

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 1, стр. 50-70
Синтез и свойства чередующихся сополимеров на основе 4H-циклопента [2,1-b:3,4-b']дитиофена и 4H-дитиено[3,2-b:2',3'-d]силола
Ф. В. Дроздов a, *, Н. М. Сурин a, С. М. Перегудова b, В. А. Труханов c, П. В. Дмитряков d, С. Н. Чвалун a, d, Д. Ю. Паращук c, С. А. Пономаренко a, e
a Институт синтетических полимерных материалов Российской академии наук
117393 Москва, ул. Профсоюзная, 70, Россия
b Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119334 Москва, ул. Вавилова, 28, Россия
c Московский государственный университет имени М.В. Ломоносова. Физический факультет
119991 Москва, Ленинские горы 1, стр. 62, Россия
d Национальный исследовательский центр “Курчатовский институт”
123098 Москва, пл. Академика Курчатова 1, Россия
e Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы 1, стр. 3, Россия
* E-mail: fedor.drozdov@gmail.com
Поступила в редакцию 18.04.2018
После доработки 11.09.2018
Принята к публикации 25.09.2018
Аннотация
Представлен синтез и сравнение свойств серии сополимеров на основе 4H-циклопента[2,1-b:3,4-b']дитиофена и 4H-дитиено[3,2-b:2',3'-d]силола, полученных с использованием прямого С–Н арилирования и кросс-сочетания в условиях реакции Сузуки. Химическое строение сополимеров доказано методами ЯМР 1Н и ЯМР 13С, молекулярно-массовые характеристики определены методом ГПХ. Исследования спектров поглощения полученных сополимеров в разбавленных растворах и тонких пленках показали, что они поглощают свет в широком диапазоне видимого спектра (300–800 нм). По данным цикловольтамперометрии оценены уровни граничных молекулярных орбиталей – высшей занятой и нижней свободной, а также ширина запрещенной зоны, которая находится в диапазоне 1.4–1.9 эВ. С использованием полученных сополимеров в качестве донорных компонентов активного слоя изготовлены образцы солнечных фотоэлементов и исследованы их фотовольтаические свойства.
Важнейшая задача современной полимерной фотовольтаики – разработка эффективных донорных компонентов, в роли которых могут выступать сопряженные полимеры. Одним из первых тиофенсодержащих полимеров, до сих пор широко используемых в качестве донорного компонента в фотовольтаике, является поли(3-гексил)тиофен (ПГT)

Фотоэлементы на его основе демонстрировали эффективность преобразования солнечного света до 3.6% [1], однако, несмотря на многочисленные попытки увеличить эффективность таких фотоэлементов, значения КПД фотовольтаических ячеек на основе ПГТ находятся около 6.0–7.7% [2, 3]. Данный факт в случае ПГТ можно объяснить как достаточно узким спектром поглощения этого полимера по сравнению со спектром солнечного излучения, так и высоколежащим уровнем ВЗМО, что ограничивает напряжение холостого хода Vxx фотоэлемента, которое для ПГТ, как правило, не превышает 0.6 В.
Большое число публикуемых в последнее время работ по фотовольтаике посвящено синтезу нового поколения узкозонных сополимеров, содержащих чередующиеся донорные и акцепторные звенья [4–9]. Такое строение полимерной цепи позволяет повысить эффективность преобразования солнечного света фотоэлементов на основе данного типа полимеров путем более точного подбора энергетических уровней сополимера относительно фуллеренового акцепторного компонента фотоэлемента за счет увеличения плотности тока короткого замыкания Jкз и улучшения спектральных характеристик полимера. Стоит отметить, что 4H-циклопента[2,1-b:3,4-b']дитиофен (ЦПДТ)

и его структурный аналог – 4H-дитиено[3,2-b:2',3'-d]силол (ДТС)

можно рассматривать как аналог мономерного звена региорегулярного политиофена ПГТ, в котором каждая пара тиофеновых колец жестко связана друг с другом в аннелированные структуры.
Из-за плоской структуры, производные ЦПДТ и его аналоги обладают пониженным окислительным потенциалом, что служит причиной уменьшения ширины запрещенной зоны Eg и смещения края спектров поглощения в красную область спектра, где испускаемая солнцем световая энергия максимальна. Именно поэтому среди исследованных тиофенсодержащих соединений циклопентадитиофеновые структуры стали одними из перспективных донорных фрагментов для получения сопряженных полимеров [10–13].
В настоящей работе была получена серия новых сополимеров на основе донорных блоков ЦПДТ и ДТС. В качестве акцепторных звеньев для чередующихся сополимеров были выбраны гетероциклические соединения с сильными электроноакцепторными свойствами: изо-пирроло[3.2-b]пиррол-2,5-дион (ИДПП)

(R – арил, алкил) и 4,4-дифтор-4H-циклопента[2,1-b:3,4-b']дитиофен (ДФЦПДТ).

Фрагмент ИДПП, как и его аналог дикетопирроло[3.4-c]пиррол (ДПП),

где R – арил, алкил, широко используемый в фотовольтаике [14–19], обладает высокой электроноакцепторной способностью. Это является важным фактором для получения эффективных донорно-акцепторных сополимеров. Однако синтез молекулы ИДПП намного проще, чем ДПП, что делает его более привлекательным для возможного применения в фотовольтаике.
В качестве еще одного акцепторного блока был выбран ДФЦПДТ. За счет присутствия дифторметиленового фрагмента в структуре ЦПДТ, характер данного блока меняется с донорного на акцепторный. Такой выбор обусловлен тем, что сополимеры на основе ДФЦПДТ, полученные нами ранее, обладали перспективными фотовольтаическими свойствами [20].
В работе были синтезированы три сополимера с акцепторным блоком ИДПП, которые содержали в качестве донорного блока ЦПДТ, битиофен (2Т) как структурный аналог блока ЦПДТ и кватротиофеновый (4Т) блок для сравнения оптических и электрохимических свойств, а также четыре сополимера с акцепторным блоком ДФЦПДТ с донорными блоками ЦПДТ и ДТС. Сополимеры на основе ИДПП были получены по методу прямого С–Н арилирования, который, как описано в литературе, позволяет синтезировать сополимеры с высокой молекулярной массой [21–24], а сополимеры на основе ДФЦПДТ получали по реакции кросс-сочетания Сузуки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.5 М растворы н-BuLi в гексане, 2-бромтиофен, N-бромсукцинимид, бром, гидразин гидрат, 2‑изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан, цитронеллол, 2-этилгексилбромид, пивалиновую кислоту, N,N-диметилкарбамоилхлорид, 1,1'-бис(дифенилфосфино)ферроцен] палладий(II) дихлорид Pd(dppf)Cl2, тетракис(трифенилфосфин) палладий(0) Pd(PPh3)4, ацетат палладия (II) Pd(OAc)2 (“Aldrich”) использовали без дополнительной очистки. Растворителями служили диэтиловый эфир (х.ч.), ТГФ (х.ч.), толуол (ч.д.а.), гексан (х.ч.), ДМФА (ч.), дихлорметан (х.ч.) и этанол (ч.). ТГФ и диэтиловый эфир сушили над гидридом кальция и перегоняли в токе аргона. ДМФА и тетраметилэтилендиамин сушили оксидом бария и перегоняли под вакуумом. Дихлорметан сушили кипячением с оксидом фосфора (V) и перегоняли в токе аргона. Остальные растворители перегоняли на роторном вакуумном испарителе непосредственно перед использованием. Для препаративной колоночной хроматографии использовали силикагель 60 (“Merck”, Германия). Для тонкослойной хроматографии применяли пластинки Sorbfil (“Сорбполимер”, Россия).
Для исследования оптических свойств полученных сополимеров использовали спектроскопически чистый ТГФ (градации “для УФ-спектроскопии”, “Acros organic”). Аналитическую ГПХ осуществляли с помощью прибора “Shimadzu” (Япония), детекторы – рефрактометр RID-10A и диодная матрица SPD-M10AVP, колонки – “Phenomenex” (США) 7.8 × 300 мм, заполненные сорбентом “Phenogel” с размерами пор 500 Å и 103 Å, элюент – ТГФ. Аналитическую ГЖХ проводили на хроматографе “Хроматэк Аналитик 5000” (Россия), детектор — катарометр, газ-носитель – гелий, колонки 2 м × 3 мм, неподвижная фаза SE-30 (5%), нанесенная на Chromaton H-AW.
Спектры ЯМР 1Н получали на спектрометре “Bruker WP-250 SY” с регистрацией на частоте 250 MГц, а спектры ЯМР 13C и ЯМР 29Si – на спектрометре “Bruker Avance 300” с регистрацией на частоте 75 и 60 MГц соответственно. Спектры поглощения измеряли в области 200–1000 нм в разбавленных растворах с концентрацией 10–5–10–6 моль/л в расчете на мономерное звено полимера во избежание самопоглощения. Спектры поглощения записывали на спектрофотометре UV-2501PC “Shimadzu” (Япония).
Термогравиметрические исследования образцов проводили в динамическом режиме в диапазоне 50–700°C с использованием системы “TG50 Mettler Toledo” в токе азота и воздуха (200 мл/мин). Точность определения массы образца до 1 мкг. Скорость нагревания составляла 10 град/мин. Фазовые и релаксационные переходы в сополимерах исследовали на приборе “DSC30 Mettler Toledo” в динамическом режиме со скоростью нагревания 20 град/мин в токе азота 50 мл/мин. Электрохимические измерения проводили в растворе электролита, содержащего 0.1 М гексафторфосфата тетрабутиламмония (Bu4NPF6) в ацетонитриле. В каждом случае пленку полимера наносили на стеклоуглеродную поверхность, используемую в качестве рабочего электрода. Вспомогательным электродом служила платиновая пластинка, расположенная в ячейке. Потенциалы измеряли относительно насыщенного каломельного электрода (н.к.э.). Скорость развертки потенциала составляла 200 мВ/с. Полимерную пленку готовили следующим образом: насыщенный раствор полимера в о-дихлорлорбензоле наносили на стеклоуглеродный диск электрода и сушили его в течение 30 мин. Микроволновый синтез проводили в системе для микроволнового синтеза “SEM Discovery System” (США). Для нанесения пленок применяли спин-коутер G3 (“Spin Coating Systems”, США). Металлические электроды напыляли с помощью вакуумного испарителя “Univex 300” (“Leybold”, США). В качестве материала катода использовали двуслойную систему иттербий–алюминий или кальций–алюминий.
Синтез мономеров
2,2'-Битиофен (2). Соединение получали по методике, описанной ранее [20], из 195 г (1.19 моля) 2-бромтиофена, 14.7 г (0.61 моля) магния и 0.853 г, 1.2 ммоля (2 мол. %) катализатора Pd(dppf)Cl2 с выходом 96% в виде бесцветной кристаллической массы с Тпл = 31°С (по лит. данным [20] Тпл = 32–22°С). ЯМР 1H (δ, м.д., CDCl3): 7.19 (м, 4Н, 2,2',4,4'-ThН), 7.01 (дд, 2H, J1 = 3.7 Гц, J2 = = 1.2 Гц, 3.3'-ThH). ЯМР 13C (δ, м.д., CDCl3): 123.7 (4-C, 4'-C), 124.3 (3-C, 3'-C), 127.7 (5-C, 5'-C), 137.3 (2-C, 2'-C)
3,3',5,5'-Тетрабромо-2,2'-битиофен (3). Соединение синтезировали по методике [20], в виде белых кристаллов из 32.7 г (0.197 моля) соединения 2 и 44 мл (0.865 моля) брома. Очистку проводили перекристаллизацией из толуола и этанола. После очистки выход продукта составил 82%, Тпл = = 138°С (по лит. данным [20] Тпл= 140°С). ЯМР 1H (δ, м.д., CDCl3): 7.04 (с, 2H). ЯМР 13C (δ, м.д., CDCl3): 112.1 (3-CBr, 3'-CBr), 114.8 (5-CBr, 5'-CBr), 129.5 (2-C, 2'-C), 133 (4-C, 4'-C).
3,3'-Дибромо-5,5'-бис-(триметилсилил)-2,2'-битиофен (4). Соединение получали по методике [20], из 21.21 г (0.044 моля) соединения 3, 36.8 мл (0.092 моля) 2.5 М раствора н-бутиллития и 11.7 мл (0.092 моля) триметилхлорсилана. После трех перекристаллизаций из этанола продукт выделяли в виде белых кристаллов с выходом 65%, Тпл = 85°С (по лит. данным [20] Тпл = 86°С). ЯМР 1H (δ, м.д., CDCl3): 7.15 (c, 2H), 0.33 (c, 18H). ЯМР 13C (δ, м.д., CDCl3): 142.9 (2-C, 2'-C), 137 (4-C, 4'-C), 133.9 (5-C, 5'-C), 112.9 (3-CBr, 3'-CBr), -0.39 (SiMe3).
2,6-Бис-(триметилсилил)-циклопента[2,1-b:3,4-b']дитиофен-4-он (5). Соединение синтезировали по методике [29], из 3.0 г (6.4 ммоля) соединения 4, 5.6 мл (14 ммоль) 2.5 М раствора н-BuLi, 2.0 мл (13 ммоль) тетраметилэтилендиамина и 0.69 г (6.4 ммоля) N,N-диметилкарбамоилхлорида. Выход 84%, Тпл = 82°С. ЯМР 1H (δ, м.д., CDCl3): 7.07 (с, 2H), 0.31 (с, 18H). ЯМР 13C (δ, м.д., CDCl3): 183.12 (C=O), 154.33 (4-C), 144.88 (5-C), 144.14 (2-C), 127.10 (3-С), -0.24 (SiMe3).
4H-Циклопента-[2,1-b:3,4-b′]дитиофен (6). К суспензии 4.54 г (13.5 ммоля) соединения 5 в этиленгликоле (200 мл) при перемешивании вводили 3.78 г (67.4 ммоля) измельченного в порошок гидроксида калия и 5.3 мл (0.108 моля) гидразингидрата. Реакционную массу нагревали до 180°С и кипятили в течение 8 ч, с обратным холодильником в инертной атмосфере. После охлаждения до комнатной температуры к реакционной массе добавляли 100 мл воды, подкисленной 25 мл 1 Н соляной кислоты и 300 мл диэтилового эфира, органический слой отделяли, а водный еще экстрагировали эфиром (2 ×100 мл). Органический слой промывали водой (2 × 200 мл) до нейтральной реакции, а затем сушили Na2SO4. Растворитель отгоняли на роторном испарителе, а осадок сушили под вакуумом 0.5 мбар. Продукт очищали колоночной хроматографией на силикагеле, используя гексан в качестве элюента. В результате получили светло-желтоватые кристаллы с выходом 90% и Тпл=73–74°C (по лит. данным [25] Тпл = = 74°С). ЯМР 1H (δ, м.д., CDCl3): 7.18 (д, 2H, J = 4.8 Гц, 4,4'Н), 7.09 (д, 2H, J = 4.8 Гц, 5,5'C), 3.54 (с, 2H).
4,4-Бис-(3,7-диметилоктил)циклопента[2,1-b:3,4-b']дитиофен (7a). К 1.63 г (9.1 ммоля) соединения 6, растворенного в 20 мл ДМСО, вводили 6.06 г (27.4 ммоля) 3.7-диметилоктилбромида-1 и 0.17 г (1 ммоль) иодида калия. Реакционную массу охлаждали на ледяной бане и добавляли одной порцией 2.18 г (39 ммоль) измельченного гидроксида калия. Реакционную массу перемешивали в течение 12 ч в инертной атмосфере, после чего охлаждали на ледяной бане и выливали в делительную воронку, содержащую 50 мл воды и 250 мл диэтилового эфира. Органическую фазу выделяли, промывали водой (2 × 100 мл) до нейтральной реакции, сушили над безводным Na2SO4, растворитель отгоняли на роторном испарителе. Избыток 3.7-диметилоктилбромида-1 удаляли отгонкой под вакуумом 0.5 мбар. Продукт очищали колоночной хроматографией на силикагеле, используя гексан в качестве элюента. Получили продукт в виде светло-желтого масла массой 3.64 г (выход 87%). ЯМР 1H (δ, м.д., CDCl3): 7.15 (д, 2H, J = 4.9 Гц, 5,5'Н), 6.92 (д, 2H, J = 4.9 Гц, 4,4'C), 1.83 (м, 4H, CН2), 1.52 (м, 2H, CH), 1.3–1.0 (уш. м, 10H, CH2), 0.83 (д, 12H, J = 6.7 Гц, 4CH3), 0.74 (д, 6H, J = 6.4 Гц, 2CH3).
4,4-Дидецилциклопента[2,1-b:3,4-b']дитиофен (7б). Соединение получали по методике, аналогичной описанной выше для соединения 7а, исходя из 1.5 г (8.4 ммоля) 4H-циклопента-[2,1-b:3,4-b′]дитиофена (6), 5.58 г (25.3 ммоля) децилбромида, 2.01 г (35.8 ммоля) гидроксида калия и 0.15 г (0.9 ммоля) йодида калия. Продукт очищали колоночной хроматографией на силикагеле с использованием гексана в качестве элюента. Получили 3.47 г желтого вязкого масла (выход 90%). ЯМР 1H (δ, м.д., CDCl3): 7.13 (д, 2H, J = 4.9 Гц, 5,5'Н), 6.65 (д, 2H, J = 4.9 Гц, 4,4'C), 1.62 (м, 4H, CH2), 1.26 (м, 29H, CH2), 0.83 (м, 9H, CH3).
2,6-Дибром-4,4-бис-(3,7-диметилоктил)-циклопента[2,1-b:3,4-b'] дитиофен (8а). К раствору 2.84 г (6.2 ммоля) соединения 7а в 90 мл ДМФА, закрытого от света, медленно прикапывали раствор 2.42 г (13.6 ммоля) N-бромсукцинимида в 5 мл ДМФА. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После окончания реакции добавляли 100 мл воды и экстрагировали диэтиловым эфиром (2 × 150 мл). Эфирный слой промывали водой до нейтральной реакции (2 × 200 мл), а затем сушили безводным Na2SO4. Растворитель отгоняли на роторном испарителе. Продукт очищали пропусканием через фильтр Шотта, заполненного силикагелем, используя гексан в качестве элюента. Получили 3.43 г ярко-желтого масла. Выход составил 90%. ЯМР 1H (δ, м.д., CDCl3): 6.95 (с, 2H, 5,5'Н), 1.82 (м, 4H, 2CН2), 0.94–1.79 (м, 16H, CH2), 0.87–0.61 (м, 4H, CH), 0.95–0.82 (м, 18H, CH3).
2,6-Дибром-4,4-дидецилциклопента[2,1-b:3,4-b']дитиофен (8б). Соединение синтезировали аналогично соединению 8а, исходя из 1.8 г (3.92 ммоля) 4,4-дидецилциклопента[2,1-b:3,4-b']дитиофена (7б) и 1.54 г (8.63 ммоля) N-бромсукцинимида. Получили 2.25 г желто-зеленого масла. Выход 93%. ЯМР 1H (δ, м.д., CDCl3): 6.96 (с, 2H, 5,5'Н), 1.8 (м, 4H, CН2), 0.90–1.84 (м, 32H, CH2), 1.05–0.80 (м, 6H, CH3).
2,6-Бис-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4,4-бис-(3,7-диметилоктил)циклопента-[2,1-b:3,4-b']дитиофен (9а). К раствору 1.32 г (2.14 ммоля) соединения 8а в ТГФ (50 мл), охлажденному до –78°С прикапывали 1.9 мл 2.5 M раствора н-BuLi в гексане (4.71 ммоля). Реакционную смесь перемешивали в течение 1 ч при температуре ниже –65°С, после чего добавляли к ней 0.96 г (5.14 ммоля) 2‑изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (I). Затем охлаждение прекращали и продолжали перемешивание при комнатной температуре еще 4 ч. После окончания реакции реакционную смесь выливали в делительную воронку, содержащую 50 мл насыщенного раствора NH4Cl и 250 мл диэтилового эфира. Органическую фазу отделяли, промывали водой до нейтральной реакции (2 × 200 мл) и сушили безводным Na2SO4. Растворитель отгоняли на роторном испарителе. Продукт массой 1.37 г выделяли в виде светло-желтого масла и использовали в последующих реакциях без дополнительной очистки. Выход составил 90%, при этом чистота продукта по данным ГПХ составила не менее 95%. ЯМР 1H (δ, м.д., CDCl3): 7.41 (с, 2H, 5,5'Н), 1.78 (м, 4H, CН2), 1.35 (с, 24H, CH3), 1.29–0.71 (м, 20H, 8CH2, 4CH), 0.83 (с, 18H, CH3).
2,6-Бис-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4,4-дидецил-циклопента-[2,1-b:3,4-b']дитиофен (9б). Соединение получали аналогично соединению 9а, исходя из 1.2 г (1.95 ммоля) 2,6-дибром-4,4-дидецил-циклопента[2,1-b:3,4-b']дитиофена (8б), 1.7 мл (4.28 ммоля) 2.5 М раствора н-BuLi и 0.8 г (4.28 ммоля) 2‑изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана. Получили 1.31 г желто-зеленого масла. Выход составил 95%. ЯМР 1H (δ, м.д., CDCl3): 7.45 (с, 2H, 5,5'Н), 1.80 (м, 4H, CН2), 1.35 (с, 24H, CH3), 0.93–1.74 (м, 32H, CH2), 1.05–0.80 (м, 6H, CH3).
4,4'-Бис-(3,7-диметилоктил)-5,5'-бис-(триметилсилил)дитиено[3,2-b:2',3'-d]силол (10a). К раствору 12.3 г (26.3 ммоля) соединения 4 в 500 мл TГФ, охлажденного до –78 °С, прикапывали 33.8 мл 1.6 М раствора н-BuLi в гексане. Через 15 мин перемешивания реакционной смеси при температуре –78°С приливали к ней 11 г (0.034 моля) бис-(3,7-диметилоктил)дихлорсилана. После этого охлаждающую баню убирали и реакционную массу перемешивали в течение 2 ч при 23°С, затем выливали в воду и экстрагировали диэтиловым эфиром. Органическую фазу отделяли, промывали до нейтральной реакции водой и сушили безводным Na2SO4, после чего растворитель упаривали. Продукт очищали колоночной хроматографией на силикагеле, используя гексан в качестве элюента. Получили 11.4 г вязкой светло-желтой массы (выход 78%). ЯМР 1H (δ, м.д., CDCl3): 7.15 (c, 2H, 5,5'Н), 1.36 (м., 16H, CH2), 1.1 (м, 4H, CH), 0.61 (м, 4H, CH2–Si), 0.30 (c, 18H).
4,4'-Дидецил-5,5'-бис-(триметилсилил)-дитиено[3,2-b:2',3'-d]силол (10б). Соединение синтезировали аналогично соединению 10а, исходя из 4.01 г (8.56 ммоля) соединения 4, 7.1 мл (17.5 ммоля) 2.5 М раствора н-BuLi и 3.27 г (8.56 ммоля) дидецилдихлорсилана. Получили 4.31 г вязкой массы темно-желтого цвета (выход 76%). ЯМР 1H (δ, м.д., CDCl3): 7.15 (c, 2H, 5,5'Н), 1.8 (м, 4H, CН2), 0.90–1.81 (м, 32H, CH2), 1.05–0.80 (м, 6H, CH3), 0.32 (c, 18H, SiMe3).
4,4'-Бис-(3,7-диметилоктил)-5,5'-дибромдитиено[3,2-b:2',3'-d]силол (11a). К раствору 1.69 г (3 ммоль) соединения 10а в 20 мл ТГФ присыпали 1.1 г (6.17 ммоля) N-бромсукцинимида. Реакционную смесь перемешивали при 23°С в течение 4 ч. После окончания реакции реакционную массу выливали в воду и экстрагировали диэтиловым эфиром. Органическую фазу отделяли, промывали водой до нейтральной реакции, сушили безводным Na2SO4 и растворитель упаривали. Продукт очищали колоночной хроматографией на силикагеле, используя гексан в качестве элюента. Получили 1.28 г вязкого масла зеленого цвета (выход 90%). ЯМР 1H (δ, м.д., CDCl3): 7.13 (c, 2H, 5,5'Н), 1.34 (м, 16H, CH2), 1.10 (м, 4H, CH), 0.61 (м, 4H, CH2–Si), 0.83 (с, 18H, CH3).
4,4'-Дидецил-5,5'-дибромдитиено[3,2-b:2',3'-d]силол (11б). Соединение синтезировали аналогично соединению 11а, исходя из 5.6 г (9 ммоль) соединения 10б и 3.54 г (19.9 ммоля) N-бромсукцинимида. Получили 5.3 г вязкой массы зеленого цвета (выход 94%). ЯМР 1H (δ, м.д., CDCl3): 7.15 (c, 2H, 5,5'Н), 1.8 (м, 4H, CН2), 0.90–1.81 (м, 32H, CH2), 1.05–0.8 (перекрывающиеся сигналы, 6H, CH3).
4,4'-Бис-(3,7-диметилоктил)-5,5'-бис-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-дитиено-[3,2-b:2',3'-d]силол (12а). Соединение получали по методике, аналогичной описанной выше для соединения 9а, исходя из 1.10 г (1.73 ммоля) соединения 11а, 1.5 мл (3.81 ммоля) 2.5 М раствора н-BuLi и и 0.71 г (3.81 ммоля) 2‑изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана. Продукт представлял собой вязкую массу зеленого цвета массой (1.14 г, выход 91%). ЯМР 1H (δ, м.д., CDCl3): 7.16 (с, 2H, 5,5'Н), 1.72 (м, 4H, CН2), 1.35 (с, 24H, CH3), 1.32–1.93 (м, 16H, CH2), 1.10–0.71 (м, 4H, CH), 1.37–0.80 (с, 18H, CH3).
4,4'-Дидецил-5,5'-бис-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-дитиено-[3,2-b:2',3'-d]силол (12б). Соединение получали по методике, аналогичной описанной выше для соединения 9а, исходя из 1.77 г (2.8 ммоля) соединения 11б, 2.5 мл (6.15 ммоль) 2.5 М раствора н-BuLi и и 1.26 г (6.15 ммоля) 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана. Продукт представлял собой вязкую массу зеленого цвета (1.78 г, выход 88%). ЯМР 1H (δ, м.д., CDCl3): 7.17 (с, 2H, 5,5'Н), 1.74 (м, 4H, CН2–Si), 1.35 (с, 24H, CH3), 0.93–1.74 (м, 32H, CH2), 1.12–0.60 (м, 6H, CH3).
3,7-Диметилоктанол-1 (13). 50 г (0.32 моля) цитронеллола растворяли в 500 мл этилового спирта, в токе аргона добавляли 1.7 г (16 ммоль) 5% Pd/C и пропускали водород при перемешивании. После того, как измеряемый объем водорода перестал меняться (13 ч), реакционную смесь отфильтровывали через фильтр Шотта, растворитель отгоняли на роторном испарителе. Продукт в виде желтого масла перегоняли под вакуумом, Ткип = 95°C (21 мбар). Масса продукта 48.5 г (выход 96%). ЯМР 1H (δ, м.д., CDCl3): 3.65 (м, 2H, CH2ОН), 1.53 (м, 2Н, СН), 1.05–1.42 (перекрывающиеся сигналы, 8Н, СН2), 0.86 (т, 9Н, J = 6.7 Гц, CH3).
3,7-Диметилоктилбромид-1 (14). К 8.84 (56 ммоля) 3,7-диметилоктанола-1 и 16.10 г (0.061 моля) трифенилфосфина в сухом дихлорметане добавляли порциями 10.93 г (0.061 моля) N-бромсукцинимида, поддерживая температуру реакционной смеси не выше 30°C. После перемешивания при комнатной температуре в течение 16 ч растворитель отгоняли на роторном испарителе, остаток экстрагировали гексаном и отфильтровывали на фильтре Шотта. После удаления гексана продукт очищали фракционной вакуумной перегонкой. Выход: 12.35 г (78%). Продукт представляет собой бесцветную прозрачную жидкость с Ткип = 91–94°C (13 мбар) (по лит. данным [26] Ткип = 103–105°C (23 мбар)). ЯМР 1H (δ, м.д., CDCl3): 3.52–3.33 (м, 2H, CH2Br), 1.85–1.67 (м, 2Н, СН2), 1.62 (м, 1Н, СН), 1.51 (м, 1Н, CH), 1.35–1.21 (м, 2Н, СН2), 1.19–1.05 (м, 4Н, СН2), 0.88 (д, J = 6.5 Гц, 3Н, СН3), 0.85 (д, J = 6.5 Гц, 6H, СН3). ЯМР 13С (δ, м.д., CDCl3): 40.00, 39.11, 36.65, 32.27, 31.59, 27.90, 24.50, 22.55, 22.66, 18.91.
Бис-(3,7-диметилоктил)дихлорсилан (15а). К 2.35 г (0.056 моля) магния в 30 мл сухого ТГФ добавляли раствор 10.5 мл (0.05 моля) 3,7-диметил-октилбромида в 30 мл сухого ТГФ, а затем кипятили 2 ч. Реакционную смесь остужали до комнатной температуры. Полученный раствор прибавляли к 4.3 мл (0.025 моля) тетрахлорсилана в 50 мл сухого ТГФ при –78°С, реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. От неорганических примесей избавлялись высаживанием их сухим гексаном и фильтрацией в аргоне. Чистый продукт массой 17.1 г получили в виде прозрачной жидкости с Ткип = 140–143°С (3.1 мбар). Выход 47%. Чистота продукта, определенная с помощью ГЖХ-хроматографии, составила 98%. ЯМР 1H (δ, м.д., CDCl3): 1.54 (м, 4H, CН), 1.32–1.93 (м, 16H, CH2), 1.10 – 0.70 (м, 4H, CH2–Si), 1.37–0.80 (с, 18H, CH3).
Дидецилдихлорсилан (15б). Соединение получали аналогично соединению 15а, исходя из 53.3 г (0.241 моля) децилбромида, 6.44 г (0.265 моль) магния и 13.8 мл (0.120 моля) тетрахлорсилана. Ткип = 198°С (5.1 мбар) (по лит. данным [27] Ткип = = 190°С, 1.33 мбар). Выход продукта в виде прозрачной жидкости составил 19.7 г (43%). ЯМР 1H (δ, м.д., CDCl3): 0.87 (с, 6H, CH3), 1.25 (с, 32H, CH2), 1.07 (т, 4H, CH2–Si).
N,N'-бис-(4-бутилфенил)оксаламид (16). Раствор 1.20 г (10 ммоль) оксалилхлорида в 20 мл дихлорметана прикапывали при 0°C в течение 20 мин к раствору 3.00 г (20 ммоль) 4-бутиланилина и 1.75 г (22 ммоль) пиридина в 50 мл сухого дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли 150 мл насыщенного водного раствора NaHCO3 и перемешивали в течение 2 ч, при этом осадок розового цвета обесцвечивался. Продукт отфильтровывали на фильтре Шотта, промывали 2 раза водой и этилацетатом, сушили под вакуумом 0.5 мбар. Продукт получили в виде порошка белого цвета массой 3.2 г. Выход 93%. ЯМР 1H (δ, м.д., CDCl3): 9.29 (с, 2H, NH), 7.56 (д, 4H, –NHC6H4, J = 10.1 Гц), 7.20 (д, 4H, –NHC6H4, J = = 10.8 Гц), 2.61 (т, 4H, J = 10 Гц, –CH2–Ph), 1.60 (м, 4H, –CH2–CH2–), 1.36 (м, 4H, –CH2–CH3), 0.93 (т, 6H, J = 10 Гц, –CH2–CH3).
Оксалил-бис-(имидоил)дихлорид (17). К смеси 3.00 г (8.5 ммоля) N,N'-бис-(4-бутилфенил)оксаламида (16) и 3.53 г (1.7 ммоля) тонкоизмельченного PCl5, добавляли 200 мл сухого толуола и кипятили в течение 8 ч с обратным холодильником. После окончания реакции растворитель отгоняли на роторном испарителе. Остаток растворяли в 400 мл сухого гексана и отфильтровывали от нерастворимых примесей. Гексан отгоняли на роторном испарителе, продукт сушили под вакуумом 0.5 мбар. Выход желтой расплывающейся на воздухе кристаллической массы составил 2.4 г (70%). ЯМР 1H (δ, м.д., CDCl3): 7.24 (д, 4H, –NHC6H4, J = 10.1 Гц), 7.09 (д, 4H, –NHC6H4, J = 10.8 Гц), 2.64 (т, 4H, J = 10 Гц, –CH2–Ph), 1.61 (м, 4H, –CH2–CH2–), 1.38 (м, 4H, –CH2–CH3), 0.94 (т, 6H, J = 9.6 Гц, –CH2–CH3).
1,4-Бис-(4-бутилфенил)-3,6-ди(тиофен-2-ил)пирроло[3,2-b]пиррол-2,5(1H,4H)-дион (18). Раствор 0.77 г (4.5 ммоля) этилтиофенацетата в 40 мл ТГФ прикапывали к раствору диизпропиламида лития (3.8 мл, 2 M раствор в ТГФ) в 10 мл ТГФ в атмосфере аргона при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч, а затем охлаждали до –78°C, затем медленно прикапывали раствор 0.8 г (2.0 ммоль) оксалил-бис(имидоил)дихлорида в 20 мл сухого ТГФ. После прибавления всего объема охлаждение прекращали и перемешивали при комнатной температуре в течение 12 ч. После образования коричневой массы реакционную смесь нейтрализовали водой, экстрагировали 150 мл дихлорметана и сушили безводным сульфатом натрия. После удаления растворителя в вакууме к сухой массе добавляли 100 мл гексана и отфильтровывали на фильтре Шотта и промывали несколько раз гексаном. Образовавшийся коричневый порошок сушили под вакуумом 0.5 мбар. Выход 0.76 г (62%). ЯМР 1H (δ, м.д., CDCl3): 7.26 (дд, J1 = 5.1 Гц; J2 = 1.0 Гц, 2H, Th), 7.20 (c, 8H, –C6H4), 6.74 (дд, J1 = 5.1 Гц, J2 = 3.8 Гц, 2H, Th), 6.52 (дд, J1 = 3.8 Гц, J2 = 1.0 Гц, 2H, Th, 2.67 (т, 4H, J = 10 Гц, –CH2–Ph), 1.64 (м, 4H, –CH2–CH2–), 1.36 (м, 4H, –CH2-CH3), 0.96 (т, 6H, J = 9.6 Гц, –CH2–CH3).
3,6-Бис-(5-бромтиофен-2-ил)-1,4-бис-(4-бутилфенил)пирроло[3,2-b]пиррол-2,5(1H,4H)-дион (19). К раствору 0.20 г (0.35 ммоля) соединения 18 в 50 мл сухого хлороформа, порциями при комнатной температуре добавляли 0.164 г (0.9 ммоля) N-бромсукцинимида. После перемешивания в течение 2 ч реакционную смесь экстрагировали 150 мл дихлорметана, сушили безводным сульфатом натрия и отгоняли растворитель на роторном испарителе. Неочищенный продукт растворяли в 150 мл диэтилового эфира, отфильтровывали, растворитель отгоняли на роторном испарителе. Продукт очищали колоночной хроматографией на силикагеле, используя хлористый метилен в качестве элюента. Полученный кирпично-красный порошок сушили под вакуумом. Выход 1.23 г (80%). ЯМР 1H (δ, м.д., CDCl3): δ = 7.19 (м, 8H, –NHC6H4), 6.68 (д, 2H, 4-Th, J = 5.0 Гц), 6.02 (д, 2H, 3-Th, J = = 5.0 Гц), 2.68 (т, 4H, J = 10 Гц, –CH2–Ph), 1.62 (м, 4H, –CH2–CH2–), 1.37 (м, 4H, –CH2–CH3), 0.96 (т, 6H, J = 9.6 Гц, –CH2–CH3).
4,4-Этилендитио-циклопента[2,1-b:3,4-b']дитиофен (20). К раствору 5.73 г (29.8 ммоля) соединения 5 и 5.62 г (59.6 ммоля) 1,2-этандитиола в 200 мл дихлорметана добавляли 11.9 г (89.4 ммоля) AlCl3 при комнатной температуре. Реакцию вели в течение 1 ч, после чего реакционную смесь выливали в делительную воронку, содержащую 400 мл ледяной дистиллированной воды и 600 мл дихлорметана. Органическую фазу отделяли, промывали дистиллированной водой (2 × 200 мл) до нейтральной реакции и сушили безводным Na2SO4. Растворитель отгоняли на роторном испарителе, остаток очищали колоночной хроматографией на силикагеле, используя толуол в качестве элюента. Получили 7.2 г (выход 90%) чистого продукта, который представлял собой порошок светло-желтого цвета. Тпл =149–150°С (по лит. данным [28] Тпл = 148–150°С). ЯМР 1H (δ, м.д., CDCl3): 7.16 (д, 2H, J = 4.9 Гц, 3-Th), 7.08 (д, 2H, J = 5.2 Гц, 2-Th), 3,68 (с, 4H, CН2).
2,6-Дибромо-4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофен (21). Соединение получали по методике [20], исходя из 1.0 г (3.7 ммоля) 4,4-этилендитиоциклопента[2,1-b:3,4-b']дитиофена, 3.31 г (0.018 моля) N-бромсукцинимида и 2.3 мл (0.018 моля) 70%-ного раствора фтороводорода в пиридине. Получили кристаллы ярко-желтого цвета массой 0.78 г. Выход 57%. Тпл = 127–132 °С (по лит. данным [20] Тпл = 127–131°C). ЯМР 1H (δ, м.д., CDCl3): 7.08 (с, 2H, 3,5-H). ЯМР 13С (δ, м.д., CDCl3): 143.1, 139.9, 124.1, 116.9, 114.1. ЯМР 19F (δ, м.д., CDCl3): –121.31.
4,4-Дифтор-циклопента[2,1-b:3,4-b']дитиофен (22). К раствору 0.4 г (1.1 ммоля) соединения 21 в 15 мл сухого ТГФ добавляли порциями 0.24 г (6.5 ммоля) LiAlH4 в токе аргона. Реакционную смесь перемешивали в течение 1 ч, затем приливали 50 мл 1 Н раствора HCl и реакционную массу экстрагировали эфиром (2 × 100 мл). Эфирный слой промывали водой, сушили сульфатом натрия, после чего растворитель отгоняли. После очистки методом колоночной хроматографии на силикагеле (элюент толуол) получили чистый продукт в виде белого порошка, выход: 0.21 г (89%). Тпл = 85–87°С (по лит. данным [28] Тпл = = 87°C). ЯМР 1H (δ, м.д., CDCl3): 7.15 (д, 2H, J = 4.8 Гц, 2,6-H), 7.07 (д, 2H, J = 4.8 Гц, 3,5-H).
Общая методика синтеза сополимеров по реакции Сузуки
К равномольной смеси сомономеров в диметоксиэтане (4 мл) добавляли Pd(PPh3)4 (5 мол. %) и 2 М водный раствор Na2СO3 (1.0 мл). Реакцию проводили в микроволновом реакторе с плавным повышением температуры от 105 до 125°C в течение 10–12 ч. После охлаждения реакционную смесь экстрагировали хлороформом (50 мл), органический слой отделяли и промывали водой. Затем к нему добавляли 100 мл этанола и центрифугировали при скорости 10 000 об/мин в течение 15 мин. Остаток сушили под вакуумом, затем отделяли низкомолекулярную часть экстрагированием в аппарате Сокслета последовательно ацетоном, этанолом и гексаном. Полученный остаток растворяли в минимальном количестве толуола и пропускали через колонку с силикагелем, нагретую до 80°C. Раствор упаривали до минимального объема и осаждали полимер, добавляя шестикратный объем этанола. Продукт сушили под вакуумом 0.5 мбар. Выход высокомолекулярной фракции полимеров 54–70%.
Общая методика синтеза сополимеров по реакции прямого С–Н арилирования
К равномольной смеси сомономеров в N-метилпирролидоне добавляли 10 мол. % Pd(OAc)2, 30 мол. % пивалиновой кислоты и пятикратный мольный избыток K2СO3. Реакцию проводили в микроволновом реакторе при 110°C в течение 10–12 ч. Полимеры выделяли и очищали аналогично методике, описанной выше для реакции Сузуки. Выход полимеров после фракционирования составил 67–87%.
Поли[{1,4-бис-(4-бутилфенил)пирроло[3.2-b]пиррол-2,5(1H,4H)-дион-3,6-диил}-со-{2,2'-битиофен-5,5'-диил}] (P1). Полимер получали по общей методике синтеза сополимеров по реакции прямого С–Н арилирования, описанной выше, из 0.88 г (1.42 ммоля) соединения 18 и 1.02 г (1.42 ммоля) соединения 19. Выход высокомолекулярных фракций продукта 75%. Mn = 44 × 103, Mw/Mn = = 2.9. ЯМР 1H (δ, м.д., CDCl3): 7.71–7.65 (перекрывающиеся сигналы, 4H, Th), 7.46–7.41 (м, 4H, Ph), 7.05–6.93 (м, 4H, Ph), 2.68 (м, 4H, CH2), 1.66–1.23 (уш. м, 8Н), 0.91 (с, 6Н, CH3).
Поли[{1,4-бис-(4-бутилфенил)пирроло[3.2-b]пиррол-2,5(1H,4H)-дион-3,6-диил}-со-{2,2':5',2'':5'',2'''-кватротиофен-5,5'''-диил}] (P2). Полимер получали по общей методике синтеза сополимеров по реакции прямого С–Н арилирования, описанной выше, из 0.3 г (1.8 ммоля) 2,2'-битиофена 1 и 1.30 г (1.8 ммоля) 3,6-бис-(5-бромтиофен-2-ил)-1,4-бис-(4-бутилфенил)пирроло[3.2-b]пиррол-2,5(1H,4H)-диона 19. Выход высокомолекулярных фракций продукта 69%. Mn = 58.5 × 103, Mw/Mn = 3.0. ЯМР 1H (δ, м.д., CDCl3): 7.73–7.05 (перекрывающиеся сигналы, 8H, Th), 7.47 (c, 4H, Ph), 6.95 (c, 4H, Ph), 2.63 (м, 4H, CH2), 1.75–0.98 (уш. м, 8Н), 0.89 (с, 6Н, CH3).
Поли[{1,4-бис-(4-бутилфенил)пирроло[3.2-b]пиррол-2,5(1H,4H)-дион-3,6-диил}-со-{4,4-дидецилциклопента[2,1-b:3,4-b']дитиофен-2,6-диил}] (P3). Полимер получали по общей методике синтеза сополимеров по реакции прямого С–Н арилирования, описанной выше, из 0.64 г (1.39 ммоля) 4,4-бис-(децил)циклопента[2,1-b:3,4-b']дитиофена и 1.00 г (1.39 ммоля) 3,6-бис-(5-бромтиофен-2-ил)-1,4-бис-(4-бутилфенил)пирроло[3.2-b]пиррол-2,5(1H,4H)-диона. Выход высокомолекулярных фракций продукта 83%. Mn = 117 × 103, Mw/Mn = = 3.5. ЯМР 1H (δ, м.д., CDCl3): 7.68–7.43 (перекрывающиеся сигналы, 6H, Th), (c, 4H, Ph), 7.08 (c, 4H, Ph), 2.68 (м, 4H, CH2), 1.65–0.83 (перекрывающиеся сигналы, 60Н).
Поли[{4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофен-3,6-диил}-со-{4,4-дидецилциклопента-[2,1-b:3,4-b']дитиофен-3,6-диил}] (Р4). Полимер получали по общей методике синтеза сополимеров по реакции кросс-сочетания Сузуки из 0.315 г (0.4 ммоля) 2,6-бис-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)-4,4-дидецилциклопента-[2,1-b:3,4-b']дитиофена 9б и 0.150 г (0.4 ммоля) 2,6-дибромо-4,4-дифтор-циклопента[2,1-b:3,4-b'] дитиофена 21. Выход высокомолекулярных фракций продукта 81%. Mn = 27.4 × 103, Mw/Mn = 2.3. ЯМР 1H (δ, м.д., CDCl3): 7.17–7.07 (перекрывающиеся сигналы, 2H, Th), 6.99 (уширенный с, 2H, Th), 1.90–1.76 (м, 4H), 1.29–1.19 (перекрывающиеся сигналы, 32H), 0.83 (с, 6H).
Поли[{4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофен-3,6-диил}-со-{4,4-бис-(3,7-диметилоктил)-циклопента-[2,1-b:3,4-b']дитиофен-3,6-диил}] (Р5). Полимер получали по общей методике синтеза сополимеров по реакции кросс-сочетания Сузуки из 0.567 г (0.8 ммоля) 2,6-бис- (4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4,4-бис-(3,7-диметилоктил)-циклопента-[2,1-b:3,4-b']дитиофена 9а и 0.270 г (0.8 ммоля) 2,6-дибромо-4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофена 21. Выход высокомолекулярных фракций продукта 79%. Mn = 21.5 × 103, Mw/Mn = 3.2. ЯМР 1H (δ, м.д., CDCl3): 7.15–6.96 (перекрывающиеся сигналы, 4H, Th), 3.68 (м, 4H), 1.35–1.09 (перекрывающиеся сигналы, 20H), 0.83 (уш. с, 18H).
Поли[{4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофен-3,6-диил}-со-{4,4'-дидецил-дитиено-[3,2-b:2',3'-d]силол-3,6-диил}] (Р6). Полимер получали по общей методике синтеза сополимеров по реакции кросс-сочетания Сузуки из 0.268 г (0.37 ммоля) 4,4'-дидецил-5,5'-бис-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-дитиено-[3,2-b:2',3'-d]силола 12б и 0.125 г (0.37 ммоля) 2,6-дибромо-4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофена 21. Выход высокомолекулярных фракций продукта 73%. Mn = 15.8 × 103, Mw/Mn = 2.4. ЯМР 1H (δ, м.д., CDCl3): 7.23–7.10 (перекрывающиеся сигналы, 4H, Th), 3.74 (м, 4H), 1.28–1.07 (перекрывающиеся сигналы, 32H), 0.76 (уш. с, 6H).
Поли[{4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофен-3,6-диил}-со-{4,4'-бис-(3,7-диметилоктил)-дитиено-[3,2-b:2',3'-d]силол-3,6-диил}] (Р7). Полимер получали по реакции кросс-сочетания Сузуки из 0.874 г (1.2 ммоля) 4,4'-бис-(3,7-диметилоктил)-5,5'-бис-(4,4,5,5-тетраметил-1,3,2-диоксо-боролан-2-ил)-дитиено-[3,2-b:2',3'-d]силола 12а и 0.407 г (1.2 ммоля) 2,6-дибромо-4,4-дифтор-циклопента[2,1-b:3,4-b']дитиофена 21. Выход высокомолекулярных фракций продукта 85%. Mn = = 13.8 × 103, Mw/Mn = 1.1. ЯМР 1H (δ, м.д., CDCl3): 7.17–6.17 (перекрывающиеся сигналы, 4H, Th), 3.65 (м, 4H), 1.43–1.12 (перекрывающиеся сигналы, 20H), 0.80 (уш. с, 18H).
Изготовление фотоэлементов и измерение вольтамперных характеристик
В условиях инертной атмосферы (аргон) готовили растворы для получения активного слоя путем смешивания сополимеров с производными фуллерена – метиловым эфиром фенил-C61-бутановой кислоты (PC61BM) и метиловым эфиром фенил-C71-бутановой кислоты (PC71BM) в массовом соотношении 1:1 или 2:1 и общей концентрацией 20 мг/мл в о‑дихлорбензоле. Растворы перемешивали на магнитной мешалке с подогревом до 75°С в течение 12 ч, а затем обрабатывали 1 ч в ультразвуковой бане. Из свежеприготовленных растворов на воздухе наносили активный слой на стеклянные подложки размером 23 × 23 × × 1.1 мм с проводящим слоем из оксида индия-олова (ITO) и комплексом поли(3,4-этилендиокситиофена) (ПЭДОТ) с полистиролсульфокислотой (ПСК) методом вращающейся подложки при скорости вращения 700 об/мин. После этого образцы помещали в вакуумную камеру, где проводили напыление верхних металлических электродов – слоев Yb или Ca (20 нм) и Al (200 нм). Изготовленные образцы имели общую конфигурацию ITO/ПЭДОТ : ПСК/активный слой/Mt/Al, где Мt = Yb, Ca. Фотовольтаические свойства – вольтамперные характеристики в темноте и при освещении – измеряли в аргоновом боксе непосредственно после изготовления фотоэлементов. В качестве источника освещения при измерении вольтамперных характеристик использовали симулятор солнечного излучения со спектром АМ1,5G, интенсивность падающего излучения составляла 100 мВт/см2. Плотность тока короткого замыкания Jкз, напряжение холостого хода Uхх, фактор заполнения FF и коэффициент полезного действия определяли из вольтамперных характеристик при освещении.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для синтеза исходных мономеров на основе ЦПДТ и ДТС ключевым соединением является триметилсилильное производное битиофена 4, полученное по отработанной методике [20] (схема 1). Среди нескольких известных методов замыкания битиофенового цикла [29–31] для получения фрагмента ЦПДТ был выбран метод циклизации 3,3'-дибромпроизводного битиофена с помощью N,N-диметилкарбамоилхлорида как наиболее удобный в препаративном плане. Литирование битиофенового производного 4 проводили в присутствии тетраметилэтилендиамина (ТМЭДА), необходимого для увеличения активности н-бутиллития. К полученному дилитиевому производному in situ добавляли 1 экв. N,N-диметилкарбамоилхлорида, что приводило к образованию кетона 5. Для введения алкильных заместителей в положение 4 циклопентадитиофенового цикла, кетогруппу кетона 5 восстанавливали по реакции Кижнера–Вольфа и затем алкилировали соответствующим алкилбромидом в присутствии йодида калия в среде ДМСО. Ди-бромпроизводные 8а и 8б получали действием 2 экв. N-бромсукцинимида в среде ДМФА. Борорганические производные 9а и 9б синтезировали в две стадии: замещение атомов брома на литий в соответствующих дибромидах 8а и 8б и обработкой полученных дилитиевых производных 2 экв. 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксабороланом (I) in situ.

Для получения дитиеносилольных мономеров (Схема 2), дибромид 4 сначала переводили в дилитиевое производное в условиях кинетического контроля для селективного замещения атомов брома в положениях 3 и 3', а затем замыкали цикл в положении 4 соответствующим диалкилдихлорсиланом, что приводило к образованию бис-(триметилсилильных) производных ДТС 10а и 10б. Замещение триметилсилильных групп в последних проводили действием N-бромсукцинимида в среде ДМФА с образованием дибромпроизводных 11а и 11б, которые очищали колоночной хроматографией на силикагеле и переводили в целевые борорганические производные 12а и 12б, используемые в последующих реакциях поликонденсации без очистки.

Для донорных блоков в качестве боковых заместителей в отличие от полученных ранее аналогичных сополимеров [15], в которых обычно используют 2-этилгексильный или октильный фрагмент, были выбраны более длинный линейный децильный и разветвленный 3,7-диметилоктильный фрагмент, введение которых в молекулу ЦПДТ и ДТС, как предполагалось, повысит упорядочение целевых сополимеров без потери их растворимости. Синтез 3,7-диметилоктилбромида 15 осуществляли из коммерчески доступного цитронеллола 13, двойную связь которого сначала восстанавливали гидрированием в присутствии палладия на угле, а затем полученный 3,7-диметилоктанол-1 14 бромировали с помощью N-бромсукцинимида с PPh3 в безводном CH2Cl2 (Схема 3).

Диалкилдихлорсиланы 15а и 15б были получены из соответствующих бромидов по реакции их магнийорганических производных, полученных in situ по реакции Гриньяра, с тетрахлорсиланом. В результате реакций, представленных на схемах 1 и 2, были получены четыре донорных блока на основе ЦПДТ и ДТС с 3,7-диметилоктильными и децильными заместителями – соединения 9а,9б и 12а,12б.
Синтез дитиенопирроло[3.2-b]пиррол-2,5(1H,4H)-дионового (ДТИДПП) акцепторного блока включал четыре стадии (Cхема 4). Сначала был получен диамид 16 из оксалилхлорида и п-бутиланилина, который сразу переводили в имидоилхлорид 17 кипячением с пентахлоридом фосфора в безводном толуоле. Согласованная циклизация имидоилхлорида 17 с 2 экв. этилового эфира тиеноуксусной кислоты в мягких условиях давала незамещенный по тиофеновым кольцам ДТИДПП 18, последующее бромирование которого позволило получить дибромпроизводное ДТИДПП – соединение 19. Таким образом, благодаря небольшому числу стадий с хорошим выходом целевых продуктов представленная схема является удобным способом получения дифункционального акцепторного блока ДТИДПП.

4,4-дифторированные производные ЦПДТ 21 и 22 (cхема 5) синтезировали по методикам, опубликованным ранее [20]. Незамещенное по 2,2' положениям тиофеновых колец производное 22 было получено для исследования возможности проведения реакции прямого С–Н арилирования с его участием.

Cополимеры Р1–Р3 с чередующимися битиофеновым (2Т), кватротиофеновым (4Т), циклопентадитиофеновым донорными фрагментами и акцепторным блоком ДТИДПП были получены поликонденсацией в условиях кросс-сочетания по методу прямого С–Н арилирования (Схема 6).
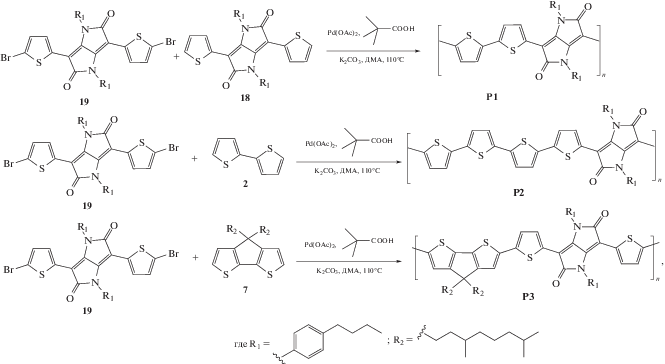
Синтез осуществляли в микроволновом реакторе в среде диметилацетамида при 110°С. Каталитическая система представляла собой ацетат палладия с пивалиновой кислотой в качестве лиганда и K2CO3 как основание. Реакции проводили до максимальных степеней конверсии мономеров, что контролировали методом ГПХ. Сополимеры очищали по стандартной методике, описанной ниже. К реакционной смеси добавляли 10%-ный раствор HCl и перемешивали при температуре 50°С в течение 2 ч. Затем органическую часть экстрагировали хлороформом, после чего полимерную фракцию высаживали избытком метанола. Полученный центрифугированием полимерный остаток помещали в аппарат Сокслета и отделяли высокомолекулярную часть от олигомерных продуктов последовательно кипячением в ацетоне, этаноле и гексане. Оставшийся продукт растворяли в минимальном количестве толуола и пропускали через колонку с силикагелем, нагретую до 80°C. Раствор упаривали до минимального объема и осаждали полимер, добавляя шестикратный объем этанола.
Для получения сополимеров P4–P7 с акцепторным 4,4-дифторпроизводным ЦПДТ была использована реакция Сузуки между дифункциональным пинаколбораном 9а, 9б и дибромидом 4,4-дифторпроизводного ЦПДТ 21 (Схема 7).

Реакцию проводили в условиях микроволнового синтеза в присутствии 10 мол. % Pd(PPh3)4 и 2 М водного раствора Na2CO3 в качестве основания. Полноту прохождения реакции оценивали методом ГПХ.
Были предприняты попытки осуществить синтез сополимеров с дифункциональным акцепторным блоком ДФЦПДТ 22 и соответствующим нефункциональным производным ЦПДТ 7 аналогично сополимеризации Р1–Р3, однако методом ГПХ было установлено отсутствие как олигомерной, так и полимерной составляющих. Изменение функциональности на донорном и акцепторном блоке на обратную в данных условиях также не приводило к поликонденсации соответствующих мономеров:


Такая инертность производного ДФЦПДТ, возможно, связана с сильным акцепторным влиянием 4,4-дифторметиленовой группы на реакционноспособность блока ЦПДТ.
Молекулярные массы и степень конверсии мономеров определяли методом ГПХ, с использованием полистирольных стандартов. Кривые ГПХ представлены на рис 1.
Рис. 1.
Кривые ГПХ для сополимеров Р1–Р3, синтезированных методом прямого арилирования, и сополимеров Р4–Р7, полученных по реакции Сузуки.

Молекулярно-массовые характеристики полученных сополимеров Р1–Р7 приведены в табл. 1.
Таблица 1.
Молекулярно-массовые характеристики, данные ТГА сополимеров
| Сопо-лимер | Mn × 10–3 | Mw × 10–3 | Mw /Mn | Td*5%, °С (азот/воздух) |
|---|---|---|---|---|
| P1 | 44.0 | 127.6 | 2.9 | 300/300 |
| P2 | 58.5 | 175.5 | 3.0 | 350/318 |
| P3 | 117.0 | 409.5 | 3.5 | 395/335 |
| P4 | 27.4 | 62.6 | 2.3 | 322/313 |
| P5 | 21.5 | 68.8 | 3.2 | 330/330 |
| P6 | 15.8 | 37.8 | 2.4 | 313/305 |
| P7 | 13.8 | 14.2 | 1.1 | 290/286 |
Для сополимеров Р1–Р3, полученных методом прямого С–Н арилирования, значения Mn и Mw оказались достаточно высокими. Это согласуется с литературными данными, в которых методом прямого С–Н арилирования удалось достичь больших значений Mn сополимеров [32, 33]. Однако высокая полидисперсность может указывать на большое число разветвлений полимерных цепей в результате низкой региоселективности в условиях данной реакции, что обусловлено наличием четырех возможных положений присоединения (С–Н) в исходном нефункциональном мономере в отличие от дифункциональных мономеров с двумя возможными положениями. Молекулярная масса сополимеров Р4–Р7, полученных по реакции Сузуки, оказалась значительно меньше, что может быть связано с невысокой чистотой (95%) использованных диборорганических производных, однако и полидисперсность данных сополимеров была несколько ниже по сравнению с полидисперсностью сополимеров Р1–Р3.
Изучение термической стабильности сополимеров методом ТГА в азоте и на воздухе показало, что для сополимеров Р1–Р3 (рис. 2а,2б) 5%-ная потеря массы (табл. 1) происходит при температурах выше 300°С и в токе азота, и на воздухе. При этом следует отметить, что замена донорных фрагментов с битиофеновых (2Т) на кватротиофеновые (4Т) и на циклопентадитиофеновые приводит к улучшению темростабильности от 300 до 395°С в инертной атмосфере и от 300 до 335°С на воздухе, а также к увеличению коксового остатка при 700°С с 52 до 63% в токе азота.
Рис. 2.
Кривые ТГА сополимеров Р1–Р3 (а, б) и Р4–Р7 (в, г). Нагревание со скоростью 10 град/мин на воздухе (а, в) и в азоте (б, г).

Сополимеры Р4–Р7 стабильны до температур 290–330°С, при которых наблюдается 5%-ная потеря массы (рис. 2в, 2г). Замена атома С на Si при переходе от в сополимеров на основе ЦПДТ к ДТС вызывает незначительное понижение термостабильности и не влияет на коксовый остаток.
По данным ДСК, для сополимеров Р1–Р3 никаких фазовых и релаксационных переходов в исследуемом диапазоне не выявлено; для сополимеров Р4–Р7 фазовые переходы не обнаружены, но в случае сополимеров Р6 и Р7 были зафиксированы скачки теплоемкости, связанные с расстекловыванием образцов при температурах 120 и 55°С соответственно. Таким образом, замена атома С на Si при переходе от в сополимеров на основе ЦПДТ к ДТС способствует увеличению подвижности молекулярной цепи, а замена н-децильных боковых заместителей на разветвленные 3,7-диметилоктильные – понижению температуры стеклования.
Для сополимеров Р1–Р7 были измерены спектры поглощения в видимой УФ-области спектра в разбавленных растворах ТГФ (10-5 моль/л), а также в тонких пленках, нанесенных на стеклянные пластины из раствора ТГФ методом вращающейся подложки. Полученные данные представлены в табл. 2, из которой следует, что сополимеры эффективно поглощают видимый свет в диапазоне 300–800 нм в растворах и в тонких пленках, при этом максимум поглощения находится в области 450–600 нм.
Таблица 2.
Данные спектроскопии в видимой УФ-области и цикловольтамперометрии
| Полимер | λмакс, р, нм | λкр, р, нм | λмакс, пл, нм | λкр, пл, нм | Eg, опт, эВ | Еок, В | ЕВЗМО, эВ | Евосст В | ЕНСМО, эВ |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 420 | 640 | 440 | 650 | 1.90 | 1.26 | –5.66 | –1.09 | –3.31 |
| P2 | 540 | 730 | 550 | 720 | 1.72 | 0.98 | –5.38 | –1.05 | –3.35 |
| P3 | 600 | 820 | 620 | 850 | 1.46 | 0.82 | –5.22 | –1.03 | –3.37 |
| P4 | 480 | 602 | 485 | 660 | 1.87 | 0.99 | –5.39 | – | –3.56 |
| P5 | 417 | 655 | 450 | 675 | 1.83 | 1.02 | –5.42 | – | –3.59 |
| P6 | 493 | 660 | 526 | 703 | 1.76 | 1.17 | –5.57 | – | –3.81 |
| P7 | 487 | 595 | 509 | 670 | 1.85 | 1.21 | –5.61 | – | –3.76 |
Примечание. λмакс, р – максимум спектра поглощения, измеренного в разбавленном растворе; λкр, р – край спектра поглощения, измеренный в разбавленном растворе; λмакс, пл – максимум спектра поглощения, измеренный в тонкой пленке; λкр, пл – край спектра поглошения, измеренный в тонкой пленке; Eg, опт – ширина запрещенной зоны, полученная из данных спектров поглощения; Еок и Евосст – окислительный и восстановительный потенциалы, полученные из данных цикловольтамперометрии. Значения энергии ЕНСМО для сополимеров Р4–Р7 найдены по разнице между Eg, опт и ЕВЗМО.
На основании спектров поглощения сополимеров Р1–Р3 на основе ИДПП (рис. 3а, 3б) можно заключить, что увеличение длины сопряжения на два тиофеновых кольца при переходе от Р1 к Р2 приводит к длинноволновому сдвигу максимумов поглощения на 120 нм в растворе и на 110 нм в тонкой пленке, а в случае аннелированной структуры ЦПДТ (Р3) смещение в красную область спектра составляет 180 нм как в растворе, так и в тонкой пленке относительно неаннелированного структурного аналога Р1. Для сополимеров на основе ДФЦПДТ (рис. 3в, 3г), по данным спектроскопии, при замещении в донорном фрагменте атома углерода на кремний при сравнении пары с одинаковыми 3,7-диметилоктильными боковыми заместителями (Р4 и Р6) и для пары с н-децильными боковыми заместителями (Р5 и Р7), максимумы поглощения смещаются в красную область на 13 и 70 нм в растворах, и на 41 и 59 нм в пленках. Это можно объяснить стабилизирующим действием d-орбитали кремния на π-сопряженную полимерную цепь. При замене децильной группы на 3,7-диметилоктильную также наблюдается батохромный сдвиг для пар Р4–Р5 и Р6–Р7, что может быть связано с особенностями агрегации макромолекул в растворе и тонких пленках.

В ряду сополимеров Р1–Р2–Р3 края спектров поглощения для пленок смещаются в красную область спектра и лежат при 650, 720 и 850 нм соответственно, в результате чего уменьшается ширина запрещенной зоны Eg от 1.91 для Р1 до 1.72 для Р2 и 1.46 эВ для Р3. Как показывают данные цикловольтамперометрии (см. ниже), такая зависимость обусловлена прежде всего повышением уровней высшей занятой молекулярной орбитали (ВЗМО). В случае сополимеров Р4–Р7 значения Eg близки к 1.8 эВ, что обусловлено более низкорасположенными уровнями нижней свободной молекулярной орбитали (НСМО) по сравнению с сополимерами Р1-Р3.
По измеренным потенциалам окисления и восстановления для сополимеров Р1–Р3 (рис. 4а) были оценены энергии ВЗМО и НСМО с использованием следующих уравнений [34]:
где заряд электрона е = 1.602 × 10–19 Кл.Рис. 4.
Кривые окисления и восстановления полимеров Р1–Р7, полученные методом цикловольтамперометрии.

Для сополимеров Р4–Р7 (рис. 4б) на цикловольтамперограммах не наблюдалось пика восстановления, поэтому энергии НСМО в этих случаях оценивались по разнице между энергией ВЗМО и оптической шириной запрещенной зоны.
Для большинства из полученных сополимеров (кроме P3) наблюдается достаточно низколежащие уровни ВЗМО – от –5.4 до –5.7 эВ (по сравнению с –5.2 для ПГТ), что связано с большим окислительным потенциалом структуры ЦПДТ относительно тиофенового кольца (рис. 5). Для сополимеров Р1–Р3 характерно монотонное повышение уровней ВЗМО с –5.66 до –5.22 эВ из-за увеличения донорной способности в ряду 2Т–4Т–ЦПДТ. При замене атома углерода на кремний для сополимеров Р6 и Р7 понижаются уровни ВЗМО: соответственно –5.61 и –5.57 эВ по сравнению с –5.42 эВ и –5.39 эВ для Р4 и Р5, что опять же обусловлено наличием π–d-сопряжения с атомом кремния.

Для оценки фотовольтаических свойств на основе каждого из синтезированных сополимеров были изготовлены образцы органических солнечных фотоэлементов. Измерения вольтамперных характеристик фотоэлементов на основе сополимеров Р1–Р3 (рис. 6а, 6б) показали сравнимые с таковыми для фотоэлементов на основе ПГТ значения Uхх, однако значения Jкз и FF оказались очень низкими в связи с плохой растворимостью полимеров в о-дихлорбензоле и достаточно высокой молекулярной массы, что в свою очередь привело к неоптимальной морфологии пленок активного слоя (рис. 7).
Рис. 6.
Фотоэлектрические характеристики фотоэлементов с активным слоем из сополимеров Р1–Р3 в смеси с PC61BM = 1 : 1 (1–3), а также Р3 в смеси с PC61BM и PC71BM в соотношении 1 : 1 (4, 6) и 2 : 1 (5, 7).

Рис. 7.
АСМ-изображения активного слоя фотоэлемента при соотношении Р3:PC61BM = 1 : 1 (а) и 2:1 (б).

Из табл. 3 следует, что несмотря на примерно равные величины Uxx, значения Jкз, как и КПД, увеличиваются при переходе от фотоэлементов на основе Р1 к Р3. Очевидно, что эти значения коррелируют со смещением спектров поглощения в длинноволновую область.
Таблица 3.
Фотоэлектрические характеристики фотоэлементов на основе сополимеров Р1–Р3
| Образец | Uxx, В | Jкз, мA/cм2 | FF, % | КПД ,% |
|---|---|---|---|---|
| P1 : PC61BM (1 : 1) | 0.466 | –1.34 | 38 | 0.24 |
| P2 : PC61BM (1 : 1) | 0.507 | –1.94 | 43 | 0.43 |
| P3 : PC61BM (1 : 1) | 0.510 | –4.49 | 45 | 1.03 |
| P3 : PC61BM (2 : 1) | 0.600 | –4.89 | 58 | 1.72 |
| P3 : PC71BM (1 : 1) | 0.576 | –5.68 | 62 | 2.03 |
| P3 : PC71BM (2 : 1) | 0.567 | –6.63 | 63 | 2.37 |
Для фотоэлементов на основе наиболее эффективного сополимера Р3 были исследованы возможности дальнейшего повышения эффективности за счет изменения состава активного слоя фотоэлементов. Так, при изменении соотношения Р3 : PC61BM от 1 : 1 до 2 : 1, как Uxx, так и Jкз ощутимо повысились, что привело к росту КПД с 1.03 до 1.72%. Замена акцепторного фуллеренового производного PC61BM на PC71BM позволила увеличить Jкз почти в 1.2 раза и удвоить значение КПД относительно показателей для фотоэлементов с тем же соотношением PC61BM. Для фотоэлементов с активным слоем Р3 : PC71BM = 2 : 1 значение Jкз возросло до –6.63 мА/см2 по сравнению с –4.89 мА/см2 для фотоэлементов с активным слоем Р3 : PC61BM. Варьируя природу фуллеренового акцептора и его соотношение с сополимером Р3, удалось повысить КПД фотоэлементов более чем в 2 раза. Такое влияние природы фуллеренового производного PC71BM связано с лучшим поглощением света в видимой области и, предположительно с лучшей морфологией активного слоя.
На основе сополимеров Р4–Р7 в смеси с PC61BM 1 : 1 были изготовлены макеты солнечных фотоэлементов и измерены их вольтамперные характеристики (табл. 4). Типичные значения Uxx = 0.4–0.5 В оказались близкими к значениям Uхх для фотоэлементов на основе полимера ПГТ, однако низкие величины Jкз и FF указывают на плохую морфологию пленки, что является причиной неоднородностей вследствие низкой растворимости (5–7 мг/мл) данных сополимеров. Для оптимизации морфологии активного слоя сразу после нанесения подложки отжигали на воздухе при 100°С в течение 10 мин, после чего на них вместе с подложками сравнения без отжига наносили контакты и измеряли вольтамперные характеристики.
Таблица 4.
Фотоэлектрические характеристики фотоэлементов на основе сополимеров Р4–Р7, изготовленные без отжига (числитель) и с отжигом при 100°С (знаменатель)
| Образец | Uxx, В | Jкз, мA/cм2 | FF, % | КПД, % |
|---|---|---|---|---|
| P4: PC61BM (1 : 1) | 0.456/0.328 | –1.64/–6.40 | 36/27 | 0.27/0.58 |
| P6: PC61BM (1 : 1) | 0.516/0.380 | –2.16/–4.78 | 36/31 | 0.41/0.57 |
| P5: PC61BM (1 : 1) | 0.486/0.464 | –5.34/–6.16 | 31/32 | 0.81/0.92 |
| P7: PC61BM (1 : 1) | 0.522/0.509 | –5.71/–7.87 | 31/29 | 0.93/1.16 |
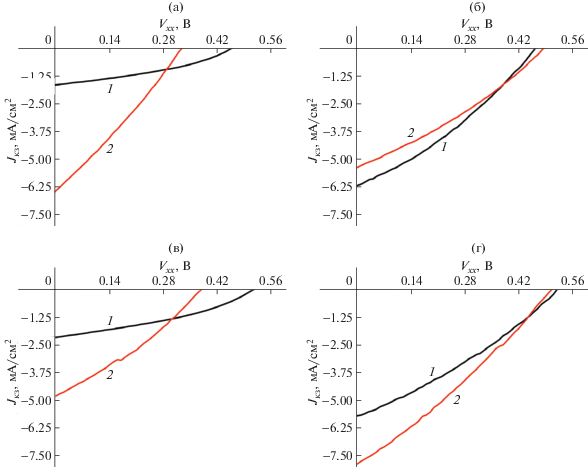
Анализируя полученные результаты, можно сделать следующие выводы: на элементах с отжигом в случае сополимеров Р4 и Р6 (рис. 8а, 8в) наблюдается уменьшение Uxx, что в совокупности с более крутыми вольтамперными характеристиками отожженных образцов свидетельствует об уменьшении параллельного сопротивления фотоэлементов. Соответствующие значения Jкз увеличиваются, что может быть следствием более оптимальной упорядоченности активного слоя. Для элементов на основе Р4 и Р6 коэффициент полезного действия при отжиге повысился, однако в случае Р5 и Р7 (рис. 8б, 8г) практически не изменился. Данный факт можно объяснить различной степенью упорядоченности сополимеров при отжиге с разными алкильными заместителями. Так, сополимеры Р4 и Р6 с линейными децильными заместителями на фрагментах ЦПДТ, кристаллизующиеся в процессе отжига. В то же время сополимеры Р5 и Р7 содержат разветвленные 2,3-диметилоктильные заместители, способность к кристаллизации которых сильно затруднена. Понижение факторов заполнения и Uxx солнечных фотоэлементов на основе сополимеров Р4 и Р6 после отжига может быть вызвано увеличением подвижности дырок в полимере за счет роста его кристалличности. Как было показано в работе [35], величина Jкз монотонно увеличивается с подвижностью, Uxx монотонно убывает с ростом подвижности дырок, а фактор заполнения сначала увеличивается, достигает максимального значения при некотором значении подвижности, затем убывает с ее ростом.
Рис. 8.
Фотоэлектрические характеристики фотоэлементов на основе сополимеров Р4 (а), Р5 (б), Р6 (в) и Р7 (г) в смеси с PC61BM = 1:1 без отжига (1) и с отжигом (2).
На основании приведенных выше результатов можно сделать вывод о том, что полученные сополимеры за счет своих оптических свойств и положения энергетических уровней имеют дальнейшие перспективы с точки зрения оптимизации их фотовольтаических свойств.
Работа выполнена при поддержке гранта Президента Российской Федерации для государственной поддержки ведущих научных школ Российской Федерации НШ-5698.2018.3.
Список литературы
Peumans P., Forrest S.R. // Appl. Phys. Lett. 2001. V. 79. P. 126.
Zhao G., He Y., Li Y. // Adv. Mater. 2010. V. 22. P. 4355.
Baran D., Ashraf R.S., Hanifi D.A., Abdelsamie M., Gasparini N., Rohr J.A., Holliday S., Wadsworth A., Lockett S., Neophytou M., Emmott C.J.M., Nelson J., Brabec C.J., Amassian A., Salleo A., Kirchartz T., Durrant J.R., McCulloch I. // Nat. Mater. 2017. V. 16. P. 363.
Cheng Y.-J., Yang Sh.-H., Hsu Ch.-S. // Chem. Rev. 2009. V. 109. № 11. P. 5868.
Xu T., Yu L. // Mater. Today. 2014. V. 17. P. 11.
Guo X., Baumgarten M., Mullen K. // Prog. Polym. Sci. 2013. V. 38. P. 1832.
Duan Ch., Huang F., Cao Y. // J. Mater. Chem. 2012. V. 22. P. 10416.
Akkuratov A.V., Troshin P.A. // Polymer Science B. 2014. V. 56. № 4. P. 414.
Chang S.-W., Muto T., Kondo T., Liao M.-J., Horie M. // Polym. J. 2017. V. 49. P. 113.
Chu T.-Y., Lu J., Beaupre S., Zhang Y., Pouliot J.-R., Wakim S., Zhou J., Leclerc M., Li Zh., Ding J., Tao Y. // J. Am. Chem. Soc. 2011. V. 133. P. 4250.
Cui C., Fan X., Zhang M., Zhang J., Min J., Li Y. // Chem. Commun. 2011. V. 47. P. 11345.
Rasmussen S.C., Evenson S.J. // Prog. Polym. Sci. 2013. V. 38. P. 1773.
Zhang Y., Zou J., Yip H.-L., Sun Y., Davies J.A., Chen K.-Sh., Acton O., Jen A.K.-Y. // J. Mater. Chem. 2011. V. 21. P. 3895.
Hendriks K.H., Heintges G.H.L., Gevaerts V.S., Wienk M.M., Janssen R.A. J. // Angew. Chem. Int. Ed. 2013. V. 52. P. 8341.
Qu S., Tian H. // Chem. Commun. 2012. V. 48. P. 3039.
Guo X., Puniredd S.R., He B., Marszalek T., Baumgarten M., Pisula W., Müllen K. // Chem. Mater. 2014. V. 26. P. 3595.
Li W., Hendriks K.H., Wienk M.M., Janssen R.A.J. // Acc. Chem. Res. 2016. V. 49. № 1. 78.
Du J., Fortney A., Washington K.E., Biewer M.C., Kowalewski T., Stefan M.C. // J. Mater. Chem. A. 2017. V. 5. P. 15591.
Dominguez R., Montcada N.F., de la Cruz P., Palomares E., Langa F. // ChemPlusChem. 2017. V. 82. P. 1096.
Drozdov F.V., Myshkovskaya E.N., Susarova D.K., Troshin P.A., Fominykh O.D., Balakina M.Yu., Bakirov A.V., Shcherbina M.A., Choi J., Tondelier D., Buzin M.I., Chvalun S.N., Yassar A., Ponomarenko S.A. // Macromol. Chem. Phys. 2013. V. 214. P. 2129.
Facchetti A., Vaccaro L., Marrocchi A. // Angew. Chem. Int. Ed. 2012. V. 51. P. 3520.
Chang S.-W., Waters H., Kettle J., Kuo Z.-R., Li Ch.-H., Yu Ch.-Y., Horie M. // Macromol. Rapid Commun. 2012. V. 33. P. 1927.
Schmatz B., Ponder J.F., Reynolds J.R. // J. Polym. Sci., Polym. Chem. 2018. V. 56. P. 147.
Ponder J.F., Schmatz B., Hernandeza J.L., Reynolds J.R. // J. Mater. Chem. C. 2018. V. 6. P. 1064.
Endo K. // Synlett. 2014. V. 25. P. 2184.
Cohen N. // Helv. Chim. Acta. 1981. V. 64. P. 1158.
Seki S. // J. Am. Chem. Soc. 2004. V. 126. P. 3521.
Ie Y. // Org. Lett. 2007. V. 9. P. 2115.
Brezezinski J.Z., Reynolds J.R. // Synthesis. 2002. V. 8. P. 1053.
Gronowitz S., Erickson B. // Ark. Kemi. 1963. V. 21. P. 335.
Jordens P., Rawson G., Wynberg H. // J. Chem. Soc. C. 1970. P. 273.
Facchetti A., Vaccaro L., Marrocchi A. // Angew. Chem. Int. Ed. 2012. V. 51. P. 3520.
Chang S.-W., Waters H., Kettle J., Kuo Z.-R., Li Ch.-H., Yu Ch.-Y., Horie M. // Macromol. Rapid Commun. 2012. V. 33. P. 1927.
de Leeuw D.M., Simenon M.M. J., Brown A.R., Einerhand R.E.F. // Synth. Met. 1997. V. 87. P. 53.
Deibel C., Wagenpfahl A., Dyakonov V. // Phys. Stat. Sol. (RRL). 2008. V. 2. P. 175.
Zhou H., Yang L., Stoneking S., You W. // Appl. Mater. Interfaces. 2010. V. 2. P. 1377.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)