверительным интервалом с вероятностью 95%).РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Полученные экспериментальные результаты представлены на рис. 1–3 и в табл. 1, 2. Как показали исследования методами ДТА и калориметрии физико-химических свойств образцов сополимера БМА–МАК с различным содержанием кислоты [18, 19], у них проявляется два или три интервала расстеклования в зависимости от состава. Два значения Тс наблюдались у сополимеров, содержавших до 20 мол. % МАК, у остальных (до 40 мол. % МАК) – три значения Тс. Это свидетельствует о многофазности статистических сополимеров БМА–МАК, что связано, во-первых, с распределением МАК между водной фазой и каплями эмульсии БМА в процессе синтеза сополимера. Во-вторых, с конверсионной неоднородностью по составу, которая возникает при полимеризации до глубоких конверсий [23]. В условиях суспензионной полимеризации [18, 19, 22] получается сополимер БМА−МАК, на поверхности гранул которого адсорбируются молекулы сополимера БМА−МАК, сильно обогащенного МАК, полученные растворной полимеризацией. Таким образом, все получаемые гранулы сополимера образуют так называемую “мягкую фазу”, которая расстекловывается при Тс1. “Оболочка”, образованная сополимером, сильно обогащенным МАК, образует “жесткую фазу”, которой соответствует расстеклование при Тс2. Морфология суспензионных частиц сополимера БМА–МАК типа ядро-оболочка подтверждается электронно-микроскопическими исследованиями подобных систем [13, 18, 31, 32].
Рис. 1.
Термограммы пластифицированных образцов сополимера БМА–МАК (7.6 мол. %) с содержанием ДМФ 0 (1), 12.6 (2) и 84.9 мас. % (3, 3 ').
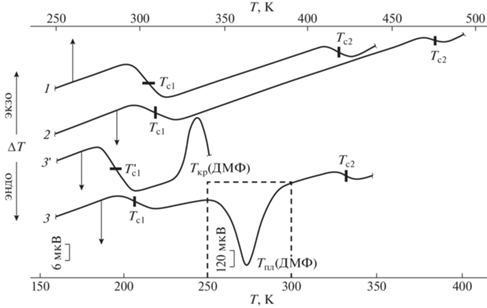
Рис. 2.
Температурная зависимость теплоемкости образца, содержащего сополимер БМА–МАК (7.6 мол. %) + 85.1 мас. % ДМФ. Физические состояния системы: АВ – стеклообразный насыщенный раствор ДМФ в сополимере + стеклообразная фаза “свободного” ДМФ; BC – высокоэластичный насыщенный раствор ДМФ в сополимере + переохлажденная жидкая фаза “свободного” ДМФ; AD – стеклообразный насыщенный раствор ДМФ в сополимере + кристаллическая фаза “свободного” ДМФ; DE – высокоэластичный насыщенный раствор ДМФ в сополимере + кристаллическая фаза “свободного” ДМФ; EFG – плавление фазы "свободного" ДМФ; GH – высокоэластичный насыщенный раствор ДМФ в сополимере + жидкая фаза “свободного” ДМФ.
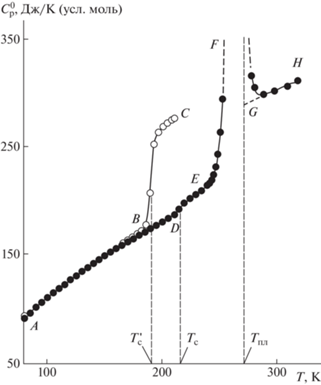
Рис. 3.
Диаграмма физических состояний системы сополимер БМА–МАК (7.6 мол. %) + ДМФ. Пояснения в тексте.
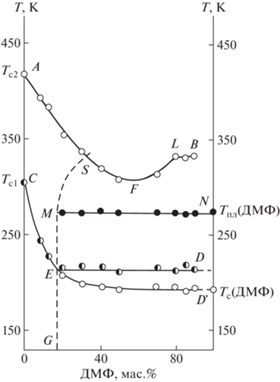
Таблица 1.
Температура физических переходов в системе сополимер БМА–МАК (7.6 мол. %) + ДМФ
| Содержание ДМФ, мас. % | Тс1, К | Тс2, К | $T_{{{\text{c}}1}}^{'}$, К | Тпл ДМФ, К |
|---|---|---|---|---|
| 0 | 306 | 419 | – | – |
| 8.6 | 243 | 393 | – | – |
| 12.6 | 228 | 389 | – | – |
| 19.9 | 215 | 353 | 208 | 272 |
| 29.8 | 217 | 336 | 200 | 274 |
| 40.1 | 216 | 319 | 195 | 272 |
| 50.3 | 212 | 308 | 196 | 272 |
| 68.8 | 213 | 313 | 194.5 | 273 |
| 79.9 | 213 | 332 | 195 | 272 |
| 84.9 | 212 | 330 | 195 | 271 |
| 89.9 | 213 | 332 | 194 | 273 |
| 100.0 | – | – | 192 | 273 |
Таблица 2.
Исходные данные и результаты опытов по определению растворимости ДМФ в образце сополимера БМА–МАК (7.6 мол. %) + ДМФ (85.1 мас. %)
| Опыт, № | Масса исходной смеси, г | Масса ДМФ в образце, г | Энтальпия плавления кристаллов “свободного” ДМФ, Дж | Масса кристаллов “свободного” ДМФ, г | ω, мас. % |
|---|---|---|---|---|---|
| 1 | 3.5577 | 3.0249 | 255.64 | 2.9267 | 15.6 |
| 2 | 3.5577 | 3.0249 | 253.74 | 2.9077 | 18.0 |
| Среднее значение: | 17.0 ± 1.0 | ||||
Проведена оценка конверсионной неоднородности сополимеров БМА–МАК (7 и 30 мол. %) по составу [18]. Для этого по формуле Мейера была рассчитана зависимость мгновенного состава мономерной смеси f2 от степени превращения. Далее по дифференциальному уравнению Майо–Льюиса были определены мгновенные составы сополимера ${{F}_{2}}$, а также рассчитан средний состав сополимера ${{\bar {F}}_{2}}$ с использованием уравнения баланса по компоненту М2. Сделанные расчеты [18] подтвердили представленные выше объяснения.
Сополимер, содержавший 7.6 мол. % МАК, представляет собой двухфазную систему и имеет два температурных интервала расстеклования со средними значениями температуры Тс1 = 306 К и Тс2 = 419 К [18, 19]. У пластифицированных образцов сополимера (<18 мас. % ДМФ) проявляются те же два температурных интервала расстеклования, средние значения (Тс1 и Тс2) которых существенно понижаются в сравнении с исходным сополимером (табл. 1; рис. 1).
У образцов, содержащих более 18 мас. % пластификатора, наблюдается расстеклование насыщенного раствора ДМФ в “мягкой фазе” сополимера и стеклообразного избытка пластификатора $T_{{{\text{c}}1}}^{'}$, который затем кристаллизуется из состояния переохлажденной жидкости (рис. 1, кривая 3'). После завершения кристаллизации образцы вновь охлаждались до 80 К. При повторном нагревании (рис. 1, кривая 3) у образцов проявляется расстеклование лишь насыщенного раствора пластификатора в “мягкой фазе” (Тс1), затем − плавление фазы избытка ДМФ, после завершения которого расстекловывается раствор пластификатора в “жесткой фазе” (Тс2).
Методом, описанным в работах [9, 18], по результатам ДТА пластифицированных образцов, содержавших более 20 мас. % ДМФ, была определена площадь эндотермического пика плавления фазы “свободного” ДМФ. Построена зависимость удельной площади пика плавления фазы “свободного” ДМФ (δ) от его общего содержания в смеси (С2, мас. %). Полученная зависимость аппроксимируется линейным уравнением
Экстраполируя ее на нулевое значение удельной площади пика, оценена концентрация насыщенного при 273 К раствора ДМФ в сополимере. Она оказалась равной ω = 18 ± 2 мас. % ДМФ.
Для более точного определения растворимости ДМФ в сополимере была измерена теплоемкость образца смеси сополимера БМА–МАК (7.6 мол. %), с 85.1 мас. % ДМФ (рис. 2). Перед измерением теплоемкости система быстро (~20 К/мин) охлаждалась в калориметре до 80 К. При последующем медленном нагревании в области от 80 до 220 К на кривой теплоемкости (кривая АВС) при 191 К проявился переход, который относится к расстеклованию насыщенного раствора ДМФ в сополимере и стеклообразного “свободного” ДМФ ($T_{{\text{c}}}^{'}$) (отрезок ВС). После этого перехода образовавшийся переохлажденный жидкий “свободный” ДМФ закристаллизовывалася. Такой процесс носит экзотермический характер и сопровождается выделением энергии в форме теплоты. После повторного охлаждения системы до 80 К и последующего медленного нагревания на кривой теплоемкости (кривая ADE) при 218 К проявился переход (отрезок DE), относящийся к расстеклованию насыщенного раствора ДМФ в сополимере (Тс). Около 273 К наблюдался разрыв (интервал EFG) на кривой $C_{p}^{^\circ }$ = f(T) – эндотермический эффект, обусловленный плавлением закристаллизовавшегося “свободного” ДМФ (Тпл = 272.83 К). По калориметрическим данным ДМФ плавится при 274.16 К [3, 26].
Методом непрерывного ввода энергии в калориметр было найдено количество энергии в форме теплоты, необходимое для плавления кристаллов фазы “свободного” ДМФ. По отношению этой величины к удельной энтальпии плавления ДМФ рассчитана масса кристаллов ДМФ. Зная массу образца и массовую долю ДМФ в нем, установлена растворимость ДМФ в сополимере (ω) при 273 К в массовых долях по формуле (2). Исходные данные и результаты двух опытов по определению предела растворимости ДМФ в сополимере БМА–МАК (7.6 мол. %) приведены в табл. 2.
Таким образом, результаты растворимости ДМФ в “мягкой фазе” сополимера БМА–МАК (7.6 мол. %) по калориметрическим данным (ω = = 17.0 ± 1.0 мас. %) и ДТА (ω = 18 ± 2 мас. %) в пределах погрешности определения совпадают между собой.
По полученным данным о температуре стеклования, температуре плавления фазы избытка ДМФ и растворимости пластификатора в сополимере была построена диаграмма физических состояний пластифицированной системы (рис. 3). Основными элементами диаграммы являются концентрационные зависимости температуры стеклования пластифицированных “мягкой” (кривая CЕD) и “жесткой” (кривая АВ) фаз. Отрезок ED' соответствует температуре расстеклования смеси насыщенного раствора пластификатора в “мягкой фазе” сополимера и стеклообразной фазы “свободного” ДМФ. Такое состояние системы получается при быстром (~20 К/мин) охлаждении. MN – линия температуры плавления (Тпл = 273 ± 1 К) фазы “свободного” ДМФ. Линия GEMS делит диаграмму на две области: слева − область значений концентрации, при которых весь введенный пластификатор растворяется (молекулярно распределяется) в “мягкой” и “жесткой фазах” сополимера; справа − кроме растворов пластификатора в фазах сополимера присутствует также фаза “свободного” ДМФ. Таким образом, линия GEMS является левой ветвью кривой взаимной растворимости. Как известно [33], растворимость зависит от температуры. Однако авторами работ [1, 3, 9, 14, 18] на основании экспериментальных данных и расчетов термодинамических характеристик систем поливинилтриметилсилан–циклогексан [1], нитрат целлюлоза–триацетин [1, 3], нитрат целлюлоза–дибутилфталат [3] и нитрат целлюлоза–нитроглицерин [3, 9, 14, 18] было показано, что изменение растворимости пластификатора в полимере при температуре ниже Тпл пластификатора невелико и составляет 2–4 мас. %. Поэтому в пределах погрешности определения растворимости пластификатора в полимере ее можно принять постоянной в указанной области температуры (отрезок GEM).
Употребление термина “свободный” по отношению к ДМФ несколько условно, поскольку в действительности существует раствор сополимера в пластификаторе. Однако концентрация сополимера в этом растворе настолько мала, что практически не влияет на температуру плавления ДМФ, которая близка к температуре плавления чистого ДМФ, а следовательно, мы имеем право называть избыток пластификатора “свободным”, не забывая при этом об условности данного понятия.
Кроме того, Тпл пластификатора остается неизменной в области двухфазности системы в соответствии с правилом фаз Гиббса [1, 33]. В этой области концентрации равновесие в системе нонвариантно. Аналогичным образом ведут себя, например, системы диацетат целлюлозы (ДАЦ)–ДМФ, ДАЦ–трифенилфосфат (ТФФ) [3], ДАЦ–диэтилфталат [1] и крахмал–вода [9, 14], в которых экспериментально удалось получить кривую взаимной растворимости компонентов с верхней критической температурой растворения.
Как видно из диаграммы (рис. 3; табл. 1), двухфазная смесь, состоящая из насыщенного раствора ДМФ в сополимере БМА–МАК и фазы “свободного” ДМФ, ведет себя по-разному в зависимости от термической предыстории. При быстром (~20 К/мин) охлаждении застекловывается как насыщенный раствор ДМФ в сополимере, так и фаза “свободного” ДМФ. Эта смесь расстекловывается в одном температурном интервале (рис. 3, отрезок ED'). Ее $T_{{{\text{c}}1}}^{'}$ ниже, чем Тс1 насыщенного раствора (табл. 1; рис. 3, отрезок ED). Можно предположить, образование кластеров из двух и более молекул ДМФ в процессе его взаимодействия с макромолекулами сополимера при значениях концентрации пластификатора, превышающих предел его растворимости в сополимере. Так, как правило, происходит в системах, в которых растворимость пластификатора в полимере невелика и пластификатор не успевает до достижения предела растворимости его в полимере понизить Тс полимера до своей собственной. Данное явление наблюдалось, например, в системах ДАЦ–ТФФ [3], крахмал–вода, грибной хитин–вода и некоторые сывороточные белки крови человека (иммуноглобулин, альбумин)–вода [9, 14].
Прослеживается также сложная концентрационная зависимость Тс2 сополимера (табл. 1; рис. 3, кривая АВ). Зависимость проходит через минимум при ~55 мас. % ДМФ (рис. 3, точка F). Объяснить такой характер изменения Тс2 можно следующим образом. Растворимость ДМФ в сополимере БМА–МАК невелика. После насыщения пластификатором “мягкой фазы” сополимера небольшие молекулы ДМФ (по сравнению с другими эфирами фталевой кислоты [3]) продолжают проникать в оболочку суспензионных частиц (“жесткая фаза”), образуя пересыщенный раствор пластификатора в сополимере и понижая Тс2. Начиная с определенной концентрации (55 ± ± 5 мас. % ДМФ), избыток пластификатора выделяется из “жесткой фазы” сополимера и Тс2 повышается (рис. 3, точка L). При охлаждении системы ниже Тпл ДМФ он кристаллизуется. Если проэкстраполировать отрезок LB до пересечения с отрезком AF, получаем точку S, которая соответствует растворимости пластификатора в “жесткой фазе” сополимера (30 ± 5 мас. % ДМФ). Похожая зависимость Тс2 = f(C2) была получена в системе сополимер БМА–МАК (25.1 мол. %) + ДОФ [18]. В данном случае такой характер концентрационной зависимости Тс2 объяснялся наличием в системе “промежуточной фазы” сополимера и перераспределением ДОФ между “жесткой” и “мягкой фазами” сополимера [18].
ЗАКЛЮЧЕНИЕ
Полученные результаты позволили сделать вывод о том, что ДМФ как пластификатор для данного сополимера обладает рядом недостатков. Его растворимость в изученном сополимере мала, а следовательно, трудно получить связующее для ТТС, обладающее необходимым комплексом свойств и не содержащее одновременно избыток пластификатора. Избыток же ДМФ может затруднить эксплуатацию ТТС, а также ее транспортировку и хранение, так как кристаллизация ДМФ при нарушении температурного режима может вызвать повреждение связующего и привести терапевтическую систему в негодность.
Напротив, как показали исследования [18], ДОФ обладает рядом достоинств по сравнению с ДМФ. Он имеет высокую растворимость (~70 мас. %) в сополимере БМА–МАК (7.6 мол. %) и не кристаллизуется при охлаждении. ДОФ понижает Тс1 сополимера до 182 К, т.е. является более эффективным пластификатором по сравнению с ДМФ. Это дает возможность получать связующее для ТТС, имеющее необходимый комплекс физико-химических свойств. При этом негативные характеристики связующего с ДМФ, отмеченные выше, в системе с ДОФ отсутствуют.
Список литературы
Rabinovich I.B., Mochalov A.N., Tsvetkova L.Ya., Khlystova T.B., Moseyeva Ye.M., Maslova V.A. // Acta Polym. 1983. Bd. 34. № 8. S. 482.
Uryash V.F., Rabinovich I.B., Mochalov A.N., Khlyustova T.B. // Thermochim. Acta. 1985. V. 93. P. 409.
Uryash V.F., Pet’kov V.I., Khlyustova T.B., Kokurina N.Yu., Larina V.N., Otvagina K.V. Cellulose Acetate: Properties, Uses and Preparation / Ed. by C. Roberson. New York: Nova Sci. Publ. Inc., 2019. Р. 1.
Платэ Н.А., Васильев А.Е. // Высокомолек. соед. А. 1982. Т. 24. № 4. С. 675.
Gebelein C.G. // J. Am. Chem. Soc.: Polym. Prepr. 1983. V. 24. № 1. P. 11.
Progress in Biomedical Polymers / Eds. by C.G. Gebelein, R.L. Dunn. New York: Springer, 2013.
Biomaterials Science: An Introduction to Materials in Medicine / Eds. by B.D. Ratner, A.S. Hoffman, F.J. Schoen, J.E. Lemons. London; New York: Elsevier Acad. Press, 2004.
Shtilman M.I. // Polymer Science A. 2010. V. 52. № 9. P. 884.
Урьяш В., Груздева А. Термодинамика биологически активных веществ. Saarbrücken: LAP Lambert Academic Pub., 2017.
Биополимеры / Под ред. Ю. Иманиси. Пер. с яп. под ред. В.В. Коршака, И.А. Ямскова. М.: Мир, 1988.
Искусственные органы / Под ред. В.И. Шумакова. М.: Медицина, 1990.
Биосовместимость / Под ред. В.И. Севастьянова. М.: Изд-во ГУП “Информационный центр ВНИИ геосистем”, 1999.
Севастьянов В.И., Соломатина Л.А., Тихобаева А.А., Собко О.М., Урьяш В.Ф. // Перспективные материалы. 2004. № 1. С. 46.
Uryash V.F., Uryash A.V., Gruzdeva A.E., Kokurina N.Yu., Larina V.N., Faminskaya L.A., Kalashnikov I.N. Physical Organic Chemistry: New Developments / Ed. by K.T. Burley. New York: Nova Sci. Pub. Inc., 2010. P. 183.
Тихобаева А.А., Саломатина Л.А., Севастьяпов В.И. // Вестн. трансплантологии и искусственных органов. 2003. № 4. С. 50.
Севастьянов В.И., Саломатина Л.А., Кузнецова Е.Г., Яковлева Н.В., Шумаков В.И. // Мед. техника. 2003. № 2. С. 21.
Dittgen M., Durrani M., Lehmann K. // S.T.P. Pharma Sci. 1997. V. 7. № 6. P. 403.
Урьяш В., Чупрова С., Кокурина Н. Термодинамика и термический анализ пластифицированных сополимеров бутилметакрилата с метакриловой кислотой – связующего для трансдермальных терапевтических систем. Saarbrücken: LAP Lambert Academic Publ., 2020.
Ur’yash V.F., Chuprova S.V., Kokurina N.Yu., Markin A.V. // Rus. J. Phys. Chem. A. 2022. V. 96. № 2. P. 246.
Рабинович И.Б., Нистратов В.П., Тельной В.И., Шейман М.С. Термодинамика металлоорганических соединений. Н.Новгород: Изд-во Нижегородского гос. ун-та, 1996.
Уэструм Э., Мак-Каллаф Дж. Физика и химия твердого состояния органических соединений / Под ред. Д. Фокса, М.М. Лэйбса, А. Вайсбергера. М.: Мир, 1967. С. 9.
Князева Т.Е., Извозчикова В.А., Урьяш В.Ф. Патент 2174522 Россия // Б.И. 2001. № 28. С. 231.
Knyazeva T.E., Myasnikova I.B., Semchikov Yu.D. // Polymer Science A. 1998. V. 40. № 8. P. 859.
Липатов Ю.С., Нестерова А.Е., Грищенко Т.М., Веселовский Р.А. Справочник по химии полимеров. Киев: Наукова думка, 1971.
Вайсбергер А., Проскауэр Э., Риддик Дж., Тупс Э. Органические растворители. Физические свойства и методы очистки. М.: Иностр. лит., 1958.
Рабинович И.Б., Новоселова Н.В., Мосеева Е.М., Бабинков А.Г., Цветкова Л.Я. // Журн. физ. химии. 1986. Т. 60. № 3. С. 545.
Douglas T.B., Furukava G.T., McCoskey R.E., Ball A.L. // J. Res. Nat. Bur. Standards. 1954. V. 53. № 1. P. 139.
McMillan J.A. // J. Chem. Phys. 1965. V. 42. № 10. P. 3497.
Garn P.D., Menis O. // J. Macromol. Chem. B. 1977. V. 13. P. 611.
Склянкин А.А., Стрелков П.Г. // Журн. прикл. механики и техн. физики. 1960. № 2. С. 100.
Van Hamersveld E.M.S., Van Es J.J., German A.L., Cuperus F.P., Weissenborn P., Hellgren A.C. // Prog. Org. Coat. 1999. V. 35. № 1. P. 235.
Tsavalas J.G., Schork F.J., Landfester K. // J. Coat. Tech. Research. 2004. V. 1. № 1. P. 53.
Аносов В.Я., Озерова М.И., Фиалков Ю.Я. Основы физико-химического анализа. М.: Наука, 1976.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)