

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 5, стр. 323-334
ОКИСЛИТЕЛЬНАЯ ПОЛИМЕРИЗАЦИЯ 3,6-ФЕНИЛЕНДИАМИНО-2,5-ДИХЛОРБЕНЗОХИНОНА
А. В. Орлов a, С. Г. Киселева a, *, Г. Н. Бондаренко a, Г. П. Карпачева a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: skisel@ips.ac.ru
Поступила в редакцию 19.04.2019
После доработки 24.05.2019
Принята к публикации 03.06.2019
Аннотация
Впервые изучена окислительная полимеризация 3,6-фенилендиамино-2,5-дихлорбензохинона. Показано, что мономер обладает высокой реакционной способностью. Его скорость полимеризации значительно выше, чем у анилина и 3,6-дианилин-2,5-дихлорбензохинона, при этом полностью отсутствует индукционный период окисления. Исследованы кинетические особенности окислительной полимеризации 3,6-фенилендиамино-2,5-дихлорбензохинона методом измерения редокс-потенциала реакционной смеси от времени. Показано, что на кинетических кривых полимеризации 3,6-фенилендиамино-2,5-дихлорбензохинона отсутствует типичный для полимеризации анилина S-образный вид. Представлен механизм реакции полимеризации, протекающий через ряд последовательных таутомерных превращений. Изучена структура полимеров методами ИК-фурье- и рентгенофотоэлектронной спектроскопии. Установлено, что в ходе реакции окислительной полимеризации протекают процессы циклизации с образованием феназиновых и феноксазиновых циклов, приводящие к формированию полисопряженного полимера лестничной структуры. Изучено влияние условий реакции – концентрации реагентов, их соотношения и рН реакционной среды на ход процесса.
ВВЕДЕНИЕ
Процесс окислительной полимеризации анилина – предмет детально изученный, что дает возможность получать полимеры с комплексом ценных свойств, варьируя условия их синтеза [1–4]. Использование таких полимеров в самых разных областях от микроэлектроники до создания газоразделительных мембран, сорбентов, химических и биосенсоров, а также в качестве антистатических, электродных, электрохромных материалов позволяет в значительной мере повысить эффективность приборов созданных на их основе [4–7]. Полимеры анилина отличает высокая устойчивость в процессах эксплуатации по сравнению с другими полисопряженными полимерами и простое регулирование их электрофизических характеристик путем обратимого изменения их кислотно-основного состояния [8, 9].
Анализ имеющихся в научной литературе данных показывает, что исследования в области окислительной полимеризации ароматических аминов ограничиваются, главным образом, полимеризацией анилина и некоторых его производных [10, 11]. Попытки улучшить физико-химические характеристики полианилина при помощи использования в процессах окислительной полимеризации различных N- и C-замещенных анилинов не привели к заметному успеху. Ароматические амины с электроноакцепторными заместителями (сульфо-, галоид-, нитрогруппы) оказались мало активны и ингибировали реакцию окислительной полимеризации [12–15]. Мономеры с электронодонорными заместителями (алкил-, алкокси-, аминогруппы) хоть и полимеризовались, однако приводили к продуктам с низкими молекулярно-массовыми и электрофизическими характеристиками за счет уменьшения межцепного взаимодействия и снижения степени упорядоченности цепей [16–18]. Некоторый успех достигнут при исследовании сополимеризации анилина и его производных. Так, сополимер анилина и метаниловой кислоты оказался электроактивным в более широком диапазоне рН рабочих растворов по сравнению с полианилином [19, 20].
Поэтому разработка новых подходов к синтезу электроактивных полисопряженных полимеров, направленных на поиск новых мономеров и способов их полимеризации, позволяющих получить материалы с характеристиками, значительно превышающими, чем у полианилина, сохраняет свою актуальность как в научном, так и в прикладном аспектах.
В последние годы удалось вовлечь в окислительную полимеризацию новые мономеры, в частности дифениламин и дифениламин-2-карбоновую кислоту, фенотиазин и феноксазин, поли-3-амино-7-метиламино-2-метилфеназина с получением электроактивных термостойких полимеров [21–26].
В работе [27] впервые показано, что 3,6-дианилин-2,5-дихлорбензохинон (ДАДХБ), представляющий собой ароматический амин, содержащий в качестве N-заместителя хлоранильную группу, не только способен вступать в реакцию окислительной полимеризации в водных растворах кислот, но и отличается более высокой реакционной способностью по сравнению с анилином. Установлено, что ДАДХБ в ходе окислительной полимеризации образует линейную полианилиновую цепь, построенную по принципу “голова к хвосту”, с хлоранильными заместителями по атому азота. Он также способен вступать в реакцию сополимеризации с анилином с образованием сополимеров статистической структуры. Электропроводность полимеров ДАДХБ сравнима с электропроводностью полианилина. Полученные на его основе нанокомпозиты с углеродными наночастицами (много-, одностенные углеродные нанотрубки) характеризуются высокими значениями электрохимической емкости и стабильностью работы при длительном циклировании в протонных и апротонных электролитах [28–30]. Стабильность работы и высокие емкостные параметры при циклировании в органическом электролите показали электродные материалы на основе нанокомпозитов, полученных в условиях in situ окислительной полимеризации 3,6-ди(аминофениламино)-2,5-дихлор-1,4-бензохинона (ДФХБ) в присутствии одностенных углеродных нанотрубок [31]. Однако сам процесс формирования полимерной цепи и ее структура исследованы не были.
В настоящей работе впервые изучена окислительная полимеризация ДФХБ, являющегося продуктом взаимодействия хлоранила с двумя молекулами м-фенилендиамина (ФДА). Исследованы кинетические особенности полимеризации в зависимости от условий синтеза. Показано формирование в условиях окислительной полимеризации полисопряженного полимера лестничной структуры.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали фенилендиамин 99 + % (“Acros Organics”, Бельгия) в чистом виде. Хлоранил (х.ч.) перекристаллизовывали из диоксана, пероксидисульфат аммония – из воды при Т = 40°С. Диоксан (х.ч.) перегоняли при температуре 374 К. Дистиллированную воду применяли дважды. Растворы HCl (ч.д.а.), NH4OH (о.с.ч.) использовали без дополнительной очистки.
ДФХБ получали реакцией алкилирования ФДА хлоранилом. К раствору ФДА 0.1 моля в 50 мл диоксана в течение одного часа прикапывали раствор 0.05 моля хлоранила в 200 мл диоксана при комнатной температуре и постоянном перемешивании. После смешения реагентов реакционный раствор еще в течение 3 ч перемешивали при комнатной температуре. Затем реакционную массу фильтровали, выделившийся коричневый осадок промывали этанолом и сушили под вакуумом при комнатной температуре до постоянной массы. Выход ДФХБ составил 78%. Структура полученного ДФХБ подтверждена методами элементного анализа (С – 55.2%, Н – 3.6%, N – 14.6%, О – 8.2%), ИК-фурье- и рентгенофотоэлектронной спектроскопией (РФЭС). Мономер растворим в ДМФА, ДМСО, N-МП и в водных растворах кислот. Мономер использовали в процессах окислительной полимеризации без дополнительной нейтрализации, поскольку реакция проводится в водных растворах соляной кислоты.
В рамках кинетических исследований условия полимеризации соответствовали следующим параметрам: [ДФХБ] = 0.03 моль/л, [Пероксидисульфат аммония]/[ДФХБ] = 1.25, [HCl] = 0.5 моль/л; Т = 20°С (стандартные условия). Мономер растворяли в водном растворе кислоты и одномоментно приливали раствор пероксидисульфата аммония. По окончании реакции раствор фильтровали, промывали 0.5 моль/л раствором HCl, водой и сушили под вакуумом при комнатной температуре до постоянной массы. Для дедопирования использовали 0.5 моль/л раствор NH4OH.
Исследование зависимостей электродвижущей силы (ЭДС) от времени в ходе окислительной полимеризации ДФХБ проводили с помощью 4-канального иономера “Эксперт-001-3(0.4)” (Россия) с точностью измерений ЭДС = = ±1.5 мВ с использованием программного обеспечения к приборам “Эксперт v2” (05/29/2015). В качестве электродов применяли редоксметрический электрод (ЭРП-105) (Россия). Измерения ЭДС осуществляли в термостатированных реакционных растворах при постоянном перемешивании. Раствор окислителя приливали одномоментно, и время его добавления отмечали как начало реакции.
Исследования методом РФЭС выполняли с использованием рентгеновского фотоэлектронного спектрофотометра “PHI5500VersaProbeII”. Источник возбуждения – монохроматическое Al Kα излучение (hν = 1486.6 эВ), мощность – 50 Вт, диаметр 200 мкм. Значения атомной концентрации определяли по обзорным спектрам (линии C1s, N1s, O1s, Cl2p) методом факторов относительной элементной чувствительности. Энергию связей Есв фотоэлектронных линий (C1s, N1s, O1s, Cl2p) находили по спектрам высокого разрешения, снятым при энергии пропускания анализатора 23.5 эВ и плотности сбора данных 0.1 эВ/шаг. Аппроксимацию спектров выполняли нелинейным методом наименьших квадратов с использованием функции Гаусса–Лоренца. Калибровку шкалы энергии связи Есв проводили по Au4f – 84.0 эВ и Cu2p3 – 932.6 эВ. Корректировку шкалы Есв осуществляли по энергии связи спектра N1s (399.9 эВ). Погрешность определения Есв составляет ±0.2 эВ, погрешность определения интенсивности пиков при аппроксимации ±5%.
ИК-спектры регистрировали в режиме НПВО на ИК-микроскопе “HYPERION-200”, сопряженном с ИК-фурье-спектрометром IFS 66 V/s “Bruker” (кристалл Ge, скан. 100, разрешение 2 см–1, диапазон 600–4000 см–1). Оптическая плотность D = lgI0/I.
Электропроводность измеряли четырехзондовым методом на таблетках, отпрессованных из порошка полимера при давлении 80 кгс/см2.
Электронно-микроскопические микрофотографии образцов полимеров получали на сканирующем электронном микроскопе с термоэмисcионным источником NVision 40 (“Carl Zeiss”, Германия). Для исследования образцы наносили в виде порошка на проводящий углеродный скотч. Ускоряющее напряжение электронной пушки составляло 1–20 кВ. Изображения получали во вторичных электронах при увеличениях до 800000× и регистрировали в оцифрованном виде на ЭВМ.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
ДФХБ отличается от изученного раннее ДАДХБ [27] тем, что содержит в мета-положениях анилиновых колец дополнительные аминогруппы. Следовало ожидать их значительного влияния на распределение электронной плотности в системе и, как следствие, на изменение реакционной способности мономера.
Кинетические особенности процесса окислительной полимеризации ДФХБ исследованы с помощью измерения зависимости редокс-потенциала реакционного раствора от времени. Потенциометрические кривые имеют простой и наглядный вид и соответствуют природе происходящих при окислительной полимеризации процессов. Кроме того, они наилучшим образом подходят для изучения процессов в сильно окрашенных гетерофазных растворах низкой концентрации [27].
На рис. 1 представлены зависимости изменения редокс-потенциала от времени для полимеризации ДФХБ при различной концентрации мономера. Для сравнения приведены кривые 4 и 5 полимеризации для ДАДХБ и анилина соответственно. Видно, что при одной и той же концентрации мономеров (кривые 2, 4, 5) скорость реакции окисления ДФХБ намного выше, чем у анилина и ДАДХБ. Если кинетические кривые полимеризации анилина и ДАДХБ похожи, то в случае полимеризации ДФХБ наблюдается практически полное отсутствие индукционного периода реакции. Кинетические кривые не имеют S-образного вида, типичного для окислительной полимеризации анилина.
Рис. 1.
Зависимость изменения редокс-потенциала от времени при окислении [ДФХБ] = 0.05 (1), 0.03 (2) и 0.01 моль/л (3); [ДАДХБ] = 0.03 моль/л (4); [анилин] = 0.03 моль/л (5). Условия стандартные.

Объяснить такой характер полимеризации можно, исходя из структуры мономера, представляющего собой систему хиноидного и ароматических колец, соединенных через группы NH. Известно, что в таких соединениях таутомерная перегруппировка двойных связей приводит к образованию симметричной хинодииминной структуры [32, 33]. В присутствии кислоты она протонируется с образованием сначала дикатиона, а затем хиноиминное кольцо перестраивается в ароматическое с образованием биполярона. Образование парамагнитных центров проходит без участия окислителя и регистрируется в ЭПР-спектрах [34]. Показаны возможные таутомерные перегруппировки для ДФАХБ:
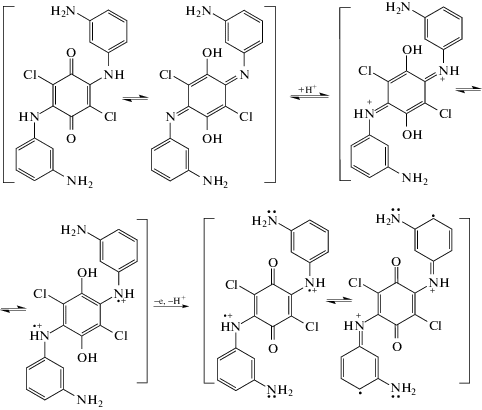
Следует отметить, что образование катион-радикальных центров инициируется без участия окислителя уже в ходе перестройки системы двойных связей хинонного и хинодииминного колец. Это значительно облегчает первоначальное накопление реакционных центров. А именно оно и определяет индукционный период реакции окислительной полимеризации [10, 35]. На кинетических кривых полимеризации наблюдается либо значительное сокращение индукционного периода, как в случае ДАДХБ [27], либо его полное отсутствие, как в случае ДФХБ. Возможность таутомерной перегруппировки хинонного кольца в хинодииминное объясняет не только сам факт полимеризации мономера с электроноакцепторным заместителем, но и значительно бóльшую скорость полимеризации по сравнению с анилином.
При добавлении окислителя образовавшаяся в результате таутомерии и протонирования ароматическая система вновь превращается в хинонную [36]. Таким образом, при полимеризации ДФХБ окислитель участвует не в одноэлектронном окислении азота, как это имеет место при окислении анилина, а окисляет гидроксильную группу. Образование хинона завершает процесс формирования реакционных центров полимеризации.
Наличие в молекуле ДФХБ дополнительной аминогруппы в мета-положении с ее неподеленной электронной парой в значительной мере увеличивает электронную плотность в пара-положении анилинового кольца (схема 1). Это приводит к увеличению его нуклеофильности и повышает реакционную способность мономера в процессе роста цепи, протекающей по типу электрофильного замещения. Это подтверждено тем, что в отличие от полимеризации анилина и ДАДХБ [27] на кинетических кривых полимеризации ДФХБ индукционный период реакции полностью отсутствует (рис. 1, кривые 1–3).
Процесс окислительной полимеризации ДФХБ визуально отличается от полимеризации анилина и ДАДХБ. Прежде всего, это отсутствие фиолетовой окраски реакционного раствора на первых этапах синтеза, что свидетельствует о незначительном количестве окисленных структур. По окончании же реакции раствор не приобретает темно-зеленой окраски, характерной для эмеральдиновой формы полианилина. Реакционная смесь в ходе синтеза быстро темнеет и выпадает мелкодисперсный порошок черного цвета, указывая на то, что окислительная полимеризация ДФХБ протекает с образованием продуктов, значительно отличающихся по структуре как от полианилина, так и от полимеров ДАДХБ. Это подтверждают данные ИК-фурье- и рентгенофотоэлектронной спектроскопии.
ИК-спектр мономера (рис. 2, спектр 1) полностью соответствует структуре дизамещенного хлоранила. Присутствие хлора в замещенном двумя фенилендиаминовыми заместителями хлоранильном ядре, хорошо подтверждают интенсивные полосы 1112 и 873 см–1 от связей С–Cl в хиноидном кольце [37, 38]. Полосы от связей С=О в спектре хлоранила при 1680–1677 см–1 в спектре мономера смещены в длинноволновую область и вместе с полосами от валентных колебаний ароматических заместителей и от деформационных колебаний в аминогруппе дают интенсивную расщепленную полосу 1624 см–1 [39, 40]. От фенилендиаминовых заместителей в спектре мономера появляются полосы от аминогрупп 3216, 3341 см–1 (–NH), 1311 cм–1 (–C–N), 1574 и 1496 см–1 (–C=C(Ph)), а также две полосы 685 и 803 см–1, хорошо сопоставимые с неплоскими деформационными колебаниями –ССН в 1,3-замещенном ароматическом кольце [41–46]. Широкие полосы в области 2000, 2810 и 2585 см–1 характеризуют аммониевый катион и указывают на солевую форму полученного мономера. Так как в процессе синтеза мономера выделяется HCl, то он частично протонирует первичную аминогруппу фенилендиаминового кольца.
Рис. 2.
ИК-спектры мономера ДФХБ (1) и полимера поли-ДФХБ (2), полученного при стандартных условиях после нейтрализации.
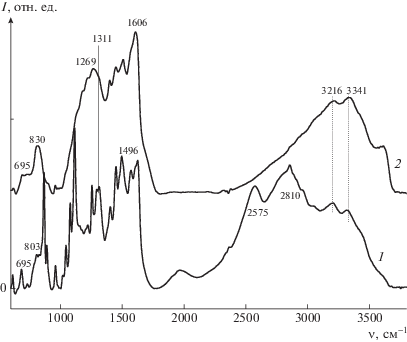
Поскольку полимеры исследовали в дедопированной форме, в ИК-спектре полимера (рис. 2, спектр 2) спектральные признаки аммониевых катионов отсутствуют. Полосы от валентных колебаний связей N–H в аминогруппах (3216, 3341 см–1), так же как и полоса от валентных колебаний связей C–N– 1311 cм–1 в спектре полимера, проявляются при тех же волновых числах, что и в спектре мономера. Меняется только относительная интенсивность этих полос, а полоса 1311 см–1 проявляется как плечо на интенсивной полосе 1269 см–1. Это указывает на то, что какая-то часть фенилендиаминовых групп остается неизменной и после окислительной полимеризации. Полоса 1624 см–1 в спектре полимера полностью исчезает, а вместо нее появляется очень интенсивная полоса 1606 см–1, которая может быть наложением полос от валентных колебаний С=С(Ph), деформационных колебаний H–N–H в аминогруппе, а также полосы от валентных колебаний в сопряженных связях C=О, –C=N-, имеющей обычно очень высокую интенсивность [47]. Спектральным признаком присутствия в структуре полимера феноксазиновых структур может служить очень интенсивная полоса 1269 см–1, относящаяся к асимметричным валентным колебаниям (Рh)С–O–C(Ph) [48]. Отсутствие в спектре полимера интенсивных полос 1112 и 873 см–1 от связей С–Cl, которые проявлялись в спектре мономера, является прямым указанием на резкое уменьшение количества хлора в структуре полимера. Слабые полосы 685 и 803 см–1 от 1,3-замещенных ароматических колец присутствуют в спектре полимера, но их малая интенсивность свидетельствует о значительном уменьшении доли 1,3-замещенных фенильных колец в составе полимера по сравнению с мономером. Новая широкая интенсивная полоса 830 см–1 в спектре полимера является указанием на присутствие большого количества 1,2,4,5-замещенных ароматических колец в полимере, являющихся результатом протекания реакций циклизации [44, 49–51].
Детальный анализ рентгенофотоэлектронных спектров продуктов подтверждают данные ИК-спектроскопии.
Как видно из табл. 1, содержание хлора снижается в 2 раза. Это указывает на то, что половина хлора удаляется в результате замещения аминогруппой с возможным образованием феназиновых колец, о чем свидетельствует детальный анализ параметров аппроксимации спектров высокого разрешения O1s, Cl2p, N1s, C1s, представленных в табл. 2.
Таблица 1.
Данные элементного анализа РФЭС (концентрация, атм. %)
| Образец | С | N | O | Cl | O/C | N/C | Cl/C |
|---|---|---|---|---|---|---|---|
| Мономер | 71.2 | 15.3 | 7.1 | 6.4 | 0.1 | 0.21 | 0.09 |
| Полимер | 72.0 | 16.7 | 8.2 | 3.1 | 0.11 | 0.23 | 0.04 |
Таблица 2.
Параметры аппроксимации спектров высокого разрешения O1s, Cl2p, N1s, C1s (РФЭС)
| Спектр | Мономер | Полимер | ||
|---|---|---|---|---|
| Eсв, эВ | Интенсивность, % | Eсв, эВ | Интенсивность, % | |
| O1s | 531.6 | 85.2 | 531.5 | 49.8 |
| 533.3 | 14.8 | 533.2 | 50.2 | |
| Cl2p | 200.86 | 87 | 200.86 | 94 |
| 197.9 | 11 | 197.4 | 4 | |
| 206.9 (Sat) | 2 | 206.9 (Sat) | 2 | |
| N1s | – | – | 398.6 | 23.1 |
| 399.9 | 86 | 399.9 | 69.9 | |
| 401.65 | 12 | 401.7 | 2 | |
| – | – | 404.1 (Sat) | 2 | |
| 406.0 (Sat) | 2 | 406.0 (Sat) | 3 | |
| C1s | 284.7 | 43 | 284.7 | 32.1 |
| 285.9 | 40.4 | 285.8 | 44.0 | |
| 286.5 | 3 | 286.4 | 16.2 | |
| 288.9 | 10.5 | 288.9 | 5.1 | |
| 291.3 (Sat) | 3 | 291.3 (Sat) | 2.5 | |
В спектре мономера O1s имеются два пика 531.6 и 533.3 эВ с отношением интенсивностей 85 : 15. Первый пик можно отнести к кислороду связи С=О, второй – к связи С–ОН [52, 53]. Это указывает на то, что мономер находится преимущественно в хинонной форме. В полимере эти два пика 531.5 и 533.2 эВ присутствуют в соотношении 50 : 50. Поскольку таутомерия в полимерных структурах невозможна, следует предположить, что в полимере ДФХБ половина кислорода остается в хинонной форме, а оставшаяся часть образует дополнительную связь со вторым углеродным атомом группу С–О–С [54].
В спектрах азота N1s мономера присутствует основной пик, относящийся к азоту, связанному с атомами кольца (399.9 эВ). Малоинтенсивный пик (401.65 эВ) указывает на присутствие небольшого количества (12%) кватернизованного азота N+–Н. В полимере ДФХБ этот пик (401.7 эВ) снижается до уровня фона. Наличие заряженного азота в мономере подтверждают и ИК-спектры. Он остается в мономере в результате синтеза, протекающего с выделением НCl. В спектре N1s полимера ДФХБ появляется плечо в области (пик 398.6 эВ – 23%), которое можно отнести к азоту феназинового цикла [55]. Спектры хлора – это спектры дуплетов 2p3/2–2p1/2 с расщеплением 1.6 эВ, обозначением Есв пика 2p3/2. Основной дублет 200.86 эВ соответствует связям C–Cl. Второй дублет относится к NH$_{2}^{ + }$Cl– (197.4 эВ), и в полимере его интенсивность снижается до 4%.
В спектрах углерода C1s при переходе от мономера к полимеру наблюдается снижение с 43 до 32% интенсивности пика 284.7 эВ, относящегося к ароматическим углеродам, не связанным с азотом, кислородом, хлором. Это уменьшение значительно больше, чем можно было ожидать, исходя из схемы анилиновой полимеризации. Следует предположить, что часть атомов углерода замещается в результате образования гетероциклов, что подтверждается увеличением интенсивности пика 286.5–286.4, относящегося к C=N и C–O–C. Увеличение суммарной интенсивности пиков 285.8 (С–N) и 286.4 эВ с 43 до 60%, и уменьшением доли C=O в 2 раза (288.9 эВ), соответствует росту полициклических структур [55].
В процессе полимеризации боковая аминогруппа, находящаяся в кольце на полианилиновой цепи, оказывается в непосредственной близости от атомов хлора хинонного цикла. Это способствует протеканию реакции нуклеофильного замещения с образованием замещенных гидрофеназиновых колец:

Однако ввиду их неустойчивости, они сразу же окисляются под действием пероксидисульфата. При этом происходит перестройка системы сопряженных двойных связей не только феназинового, но и хинонного колец. Активация кислорода хинонного кольца за счет повышения его электрофильности и увеличение нуклеофильности пара-положения бензольного кольца, расположенного по другую сторону от феназинового цикла, влечет образование феноксазинового цикла ортохиноиминной структуры (схема 3). В результате конечный полисопряженный полимер имеет лестничную структуру, в которой, наряду с феназиновыми звеньями, присутствуют феноксазиновые. При этом наблюдается уменьшение содержания атомов хлора по сравнению с мономером в 2 раза, а соотношение карбонильного кислорода хинона к кислороду эфирной группы в феноксазиновом цикле составляет 50 : 50.
Таким образом, из анализа данных, полученных с помощью ИК-фурье-спектроскопии и РФЭС, можно сделать вывод о том, что в процессе окисления ДФХБ образуется лестничная полимерная структура, в которой, наряду с обычной для полимеризации ароматических аминов полианилиновой цепью, присутствуют феназиновые и феноксазиновые гетероциклические фрагменты:
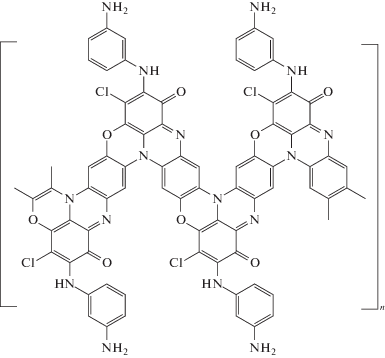
Следует подчеркнуть важную особенность протекания процесса окислительной полимеризации ДФХБ. Несмотря на наличие в структуре мономера двух равноценных реакционноспособных групп, рост цепи проходит с участием только одной. Это может быть обусловлено особенностями механизма реакции окисления. Осуществляемая на начальном этапе полимеризации таутомерная перестройка электронной структуры мономера не приводит к нарушению симметрии молекулы. Но, как только происходит рекомбинация катион-радикала атома азота с пара-положением ядра ароматического амина другой молекулы и начинается рост цепи, симметрия системы нарушается. Таутомерные перегруппировки – хинон–хинодиимин становятся невозможными. Вторая ФДА-группа этой уникальной системы теряет свою активность, превращаясь в пространственно затрудненный амин, в котором присутствует электроноакцепторный заместитель, подавляющий его реакционную способность. Полициклическая природа образующегося полисопряженного полимера препятствует его допированию по типу полианилина с образованием электропроводящей поляронной структуры. Это проявляется в его низкой электропроводности по сравнению с полианилином и ДАДХБ, а также приводит к отсутствию характерных для полианилина структур цветовых переходов синий–зеленый в процессах кислотно-основной обработки. Кроме того, в ИК-спектрах полимера практически отсутствуют полосы, характерные для окисленной формы полианилина.
Отсутствие зеленой или сине-фиолетовой окраски реакционной смеси в процессе синтеза указывает на то, что реакции циклизации протекают непосредственно в ходе роста полимерных цепей, и по окончании реакции полимер уже имеет полициклическую структуру.
Изучено влияние параметров синтеза на скорость реакции и характеристики полученных продуктов. На рис. 3 представлены потенциометрические кривые окислительной полимеризации в зависимости от концентрации ДФХБ. Видно, что с уменьшением концентрации мономера (кривые 1–3) скорость реакции уменьшается. При этом кривые однотипны и сильно отличаются от аналогичных для анилина и ДАДХБ. Нет ни выраженного индукционного периода, ни характерного для автокатализа S-образного вида. Реакция во всех случаях начинается практически сразу после добавления раствора окислителя и заканчивается в течение первых 5–10 мин.
Рис. 3.
Зависимость изменения редокс-потенциала от времени при окислении ДФХБ при соотношении [Пероксидисульфат аммония] : [ДФХБ] = 0.5 (1), 0.75 (2), 1.25 (3) и 3.75 (4). Условия стандартные.
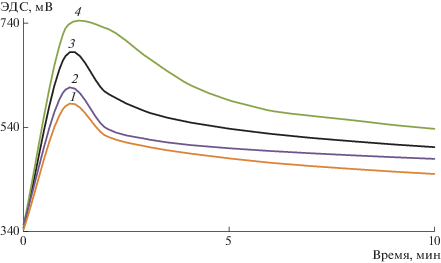
Влияние соотношения окислитель : мономер при окислительной полимеризации ДФХБ (рис. 3) не имеет ярко выраженного экстремального характера в отличие от полимеризации анилина и ДАДХБ [27]. Наибольшая скорость реакции наблюдается при стехиометрическом соотношении окислитель : мономер = 1.25 (кривая 3). При уменьшении концентрации окислителя до отношения 0.75 и 0.5 скорость реакции снижается. Однако при возрастании концентрации окислителя не наблюдается характерного при полимеризации анилина и ДАДХБ значительного увеличения индукционного периода. Лишь при отношении 3.75 (кривая 4) видно некоторое снижение скорости реакции при сохранении индукционного периода. Это объясняется тем, что значительная часть окислителя расходуется на процессы циклизации. А поскольку индукционный период реакции не меняется даже при значительном избытке окислителя, это свидетельствует в пользу того, что дополнительный расход окислителя необходим уже на первых ступенях процесса. Значит, процессы циклизации протекают паралельно с ростом полимерной цепи. Однако избыток окислителя не приводит к участию в процессе полимеризации второй ариламино- группы молекулы мономера. По данным ИК-спектроскопии и РФЭС этот участок молекулы оказывается не затронутым в реакции окисления, т.е. механизм полимеризации ДФХБ отличается от полимеризации анилина. И если вторая замещенная ариламино-группа не вовлечена в процесс таутомерной перегруппировки системы, она остается лишь ароматическим амином с сильным электроноакцепторным заместителем, не способным к окислительной полимеризации.
Влияние рН на скорость реакции представлено на рис. 4. Сравнение кинетических кривых показало, что скорость реакции растет с уменьшением рН. Растет и интенсивность пика (кривые 1–3). Это указывает на то, что концентрация катион-радикальных центров напрямую связана с концентрацией кислоты в реакционном растворе в соответствии со схемой (1). Однако при концентрации кислоты выше 0.5 моль/л HCl (кривая 4) начинает расти индукционный период и значительно снижается скорость реакции. Это связано с тем, что избыток кислоты протонирует первичные аминогруппы фенилендиаминовых колец, превращая их тем самым в электронно-акцепторные заместители, снижающие электронную плотность в пара-положении анилинового кольца. Его нуклеофильность уменьшается, и реакционная способность снижается.
Рис. 4.
Зависимость изменения редокс-потенциала от времени при окислении ДФХБ в воде (1) и [HCl] = 0.1 моль/л (2), 0.5 моль/л (3), 1 моль/л (4).

Полимеры ДФХБ представляют собой черные мелкодисперсные порошки растворимые в N-МП, ДМФА, ДМСО с образованием темно-коричневых растворов. При этом цвет раствора не изменяется в зависимости от того, протонированная эта форма или нет. Таким образом, он отличается от полианилина и поли-ДАДХБ, растворы которых в допированной форме имеют зеленную окраску, а в дедопированной – синюю. Электропроводность поли-ДФХБ, полученного при стандартных условиях, составила 8.2 × 10–5 См/см. Более низкие значения электропроводности по сравнению с поли-ДАДХБ и полианилином связаны с тем, что полициклическая структура основной цепи препятствует образованию хинодииминных звеньев, что, в свою очередь, делает маловероятным образование поляронной структуры, ответственной за электропроводность полимера в допированном состоянии.
По данным рентгенофазового анализа, полимеры ДФХБ аморфны. Исследование морфологии полученных полимеров показало, что у них рыхлая глобулярная структура (рис. 5а). Однако при более детальном рассмотрении заметно, что частицы поли-ДФХБ имеют слоистую текстуру (рис. 5б), что, вероятно, определяется лестничной структурой полимера и его плоским строением.

ЗАКЛЮЧЕНИЕ
Изучение окислительной полимеризации ДФХБ, наряду с проведенными ранее исследованиями полимеризации ДАДХБ, позволяют сделать вывод, что диарилдиаминодихлорбензохиноны можно назвать подходящими мономерами для окислительной полимеризации в водных растворах кислот. Их скорость полимеризации значительно выше, чем у анилина. Несмотря на то что формально они являются производными анилина с электроноакцепторными заместителями у атома азота, их реакционная способность выше, чем у анилина. Объясняется это иным строением интермедиатов, которые участвуют в реакции окисления. Способность мономеров к таутомерным перегруппировкам с формированием хинодииминных фрагментов, образующих катион-радикальные центры в отсутствии окислителя, – ключевой момент механизма роста полимерной цепи. Наличие дополнительной аминогруппы в ароматическом ядре не только не мешает полимеризации, но и повышает скорость реакции за счет увеличения нуклеофильности в пара-положении анилинового кольца. В результате, кинетические кривые окислительной полимеризации ДФХБ кардинально отличаются от кривых полимеризации анилина и ДАДХБ: полностью отсутствует индукционный период, и кривые не имеют S-образного вида, характерного для автокаталитического процесса.
Однако уникальность данного мономера этим не исчерпывается. Наличие дополнительной аминогруппы в мета-положении ароматического кольца приводит к тому, что в процессе роста цепи она занимает положение, благоприятствующее взаимодействию с хлором хлоранильной группы соседнего звена, в результате чего образуются гидрофеназиновые звенья. Благодаря их нестабильности и присутствию окислителя, в реакционной среде происходит их окисление до феназиновых с одновременным образованием феноксазиновых циклов. Таким образом, формируется полимер лестничной структуры. При этом пара-хиноиминные фрагменты перегруппировываются в орто-хиноиминные.
Циклизация, протекающая непосредственно в ходе роста полимерной цепи, приводит к тому, что количество окислителя, необходимое для этого процесса, превышает расчетное стехиометрическое для анилина, а его избыток не только не замедляет процесс, но до определенного предела ускоряет процесс полимеризации.
Кроме того, наличие дополнительной аминогруппы позволяет до некоторой степени регулировать скорость реакции полимеризации путем контроля рН реакционной среды. При низких значениях рН свободная аминогруппа протонируется, превращаясь в электроноакцепторный заместитель. При этом скорость окисления начинает снижаться, но реакция роста цепи не прекращается.
Установлено, что в зависимости от типа заместителя диарилдиаминодихлорбензохиноны могут формировать в поцессах окислительной полимеризации полисопряженные полимеры полианилиновой или лестничной структуры.
Работа выполнена в рамках государственного задания Института нефтехимического синтеза РАН.
Список литературы
Cao Y., Andretta A., Heeger A.J., Smith P. // Polymer. 1989. V. 30. № 12. P. 2305.
Okamoto H., Okamoto M., Kotaka T. // Polymer. 1998. V 39. № 18. P. 4359.
Monkman A.P., Adams P.N., Laughin P.J., Holland E.R. // Synth. Met. 1995. V. 69. № 1–3. P. 183.
Ciric-Marjanovic G. // Synthetic Met. 2013. V. 177. № 1. P. 1.
Eftekhari A., Li L., Yang Y. // J. Power Sourc. 2017. V. 347. P. 86.
Zarrintaj P., Ahmadi Z., Vahabi H., Ducos F., Saeb M.R., Mozafari M. // Mater. Today Proc. 2018. V. 5. № 7. P. 15852.
Orlov A.V., Kiseleva S.G., Karpacheva G.P., Teplyakov V.V., Syrtsova D.A., Starannikova L.E., Lebedeva T.L. // J. Appl. Polym. Sci. 2003. V. 89. № 5. P. 1379.
Stejskal J., Sapurina I., Trchova M., Konyushenko E.N. // Macromolecules. 2008. V. 41. № 10. P. 3530.
Mu S., Chen C., Xue H. // J. Electroanal. Chem. 2014. V. 724. P. 71.
Mezhuev Ya.O., Korshak Yu.V., Shtilman M.I. // Russ. Chem. Rev. 2017. V. 86. № 12. P. 1271.
Strakhov I.S., Mezhuev Ya.O., Korshak Yu.V., Shtil’man M.I. // Russ. J. General Chem. 2016. V. 86. № 12. P. 2682.
Alva K.S., Kumar J., Marx K.A., Tripathi S.K. // Macromolecules. 1997. V. 30. № 14. P. 4024.
Soledad P., Andrade E.M., Molina F.V. // React. Funct. Polym. 2009. V. 69. № 3. P. 197.
Oztekina Y., Toka M., Nalvuranc H., Kiyaka S., Govera T., Yazicigil Z., Ramanaviciene A., Ramanavicius A. // Electrochim. Acta. 2010. V. 56. № 1. P. 387.
Mateos M., Prest R.M., Suisse J.M., Bouvet M. // Mater. Today Proc. 2019. V. 6. № 3. P. 328.
Falcou A., Longeau A., Marsacq D., Hourquebie P., Duchne A. // Synth. Met. 1999. V. 101. № 1–3. P. 647.
Gazotti W.A., jr., De Paoli M.-A. // Synth. Met. 1996. V. 80. № 3. P. 263.
Межуев Я.О., Коршак Ю.В., Штильман М. И., Коледенков А.А., Устинова М.С., Семенова И.Н. // Пласт. массы. 2011. № 2. С. 22.
Lee R.-H., Lai H.-H., Wang J.-J., Jeng R.-J., Lin J.-J. // Thin Solid Films. 2008. V. 517. № 2. P. 500.
Xu Y., Dai L., Chen J., Gal J.-Y., Wu H. // Eur. Polym. J. 2007. V. 43. № 5. P. 2072.
Ozkan S.Z., Karpacheva G.P., Eremeev I.S. // Adv. Nanotechnol. 2015. V. 13. P. 161.
Orlov A.V., Ozkan S.Zh., Bondarenko G.N., Karpacheva G.P. // Polymer Science B. 2006. V. 48. № 1–2. P. 5.
Ozkan S.Zh., Bondarenko G.N., Orlov A.V., Karpacheva G.P. // Polymer Science B. 2009. V. 51. № 5–6. P. 149.
Ozkan S.Zh., Karpacheva G.P., Bondarenko G.N. // Russ. Chem. Bull. Int. Ed. 2011. V. 60. № 8. P. 1651.
Ozkan S.Zh., Karpacheva G.P., Orlov A.V., Dzyubina M.A. // Polymer Science B. 2007. V. 49. № 1–2. P. 36.
Ozkan S.Z., Karpacheva G.P., Bondarenko G.N., Kolyagin Y.G. // Polymer Science B. 2015. V. 57. P. 106.
Kiseleva S.G., Orlov A.V., Bondarenko G.N., Karpacheva G.P. // Polymer Science B. 2018. V. 60. № 6. P. 717.
Abalyaeva V.V., Tkachenko L.I., Nikolaeva G.V., Orlov A.V., Kiseleva S.G., Efimov O.N., Karpacheva G.P. // Polymer Science B. 2017. V. 59. № 4. P. 459.
Орлов А.В., Киселева С.Г., Карпачева Г.П., Николаева Г.В., Ткаченко Л.И., Ефимов О.Н., Абаляева В.В. Пат. 2016118436 Россия. 2017.
Abalyaeva V.V., Nikolaeva G.V., Kabachkov E.N., Kiseleva S.G., Orlov A.V., Efimov O.N., Karpacheva G.P. // Polymer Science B. 2018. V. 60. № 6. P. 511.
Tkachenko L.I., Nikolaeva G.V., Orlov A.V., Kiseleva S.G., Efimov O.N., Karpacheva G.P. // Russ. J. Appl. Chem. 2019. V. 92 (in press).
Akutagawa T., Nakamura T. // Cryst. Growth Design. 2006. V. 6. № 1. P. 70.
Surwade S.P., Dua V., Manohar N., Manohar S.K., Beck E., Ferraris J.P. // Synth. Met. 2009. V. 159. № 5–6. P. 445.
Stejskal J., Bober P., Trchová M., Horsk’y Ji., Pilaˇr J., Walterová Z. // Synth. Met. 2014. V. 192. P. 66.
Gospodinova N., Terlemezyan L. // Prog. Polym. Sci. 1998. V. 23. № 8. P. 1443.
Wang J., Wang S. // Chem. Eng. J. 2018. V. 334. P. 1502.
Bazzi H.S., Mostafa A., AlQaradawi S.Y., Nour El-M. // J. Mol. Struct. 2007. V. 842. P. 1.
Prasad R.L., Kushwaha A., Kumar S.M., Yadav R.A. // Spectrochim. Acta. A. 2008. V. 69. P. 304.
Refat M.S., El-Zayat L., Yesilel O.Z. // Polyhedron. 2008. V. 27. P. 475.
Benyoucef A., Huerta F., Vá zquez J.L., Morallon E. // Eur. Polym. J. 2005. V. 41. P. 843.
Bhat N.V., Seshadri D.T., Phadke S.R. // Synth. Met. 2002. V. 130. P. 185.
Hwang G.-W., Wu K.-Y., Hua H.-T., Chen S.-A.// Synth. Met. 1998. V. 92. P. 39.
Sivakumar R., Saraswathi R. // Synth. Met. 2003. V. 138. P. 381.
Ichinohe D., Muranaka T., Sasaki T., Kobayashi T., Kise H. // J. Polym. Sci. Polym. Chem. 1998. V. 36. P. 2593.
Ren Sh., Yang X., Zhao X., Zhang Y., Huang W. // J. Appl. Polym. Sci. 2016. V. 133. № 18. 43368.
Fatuch J.C., Soto-Oviedo M.A., Avellaneda C.O., Franco M.F., Romao W., De Paoli M.-A., Nogueira A.F. // Synth. Met. 2009. V. 159. P. 2348.
Tucceri R., Arnal P.M., Scian A.N. // J. Spectroscopy. 2013. V. 2013. 951604.
Marjanović B., Yuranić I., Ćirić-Marjanović G., Pašti I., Trchova M. // React. Funct. Polym. 2011. V. 71. P. 704.
Resada N., Park Ju., Ryu K. // Korean J. Chem. Eng. 2010. V. 33. № 10. P. 3011.
Zhang L., Chai L., Wang H., Yang Z. // Mater. Lett. 2010. V. 64. P. 1193.
Blaha M., Trchova M., Moravkova Z., Humpolicek P., Stejskal J. // Mater. Chem. Phys. 2018. V. 205. P. 423.
Kang E.T., Neoh K.G., Tan T.C., Khor S.H., Tan K.L. // Macromolecules. 1990. V. 23. № 11. P. 2918.
Carbone M.E., Ciriello R., Granafai S., Guerriari A., Salvi A.M. // Electrochim. Acta. 2014. V. 144. P. 174.
Al-Mashat L., Shin K., Kalantar-Zadeh K., Plessis J.D., Kojima H.R.W., Kaner R.B., Li D., Gou X., Ippolito S.J., Wodarski W.W. // J. Phys. Chem. C. 2010. V. 114. № 39. P. 16168.
Kang E.T., Neoh K.G., Tan K.L. // Prog. Polym. Sci. 1998. V. 23. P. 277.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)