

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 2, стр. 132-143
ОКИСЛИТЕЛЬНАЯ ДЕСТРУКЦИЯ ХИТОЗАНА И ЕГО СТАБИЛЬНОСТЬ
Б. А. Комаров a, *, Г. В. Малков a, С. Г. Васильев a, С. А. Баскаков a, Г. А. Эстрина a, Л. Л. Гурьева a, В. И. Волков a, М. А. Фролова b, А. И. Албулов b
a Институт проблем химической физики Российской академии наук
142432 Черноголовка, пр. акад. Семенова, Россия
b Всероссийский научно-исследовательский и технологический институт биологической промышленности
141113 Московской обл., Щелково, Россия
* E-mail: komarov@chgnet.ru
Поступила в редакцию 25.09.2018
После доработки 20.11.2018
Принята к публикации 06.12.2018
Аннотация
Изучены свойства хитозана, деструктированного под действием перекиси водорода. Методами ИК-фурье-спектроскопии, ВЭЖХ и спектроскопии ЯМР 13С установлено, что процесс окислительной деструкции хитозана сопровождается реакциями сшивки, приводящими к образованию нерастворимой фракции полимера. Обсуждается механизм процесса сшивки и получение хитозана, способного сохранять растворимость в течение длительного времени.
ВВЕДЕНИЕ
Хитин – один из наиболее распространенных полисахаридов в природе. Деацетилированная его форма хитозан и различные их производные нашли широкое применение в медико-биологических приложениях, в агробиологии, биотехнологии и экологии [1–3]. Известно применение хитозана в технологии предварительной обработки арамидного волокна с целью улучшения прочностных свойств эпоксидного композита [4], а также в качестве носителя для катализатора селективного гидрирования непредельных и нитроорганических соединений [5]. Использование хитозана для нековалентного функционализирования углеродных нанотрубок способствует устойчивости к агломерации наночастиц Pt–Ru, применяемых в электрокаталитическом окислении метанола [6]. Недавно показано повышение эффективности в очистке воды и микрокапсулировании медицинских препаратов при использовании хитозана в виде наночастиц или нанокомпозитов [7, 8]. Нанокомпозиты на основе хитозана применяют для регенеративной медицины и тканевой инженерии костей и хрящей [9, 10].
Особый интерес представляют хитозан-графеновые нанокомпозиты. Сочетание с графеном (или его производными) улучшает не только механические, но также теплофизические и электрические свойства [11, 12]. Объединение положительных свойств графена, углеродных нанотрубок и хитозана при разработке электрохимических сенсоров привело к возможности селективного определения нескольких веществ одновременно в широком диапазоне концентраций [13].
Производство хитозана осуществляется путем высаживания его из растворов после деструкции с последующей многократной промывкой и сушкой [14]. При длительном хранении производственных партий изменяются характеристики хитозана в зависимости от способов очистки и осушки. Происходит самопроизвольная деградация хитозана, приводящая к уменьшению молекулярной массы, растворимости и появлению нерастворимой фракции. В связи с этим проблема исследования процесса, приводящего к этим изменениям, и поиска условий и методов его устранения является актуальной, решению которой посвящено много работ.
В работе [15] показано, что в хитозане, произведенном различными фирмами, содержится от 6 до 18% хитозан-белковых комплексов. Авторы отмечают, что при длительном хранении при 20–25°С в хитозане исчезает поглощение в УФ-области при 235 нм, но не анализируют причины таких изменений.
Выделение хитозана, получаемого любым из описанных методов химического или ферментативного гидролиза, обязательно связано со стадиями переосаждения, фильтрации, промывки и распылительной или лиофильной сушки. Обнаружено [14], что лиофильное высушивание целевого продукта после промывки двукратным количеством воды практически полностью удаляет адсорбированные молекулы перекиси водорода при использовании ее для деструкции хитозана. Отсутствие перекиси водорода в объеме частиц полимера положительно влияет на длительность сохранения его свойств.
В работе [16] предложено деструктировать хитозан по радикальному механизму под действием веществ, способных генерировать радикалы. Для инициирования радикального процесса наиболее часто используют перекись водорода и окислительно-восстановительную пару перекись водорода–аскорбиновая кислота. В ИК-фурье-спектрах поглощения низкомолекулярной фракции деструктированного хитозана [17] обнаружена полоса поглощения 1667 см–1, обусловленная валентными колебаниями карбонильной группы кетона, которая по структуре близка к карбонильной группе циклогексанона с характерной очень сильной полосой поглощения в УФ-области – 285 нм.
Исследование окислительной деструкции хитозана в жестких условиях [18] показало, что наименее прочными являются гликозидные связи, а окисление аминогрупп можно предотвратить путем их защиты хелатообразованием с ионами меди либо посредством солеобразования с кислотами.
Несмотря на практическую важность, в литературе отсутствуют сведения о влиянии длительного хранения на структуру и свойства растворимого в подкисленных водных средах хитозана. Поскольку очевидно, что изменения свойств полимера при длительном сроке хранения вызваны его окислительной деструкцией, настоящая работа посвящена изучению изменения свойств хитозана в процессе его деструкции под действием перекиси водорода умеренной концентрации и в условиях защиты аминогрупп солеобразованием. Методами вискозиметрии, ВЭЖХ, ИК-спектроскопии и спектроскопии ЯМР 13С исследованы растворимость, молекулярные характеристики и структура сшивок между макромолекулами, обусловливающих образование нерастворимых фракций в процессе окислительной деструкции и во время длительного хранения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали следующие реактивы: ледяную уксусную кислоту, гидроокись натрия, перекись водорода без дополнительной очистки.
Образцы очищенного и обесцвеченного хитина (для спектров ЯМР) и хитозана предоставлены Всероссийским научно-исследовательским и технологическим институтом биологической промышленности: 1 – чешуйчатый полученный по традиционной методике [1] хитозан со средневязкостной молекулярной массой Мη = 700 × 103; 2 – чешуйчатый хитозан с Мη = 108 × 103, полученный в результате гетерогенной окислительной деструкции хитозана с Мη = 180 × 103 в мягких условиях [19] с двукратной промывкой водой и лиофильной сушкой, и образцы хитозана 3 и 4, полученные аналогичным образом, но воздушно высушенные после промывки водой в тонком слое при Т ≤ 50°С. После сушки эти образцы имели величины Мη = 87 × 103 и 48 × 103 соответственно (табл. 1). Степень деацетилирования образцов хитозана составляла 75–80%.
Таблица 1.
Характеристическая вязкость [η], средневязкостная молекулярная масса Мη и константа K' в уравнении Хаггинса для хитозана с различным временем хранения τхр и длительности растворения τр
| Образец, № | Мисх × 10–3 (год) | τхр, год | τр, сутки | Нерастворимая часть полимера, мас. % | Влажность полимера, мас. % | [η], дл/г | Мη × 10–3 | K' |
|---|---|---|---|---|---|---|---|---|
| 1 | 700 (2014) | 1 | 1 | 8.5 | 12.8 | 7.30 | 365 | 1.45 |
| 2 | 7.1 | 12.8 | 6.60 | 320 | 0.51 | |||
| 3 | 5.7 | 12.8 | 5.70 | 280 | 0.48 | |||
| 2 | 108 (2005) | 10 | 0.5 | 0 | 8.0 | 2.55 | 106 | 0.57 |
| 3 | 87 (2005) | 10 | 1 | 43.4 | 10.5 | 1.30 | 47.4 | 0.69 |
| 4 | 48 (2005) | 10 | 1 | 40.6 | 10.1 | 0.87 | 29.5 | 1.02 |
Влажность образцов хитозана определяли по разнице масс до и после высушивания в термо-шкафу при 105°С в течение 5 ч и охлаждения в эксикаторе с осушителем.
Нерастворимую долю хитозана выделяли путем растворения в смеси 2%-ной уксусной кислоты и 0.2 М уксуснокислого натрия в дистиллированной воде при мольном отношении уксусной кислоты и аминогрупп NH2 в навеске хитозана ≥4.5 при постоянном перемешивании в течение 24 ч при комнатной температуре. Количественно нерастворимую часть определяли после фильтрования, высушивания на воздухе и взвешивания.
Термоокислительную деструкцию образцов хитозана проводили под действием перекиси водорода при отношении [Н2О2] : [хитозан] ≤ 0.2 в дистиллированной воде в присутствии добавок уксусной кислоты в эквимольном количестве с аминогруппами хитозана для их защиты [18] и разупорядочения структуры полимера в закрытых сосудах при 80°С в течение 1 ч (табл. 2).
Таблица 2.
Молекулярные характеристики образцов хитозана в процессе поэтапной гомогенной деструкции под действием перекиси водорода
| Образец, № | [η], г/дл | Мη × 10–3 | Компоненты, мас. % | Данные ВЭЖХ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| хитозан | НАс | Н2О2 | Mп × 10–3 | Mw × 10–3 | Mмакс × 10–3 | Ð | S*, % | |||
| 2 | 2.55 | 106 | Исходный | 0 | 0 | 7.2 | 49.7 | 48.8 | 6.9 | 84 |
| – | – | 0.13 | – | 16 | ||||||
| 2-1 | 0.43 | 13 | 3.54 | 1.10 | 0.22 | – | – | 209 | – | 0.4 |
| 3 | 22.9 | 13.4 | 7.6 | 88 | ||||||
| – | – | 0.13 | – | 11.6 | ||||||
| 2-2 | – | – | 1.78 | 0.55 | 0.36 | 1.4 | 5 | 2.9 | 3.5 | 90.6 |
| – | – | 0.13 | – | 9.4 | ||||||
| 3-1 | 1.75 | 68 | 3.8 | 1.0 | 0.24 | – | – | 149 | – | 21.4 |
| 4.6 | 23.8 | 42.3 | 5.17 | 65.9 | ||||||
| – | – | 0.13 | – | 12.7 | ||||||
| 3-2 | 0.35 | 10 | 2.1 | 0.55 | 0.35 | 2.8 | 21.5 | 10.4 | 7.6 | 89.5 |
| – | – | 0.13 | – | 10.5 | ||||||
| 4-1 | 1.0 | 35 | 4.1 | 1.0 | 0.24 | – | – | 122 | – | 12.5 |
| 3.4 | 22.3 | 23 | 6.5 | 75.0 | ||||||
| – | – | 0.13 | – | 12.5 | ||||||
| 4-2 | 0.39 | 12 | 2.1 | 0.55 | 0.35 | – | – | 98 | – | 3.70 |
| 2.6 | 16.1 | 9.5 | 6.2 | 82.50 | ||||||
| – | – | 0.13 | – | 13.80 | ||||||
ИК-спектры получали на ИК-фурье-спектрометре “Perkin Elmer Spectrum 100” снабженного приставкой МНПВО на германиевых призмах в диапазоне волновых чисел 4000–675 см–1. Для анализа были выбраны образцы хитозана (табл. 1, образцы 2, 3 и 4) разной морфологии – тонкие чешуйки и различные объемные образования.
Величины Мη образцов хитозана определяли по вискозиметрической методике [20]. Диаметр капилляра модифицированного вискозиметра Уббелоде равен 0.5 мм. В качестве растворителя для изоионного разбавления использовали раствор 2%-ной уксусной кислоты и 0.2 М уксуснокислого натрия в дистиллированной воде, время истечения растворителя 130 с при 25°С. Экспериментальные данные обрабатывали в соответствии с уравнением Хаггинса: ηуд/с = [η] + K'[η]2с, где с – концентрация хитозана в растворителе в г/дл. По тангенсу угла наклона прямой зависимости определяли константу Хаггинса K'. Величины Мη образцов хитозана рассчитывали по уравнению Марка–Куна–Хаувинка: [η] = 1.38 × 10-4(Мη)0,85 [20].
Молекулярные характеристики полимеров изучали методом ВЭЖХ на приборе “Waters” с двумя детекторами: рефрактометром и УФ-спектрофотометром в области 200–600 нм. Использовали колонку ULTRAHYDROGEL-250 размером 7.5 × 300 мм. Элюентом служил бидистиллят с аммиачно-ацетатным буфером концентрации 0.5 моль/л. ММР и ММ фракций образцов хитозана вычисляли с помощью программы EMPOWER по калибровочному графику, полученному по образцам с известными молекулярными характеристиками. Концентрацию полимерных фракций оценивали по площади соответствующих пиков на рефрактометрической (РФ) хроматограмме.
Твердотельные спектры ЯМР снимали на спектрометре “Bruker Avance III” с рабочей частотой 400.22 и 100.64 МГц для ядер 1H и 13C соответственно. Для получения спектров использовали вращение образцов под магическим углом с частотой 14 кГц и кросс-поляризации 13C{1H} (CP-MAS). Время контакта составляло 1 мс, период повторения последовательности 6 с. Образцы хитина и хитозана в виде мелкодисперсного порошка помещались в роторы из диоксида циркония с внешним диаметром 3.2 мм.
Методом ЯМР исследовали хитин, растворимый хитозан – образец 2 и нерастворимую часть хитозана 3, полученную удалением растворимого полимера путем перемешивания в течение 1 суток суспензии хитозана (1.3 мас. %) в водной уксусной кислоте (0.56 мас %). При фильтровании образец промывали раствором NaOH при рН 9, затем 20-кратным количеством дистиллированной воды. Получили две фракции нерастворимого образца 3 с размером частиц больше 160 мкм и больше 41 мкм. Порошок сушили тонким слоем на воздухе.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Растворимость и средневязкостная Мη исходных образцов хитозана
Из табл. 1 видно, что с увеличением времени растворения в рабочем растворе чешуйчатого хитозана (образец 1) величина Мη и количество нерастворимой фракции уменьшаются. Существенно, что после отсеивания фракции с крупными чешуйками более 2.0 мм (около 10%) количество нерастворимой фракции уменьшилось с 8.5 до 4.2 мас. %. А количество нерастворимого полимера во фракции с крупными частицами увеличилось до 20%. Также было обнаружено, что после термостатирования отсеянного образца 1 при 105°С в течение 5 ч количество нерастворимой фракции хитозана, как и следовало ожидать, увеличилось с 4.2 до 13.2 мас. %.
Важно отметить, что определенная Мη исходного образца 1 (табл. 1) значительно меньше паспортных данных 700 × 103. Если предположить, что зависимость уменьшения величины Мη от времени растворения (табл. 1, образец 1) имеет экспоненциальный характер (Мt = М0 × e–kt), то несложные расчеты с экстраполяцией линейной зависимости логарифма Мη от времени растворения к нулевому времени привели к исходному значению Мη хитозана этого образца, равному ~420 × 103. Так как природа активной примеси в объеме частиц полимера и ее концентрация не известны, по зависимости: lnMt = lnM0 – kэфф × t оценена эффективная константа гомогенного гидролиза хитозана при 20°С – 15 × 10–7 с–1. Константа скорости гомогенной деструкции хитозана [18] при 20°С под действием перекиси водорода (0.02 мас. %) в водной уксусной кислоте (2.0 мас. %) для диапазона концентраций хитозана 1–4 мас. % равна 0.7 × 10–7 с–1, что в 20 раз меньше константы, оцененной в нашем случае. Исследования гомогенного кислотного гидролиза в работах [21, 22] и неизменность величин Мη при растворении хитозана в растворе уксусной кислоты в течение 7 суток [23] показали, что при температуре 20°С кислотная деструкция макромолекул хитозана практически отсутствует, причем, независимо от природы кислоты.
Наблюдаемое изменение содержания нерастворимой части во фракциях с частицами образца 1 разного размера, а также снижение Мη при увеличении времени растворения могут быть обусловлены действием активной примеси, представляющей собой белковые и хитин-хитозан-белковые структуры со сложным микроэлементным составом, которые не полностью удаляются при получении высокомолекулярного хитозана (Мη = = 700 × 103) по традиционному способу получения [1]. Количество этих примесей находится в прямой зависимости от размера частиц полимера, а деструктирующее их действие проявляется как в растворе, так и в конденсированном состоянии при нагревании. Причем деструктирующая активность этих примесей в гомогенных условиях на порядок выше, чем у перекиси водорода, что подтверждается проведенными оценочными расчетами эффективных констант деструкции.
Известно, что перекись водорода способна деструктировать белки [17, 18, 24], поэтому при деструкции хитозана под действием перекиси водорода при получении полимера с М ≤ 1 × 105 активная примесь с пептидными связями практически исчезает. Однако и в данном случае при длительном хранении возможно образование нерастворимых фракций. Например, в образцах 3 и 4 появилась нерастворимая фракция в количестве 43.4 и 40.6 мас. %, а величина Мη растворимой фракции уменьшилась в ~2 два раза. Образец хитозана 2, высушенный лиофильно при получении, после хранения в аналогичных условиях и через 10 лет полностью растворим. Величина Мη изменяется незначительно (табл. 1). Существенно, что растворимость этого образца не изменяется и после термообработки при 105°С в течение 5 ч. Указанные различия обусловлены действием молекул перекиси водорода в набухшем полимере, которые могут быть удалены только при лиофильной сушке, что наглядно продемонстрировано на примере образца 2.
Молекулярные характеристики хитозана
Исследования методом ВЭЖХ образцов хитозана после длительного хранения и поэтапной гомогенной окислительной деструкции под действием перекиси водорода позволили выявить общие закономерности изменения ММ и ММР хитозана в данном процессе (табл. 2; рис. 1, 2 и 3). Обнаружено, что растворимый исходный образец 2 состоит из двух фракций: полимерной (84%) и низкомолекулярной 16% [25]. Окислительная деструкция этого образца (образец 2-1, табл. 2) привела к количественному изменению двух фракций и появлению новой высокомолекулярной фракции с максимальной молекулярной массой Ммакс равной 209 × 103, в количестве 0.4%. Это свидетельствует о том, что наряду с деструкцией, вероятно, происходит образование разветвленных макромолекул с большей ММ. Более глубокая термоокислительная деструкция указанного образца (табл. 2, образец 2-2), как и следовало ожидать, привела к полному исчезновению высокомолекулярной фракции, уменьшению полидисперсности и величины Ммакс основной фракции при одновременном увеличении ее количества.
Рис. 1.
Зависимость приведенной вязкости растворов хитозана 3-1 (табл. 2), выдержанного после деструкции в течение 4 суток при комнатной температуре (а) и растворимого хитозана 2 (табл. 1 и 2) (б) от концентрации полимера в одинаковых диапазонах при изоионном разбавлении.

Рис. 2.
ИК-фурье-спектры образцов хитозана 2, 3 и 4 (табл. 1). Образец 2а отличается более высокой степенью дисперсности.
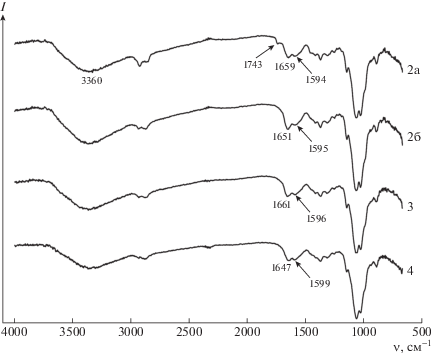
Рис. 3.
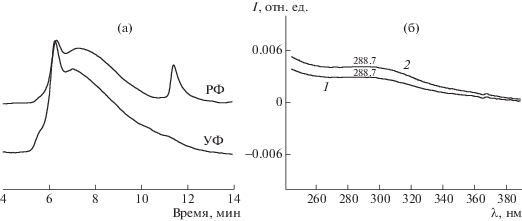
РФ- и УФ-хроматограммы при 240 нм (а) и УФ-спектры (б) в максимуме высокомолекулярной с временем выхода 6,4 мин (1) и основной фракции с временем выхода 7.4 мин (2) образца хитозана 3-1 после первого этапа деструкции.
Деструкция исходных образцов 3 и 4 (табл. 1) с большим содержанием нерастворимой фракции позволила растворить их в аммиачно-ацетатном буфере и провести ВЭЖХ-исследование деструктированных образцов (табл. 2, образцы 3-1 и 4-1). Важно отметить, что в данном случае также образуется высокомолекулярная фракция, но существенно в большем количестве (21.4 и 12.5%) по сравнению с образцом 2. После второго этапа деструкции (табл. 2, образцы 3-2 и 2-2) полностью исчезает высокомолекулярная фракция и образуется двухфракционный состав с близким количественным содержанием. Второй этап деструкции образца 4 примерно в одинаковых условиях не приводит к полному исчезновению высокомолекулярной фракции: остается 3.7% фракции с Ммакс = 98 × 103 (табл. 2, образец 4-2). Это может быть связано с большей плотностью сшивок в нерастворимой фракции образца 4, создающей повышенные стерические затруднения диффузии молекул перекиси водорода.
Отметим, что после первого этапа деструкции до растворимого состояния образец 3-1 (табл. 2) характеризуется наибольшим содержанием (214%) высокомолекулярной фракции (Ммакс = 149). Очевидно, что деструкция нерастворимой (сшитой) фракции хитозана (43.4%, образец 3 из табл. 1) приведет к образованию в первую очередь растворимых разветвленных макромолекул.
Структура макроцепей растворенного хитозана
О структуре растворенных макроцепей хитозана можно получить представление по величине константы Хаггинса K' [25]. Известно, что константа Хаггинса отражает степень взаимодействия молекул растворителя с макромолекулами и зависит от их конформации и проницаемости глобул полимерных цепей. Увеличение K' может означать “ухудшение” термодинамического качества растворителя и/или возрастание числа случайных контактов макромолекул, самоорганизацию их или наличие разветвленных цепей. В нашем случае при неизменности качества растворителя рост величины K' может быть обусловлен повышением степени разветвленности макроцепей. В образце 1 с увеличением времени растворения и с ростом степени деструкции, которой подвергаются прежде всего высокомолекулярные разветвленные макромолекулы, что приводит к снижению их степени разветвленности, значение K' падает от 1.45 до 0.48 (табл. 1). В образцах 3 и 4 величина K' также определяется степенью разветвленности макромолекул, которая выше у образца 4, и K' увеличивается от 0.69 до 1.02.
Состояние в растворе разветвленных макромолекул хитозана, имеющего природу катионного полиэлектролита при изоионном разбавлении может существенно отличаться от состояния линейных цепей. С этой целью было проведено исследование изменения приведенной вязкости соответствующих растворов хитозана в одинаковом концентрационном диапазоне (рис. 1). Как и следовало ожидать, образец хитозана 2, у которого вообще отсутствует высокомолекулярная фракция (табл. 2) имеет прямолинейную зависимость приведенной вязкости от концентрации полимера вплоть до 20-кратного изоионного разбавления (рис. 1б). Для полимера, содержащего разветвленные цепи в количестве ~20% (табл. 2, образец 3-1), при концентрации меньше 0.5 г/дл наблюдаются отклонения от прямолинейной зависимости в сторону больших значений приведенной вязкости (рис. 1а). Данное явление может быть обусловлено уменьшением концентрации низкомолекулярного электролита (ацетата натрия) в объеме разветвленной макромолекулы хитозана, что приводит к увеличению степени диссоциации ионногенных групп в звеньях цепи – ацетатаммонийных. В связи с этим усиливается взаимное отталкивание одноименных (аммонийных) групп ближайших разветвлений и происходит разворачивание цепей, сопровождающегося возрастанием общего объема разветвленной макромолекулы в растворе. Увеличение размера разветвленных макроцепей способствует относительному увеличению времени истечения раствора и к увеличению приведенной вязкости (рис. 1а). При концентрации разветвленных цепей меньше 10% и в разбавленных растворах (меньше 0.5 г/дл) эффект полиэлектролитного набухания при изоионном разбавлении практически не проявляется.
Классический полиэлектролитный эффект наблюдали в работе [26] на примере разбавления водой раствора хитозана в 1%-ной уксусной кислоте. В данном случае увеличение приведенной вязкости происходило при концентрациях хитозана меньше 0.3 г/дл. Обнаруженный нами эффект роста приведенной вязкости при изоионном разбавлении согласуется с современной теорией конформации заряженных макромолекул [27]. Теоретически предсказан и экспериментально подтвержден эффект уменьшения величины относительного сжатия полииона как функции ионной силы раствора при увеличении степени разветвления. При постоянной ионной силе раствора разветвленная макромолекула тем меньше сжимается, чем больше степень ее разветвления.
В ИК-фурье-спектрах (рис. 2) исходных образцов хитозана 2, 3 и 4 (табл. 1) присутствует широкая полоса поглощения при 3400–3300 см–1, соответствующая валентным колебаниям гидроксильных групп νОН в молекуле хитозана. Наиболее интересной с точки зрения оценки химической структуры хитозана является область 1500–1800 см–1. В спектрах всех образцов видны две полосы поглощения: 1647–1661 см–1, ответственная за валентные колебания связи νC=O амидной группы (так называемая амидная полоса I), и полоса поглощения с максимумом 1595–1599 см–1, соответствующая деформационным колебаниям связи δN–H в аминогруппах. Анализ спектров (рис. 2) показал, что величина отношения интенсивностей этих двух полос на всех спектрах разная. По литературным данным [28, 29], в результате деацетилирования хитина амидная полоса поглощения уменьшается, а интенсивность полосы поглощения аминогрупп увеличивается. Однако такой упрощенный подход может дать искаженные результаты по степени деацетилирования хитина из-за суперпозиции в области 1550–1680 см–1 полосы группы NН2 и амидной полосы II (1560 см–1). Кроме того, полоса 1640 см–1 может быть обусловлена деформационными колебаниями групп ОН макромолекул полисахарида и воды, присутствующей в образцах. Отмеченные особенности не позволяют оценивать изменения величины степени деацетилирования по ИК-фурье-спектрам [30].
Методом МНПВО регистрируются спектры отражения с поверхности частиц образца, проникая вглубь лишь на половину длины волны падающего излучения (<7 мкм), поэтому возможны некоторые отличия спектров и соответственно химической структуры в более глубоких и приповерхностных слоях полимерных частиц из-за контакта последних с кислородом воздуха при длительном хранении образцов. Например, в ИК-фурье-спектрах образца 2 в отличие от образцов 3 и 4, содержащих больше 40% нерастворимой фракции, для высокодисперсных частиц (рис. 2, спектр 2а) обнаружена полоса поглощения 1743 см–1, относящаяся к валентным колебаниям альдегидных и/или карбоксильных групп С=О. Образование этих групп связано с окислительными реакциями, происходящими прежде всего в доступных поверхностных слоях в процессе длительного хранения под действием кислорода воздуха. Таким образом, исследования методом ИК-спектроскопии показывают, что окислительному воздействию кислородом воздуха подвергаются все образцы хитозана, а образование нерастворимых фракций в образцах 3 и 4 обусловлено только действием молекул перекиси водорода, адсорбированных в объеме частиц полимере и не удаленных в процессе сушки.
Анализ УФ- и РФ-хроматограмм (рис. 3а и 4а) образцов хитозана 3-1 и 4-2 (табл. 2) позволил выявить некоторые особенности окислительной деструкции хитозана под действием перекиси водорода. На УФ-хроматограммах при 240 нм отсутствует пик низкомолекулярной фракции в отличие от РФ-хроматограмм (время выхода 11.5 мин), что означает отсутствие в низкомолекулярной фракции карбонильных (или карбоксильных) групп, имеющих характерное поглощение в области 220–240 нм. Следовательно, с учетом данных ВЭЖХ эта фракция может представлять собой мономерное звено хитозана – α-D-глюкозамин – моносахарид в циклической форме, не имеющий поглощения в указанной УФ-области.
Симбатность УФ-спектров (рис. 3б) деструктированного образца 3-1 во всем измеренном диапазоне высокомолекулярной (время выхода 6.4 мин, Ммакс = 149 × 103) и основной фракции (время выхода 7.4 мин, Ммакс = 42.3 × 103) (рис. 3а) свидетельствует об одной и той же природе полимера. Различие фракций по молекулярным массам (в ~3.5 раза) обусловлено, по-видимому, степенью разветвленности макромолекул. Существенно, что, как для высокомолекулярной, так и для основной фракции характерно наличие слабого поглощения с максимумом 288.7 нм (рис. 3б). Это поглощение может быть связано с кетонной группой, аналогичной циклогексанону с максимумом поглощения при 285 нм.
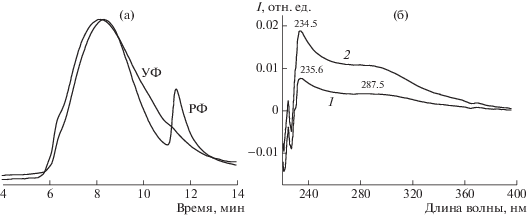
Более глубокая окислительная деструкция приводит к увеличению интенсивности поглощения в УФ-области спектров (рис. 4б). Полоса поглощения ~235 нм характерна как для высокомолекулярной фракции (Ммакс = 98 × 103, содержание 3.7%), так и для основной (Ммакс = 9.5 × 103, содержание 82.5% (табл. 2, образец 4-2), а различие интенсивности в областях ~235 и ~287.5 нм обусловлено разным количественным содержанием этих фракций. Полоса поглощения ~235 нм отражает наличие окисленных групп у шестого атома углерода мономерного цикла хитозана при глубокой степени окисления с образованием альдегидной и/или карбоксильной групп (табл. 2, образец 4-2; рис. 4б).
Рис. 4.
РФ- и УФ-хроматограммы при 240 нм (а) и УФ-спектры (б) в максимуме высокомолекулярной с временем выхода 6.4 мин (1) и основной фракции с временем выхода 8.0 мин (2) образца хитозана 4-2 после второго этапа деструкции.
Известно, что при гетерогенной деструкции хитозана в мягких условиях [19] при получении полимера с Мη = (25–30) × 103 образуется не больше 3% глюкозамина. Однако в настоящей работе установлено, при условии удаления перекисных групп из объема набухших частиц свежеполученного полимера путем лиофильной сушки (табл. 2, образец 2) в результате окислительной деструкции в поверхностных слоях частичек хитозана в течение длительного времени образуется 16% глюкозамина и полимер остается растворимым в подкисленных водных средах. Данный факт свидетельствует о разрыве гликозидной связи концевых звеньев с образованием глюкозамина. При наличии в исходном полимере высокомолекулярной фракции концентрация глюкозамина увеличивается в процессе гомогенной окислительной деструкции (табл. 2, образец 4-2). Это может быть связано с тем, что в растворе наряду с разрывом связей между серединными звеньями цепи происходит и окислительное отщепление мономерного звена – глюкозамина. Отметим, что глюкозамин, как моносахарид, может участвовать в восстановлении азометинов с образованием полимерных вторичных аминов, представляющих собой по существу разветвление цепи. Ниже представлена схема образования глюкозамина.
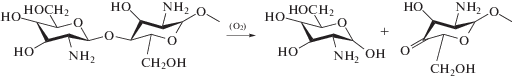
В макроцепи каждое последующее звено развернуто на 180° по сравнению с предшествующим [21]. Наибольшей стерической доступностью для активных форм кислорода обладает атом водорода у четвертого атома углерода последующего звена. Отметим, что стерические затруднения появляются в случае связи со звеном N-ацетилглюкозамина. Поэтому ни при гетерогенном окислении кислородом воздуха в приповерхностных слоях частиц полимера, ни при гомогенном окислении под действием перекиси водорода N-ацетилглюкозамин не образуется, о чем свидетельствует отсутствие поглощения группы С=О на УФ-хроматограммах низкомолекулярной фракции.
Природа сшивок нерастворимого хитозана
Процесс окислительной деструкции, сопровождающейся реакциями сшивки, подтверждается исследованиями методом ЯМР 13С (рис. 5). Спектр хитина на рис. 5 соответствует спектрам α-хитина, представленным в литературе [31, 32]. В спектре ЯМР 13С растворимого исходного хитозана 2 наблюдаются существенные отличия от спектра хитина у атомов углерода С1, С4 и С6 в цикле сахарида. В спектре хитозана наблюдается уширение линий этих атомов углерода, а также слияние сигналов С3 и С5. Максимумы сигналов С1 и С2 смещаются в сторону слабых полей в среднем на ~1.3 и 2.0 м.д. соответственно (табл. 3). Наиболее существенные изменения происходят у атома С4, сигнал которого значительно уширяется по сравнению с хитином. Вместо одного сигнала 83.6 м. д. у хитина в растворимом образце хитозана (рис. 5, спектр 2) появляется набор перекрывающихся линий в диапазоне 80–88 м.д. Учитывая чувствительность атомов углерода в спектрах ЯМР 13С к заместителям, можно предположить, что вследствие образования азометиновой связи с последующим ее химическим восстановлением к атому С4 присоединены только мономер и/или низкомолекулярные олигомеры, что существенно не ухудшает растворимость хитозана.
Рис. 5.
Спектры ЯМР 13C хитина (1), образца хитозана 2 (табл. 1 и 2) (2) и нерастворимой фракции хитозана, выделенной из образца 3 (табл. 1) (3).
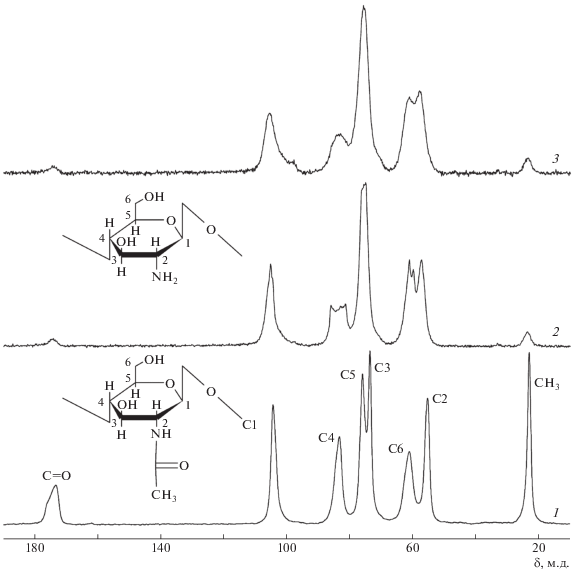
Таблица 3.
Химические сдвиги ЯМР 13C хитина и хитозана
| Образец | Химические сдвиги, м.д. | |||||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | C=O | CH3 | |
| 1 (хитин) | 104.2 | 55.4 | 73.6 | 83.6 | 75.9 | 61.2 | 174.1 | 23.0 |
| 2 | 105.2 | 57.3 | 75.7* | 83.6 | 75.7* | 61.2 | 174.2 | 23.6 |
| 3 | 105.7 | 57.8 | 75.7* | 83.5 | 75.7* | 61.2 | 174.2 | 23.6 |
Примечание. Номера образцов соответствуют номерам спектров на рис. 5.
В случае присоединения концевой группы С4=О, образующейся [17] в результате окислительного разрыва 1,4-гликозидной связи, к серединной аминогруппе длинных цепей появляются разветвления и это приводит к еще большему уширению сигнала атома С4 (рис. 5, спектр 3), который проявляется в виде одного пика. В этом случае возникают длинноцепочечные разветвления повышенной концентрации в единице объема вплоть до формирования нескольких узлов связи на одной цепи, что и обусловливает нерастворимость полимера (нерастворимая часть образца).
При гетерогенной термоокислительной деструкции хитозана степень деацетилирования практически не изменяется [19]. На примере сахарозы и аденозин-5′-монофосфата показано, что принципиальных ограничений использования интегральных интенсивностей линий в спектрах ЯМР 13С не существует [33]. Применимость количественной интерпретации спектров ЯМР 13С проверялась на хитине по соотношению интенсивностей линий кольца сахарида и группы CH3. Это соотношение с хорошей точностью соответствовало 6 : 1. Интенсивность линии C=O занижена по сравнению с CH3 из-за различия динамики кросс-поляризации данных групп [34]. Скорость кросс-поляризации зависит от величины диполь-дипольного взаимодействия между ядрами 1H и 13C, которое зависит от расстояния ~1/r3, и количества протонов в ближайшем окружении. Однако отношение интенсивностей указанных групп можно использовать для определения содержания групп C=O в образцах хитозана. Принимаем, что аминогруппы в хитине отсутствуют, поэтому для хитина характерно С=О : СН3 = 0.47. При условии отсутствия образования групп С=О в процессе деструкции отношение интегральных интенсивностей С=О : СН3 для хитозана со степенью деацетилирования 80% должно быть около 0.094%. Измерения показывают, что в хитозане образуется существенно больше групп С=О, причем как в растворимом полимере, так и в сшитом. Этот факт свидетельствует о том, что группы С=О преимущественно находятся на поверхности нерастворимых частиц хитозана. Отметим, что в сшитом хитозане относительное содержание групп С=О коррелирует с величиной поверхности частиц в единице объема: при уменьшении диаметра частиц в ~4 раза (поверхность частиц в единице объема увеличивается также в 4 раза) относительное содержание групп С=О возрастает на ~35%. Данный факт свидетельствует о том, что окислению подвержены прежде всего поверхностные макромолекулы в частицах хитозана при контакте с кислородом воздуха.
Детальное рассмотрение показывает, что во всех спектрах хитозана имеется сигнал ~98 м.д. (в хитине проявляется слабо). Это правое “плечо” сигнала атома углерода С1 может быть обусловлено раскрытой формой концевой группы, образующейся в результате циклооксотаутомерии, хорошо известной для моносахаридов с открытой формой в количестве не больше 0.02% [35]. Отметим, что этот сигнал более выражен в образцах хитозана с большим содержанием нерастворимой фракции, особенно в образцах, максимально освобожденных от растворимой фракции (рис. 5, спектр 3). Это может быть связано с большей стабильностью концевых раскрытых форм в сильно разветвленном и частично сшитом хитозане, характеризующемся пониженной молекулярной подвижностью.
Полученные данные свидетельствуют о том, что деструкция хитозана по радикальному механизму сопровождается процессами сшивки. Известно, что при окислительной деструкции каучука СКД в органической среде образуется высокомолекулярная фракция по механизму рекомбинации макромолекулярных радикалов [36]. В нашем случае в водной среде в процессе термоокислительной деструкции наиболее вероятна рекомбинация макрорадикала с очень подвижным гидроксильным радикалом.
Реакции, приводящие к сшивке макромолекул хитозана, можно представить в виде схемы:
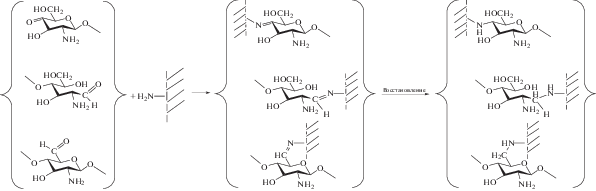
Наиболее вероятны реакции верхнего ряда в схеме, так как кетонные группы [17] образуются в каждом акте разрыва 1,4-гликозидной связи полимерной цепи, а их концентрация зависит от интенсивности процесса окислительной деструкции. Второй путь реакции (средний ряд) обусловлен наличием концевых восстанавливающих групп и степенью их циклооксатаутомерии. Роль концевых групп возрастает с уменьшением длины цепи. Третий путь реакций характерен в основном для глубоких степеней окислительной деструкции, когда происходит окисление у шестого атома углерода сахаридного цикла. Химическое восстановление азометинов возможно не только под действием низкомолекулярных продуктов деструкции [37], но и с участием моносахарида – глюкозамина.
ЗАКЛЮЧЕНИЕ
Окислительная деструкция с сопутствующими реакциями сшивки может происходить как в растворе, так и в конденсированном состоянии, чему способствует определенная степень влажности при длительном хранении с доступом кислорода воздуха и/или наличие активных веществ в объеме частиц полимера. В качестве активных веществ могут быть белковые, хитин-хитозан-белковые структуры (преимущественно при молекулярной массе хитозана М ≥ 400 × 103), а также не удаленные адсорбированные молекулы перекиси водорода.
Обнаружено проявление полиэлектролитного эффекта разветвленных макроцепей при изоионном разбавлении.
Полученные данные позволяют предложить наиболее вероятный, по мнению авторов, механизм образования глюкозамина в процессе окисления под действием кислорода воздуха в конденсированном состоянии и/или в условиях гомогенного или гетерогенного окисления под действием перекиси водорода.
Установлены основные реакции сшивки макромолекул хитозана при окислительной деструкции или при длительном хранении с доступом кислорода воздуха.
Показано, что тщательная очистка химически деструктированного до задаваемых молекулярных масс хитозана, обязательная лиофильная сушка и хранение без свободного доступа кислорода воздуха способствуют длительному сохранению растворимости хитозана. Соблюдение этих условий важно для применения хитозана в различных областях, особенно в пищевой промышленности, косметике и медицине.
Работа выполнена с использованием оборудования Аналитического центра коллективного пользования Института проблем химической физики РАН по теме государственного задания (номер государственной регистрации 0121056328).
Список литературы
Хитозан / Под редакцией К.Г. Скрябина, С.Н. Михайлова, В.П. Варламова. М.: Центр “Биоинженерия” РАН, 2013.
Скрябин К.Г., Тихонович И.А., Варламов В.П. // Наука в России. 2014. № 6. С. 4.
Ильгамов М.А. // Изв. Уфимского научного центра РАН. 2016. Т. 1. № 3. С. 5.
Комаров Б.А., Розенберг Б.А., Лебедева О.В., Колесова Л.И., Волошинова Р.З., Машинская Г.П., Гамзазаде А.И., Рогожин С.В. // А.с. 249716.1985.
Булатов А.В., Летучий Я.А., Хидекель М.Л., Комаров Б.А., Розенберг Б.А., Гамзазаде А.И., Рогожин С.В. // А.с. 1235042 от 10.11.1984.
Wu B., Zhang Y., Kuang Y., Yu Y., Zhang X., Chen J. // Chem. Asian J. 2012. V. 7. P. 190.
Khatami S.Y., Hejri Z.J. // Appl. Environ Biol. Sci. 2015. V. 4. № 11S. P. 150.
Patil R., Pande V., Sonawane R. // Adv. Pharm. Bull. 2015. V. 5. № 2. P. 169.
Pina S., Oliveria J.V., Reis R.L. // Adv. Mater. 2015. V. 27. P. 1143.
Sambudi N.S., Sathyamurthy M., Lee G.M., Park S.B. // Compos. Sci. Techn. 2016. V. 106. P. 76.
Terzopoulou Z., Kyzas G.Z., Bikiaris D.N. // Materials. 2015. V. 8. P. 652.
Liu C., Zhang J., Yifeng E., Yue J. // Electron. J. Biotechn. 2014. V. 17. P. 183.
ShiehY.-T., Jiavg H-F. J. // Electroanal. Chem. 2015. V. 736. P. 132.
Немцев С.В., Ильина А.В., Шинкарев С.М., Албулов А.И., Варламов В.П. // Биотехнология. 2001. № 6. С. 37.
Хабаров В.Б., Пронин А.Я., Самуйленко А.Я., Буряк А.К., Гринь А.В. // Докл. РАСХН. 2009. № 4. С. 58.
Shih-Chang Hsu, Trong-Ming Don, Wen-Yen Chiu // Polym. Degrad. Stab. 2002. № 75. P. 73.
Федосеева Е.Н., Смирнова Л.А., Сорокина М.А., Пастухов М.О. // Журнал прикладной химии. 2006. Т. 79. Вып. 5. С. 857–861.
Галиаскарова Г.Г. Автореф. дис. … канд. хим. наук. Уфа: ИОХ РАН, 1997.
Комаров Б.А., Албулов А.И. // Пат. Россия 2215749. 2001.
Gamzazade A.I., Shlimak V.M., Sklyar A.M., Shtykova E.V., Rogoschin S.V., Pavlova S.A. // Acta Polymerica. 1985. V. 36. P. 420.
Левитин С.В. Автореф. дис. … канд. техн. наук. М.: Моск. гос. ун-т дизайна и технологии, 2015.
Бойко И.С., Подколодная О.А., Лысачок С.Г., Шмаков С.Л. // Изв. Сарат ун-та. Химия. Биология. Экология. 2015. Т. 15. №. 4. С. 21.
Гамзазаде А.И., Скляр А.М., Павлова С.-С.А., Рогожин С.В. // Высокомолек. соед. А. 1981. Т. 23. № 3. С. 594.
Курченко В.П., Буга С.В., Петрашкевич Н.В., Буткевич Т.В., Ветошкин А.А., Демченков Е.Л., Лодыгин А.Д., Зуева О.Ю., Варламов В.П., Бородин О.И. Тр. Башкирского гос. ун-та. 2016. Т. 11. Ч. 1. С. 110.
Рафиков С.Р., Павлова С.А., Твердохлебова И.И. Методы определения молекулярных весов и полидисперсности высокомолекулярных соединений. М: АН СССР, 1963. С. 7.
Кулиш Е.И., Туктарова И.Ф., Чернова В.В., Абзальдинов Х.С., Заиков Г.Е. Вестн. Казанского технол. ун-та. 2013. Т. 16. № 14. С. 140.
Борисов О.В. Дис. … д-ра. физ.-мат. наук. СПб.: ИВС РАН, 2014.
Paulino A.T., Simionato J.I., Garcia J.C., Nozaki J. // Carbohydr. Polymers. 2006. V. 64. P. 98.
Brugnerotto J., Lizardi J., Goycoolea F.M., ArguEelles-Monal W., Desbrieeres J., Rinaudo M. // Polymer. 2001. V. 42. P. 3569.
Кучина Ю.А., Долгопятова Н.В., Новиков В.Ю., Сагайдачный В.А., Морозов Н.Н. // Вестн. МГТУ. 2012. Т. 15. № 1. С. 107.
Tanner S.F., Chanzy H., Vincendon M., Roux J.C., Gaill F. // Macromolecules. 1990. V. 23. P. 3576.
Kono H. // Biopolymers. 2004. V. 75. P. 255.
Шашков А.С., Чижов О.С. // Биоорган. химия. Обзорные статьи. 1976. Т. 2. № 4. С. 469.
Heux L., Brugnerotto J., Desbrieres J., Versali M.-F., Rinaudo M. // Biomacromolecules. 2000. V. 1. P. 746.
Физер Л., Физер М. Органическая химия. М.: Химия, 1970.
Попов А.П. Дис. … канд. техн. наук. Воронеж: Воронежский гос. ун-т инженерных технологий, 2015.
Помогайло А.Д., Джардималиева Г.И. Металлополимерные гибридные нанокомпозиты. М.: Наука, 2015. С. 71.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)