

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 6, стр. 428-438
МАТРИЧНАЯ ПОЛИМЕРИЗАЦИЯ ТРИМЕТИЛМЕТАКРИЛОИЛОКСИЭТИЛАММОНИЙ МЕТИЛСУЛЬФАТА В МИЦЕЛЛЯРНЫХ РАСТВОРАХ ДОДЕЦИЛСУЛЬФАТА НАТРИЯ
Ю. В. Шулевич a, *, Ю. А. Захарова b, П. М. Толстой c, М. А. Вовк c, Е. Г. Духанина a, Д. С. Быков a, А. В. Навроцкий a, И. А. Новаков a
a Волгоградский государственный технический университет
400005 Волгоград, пр. Ленина, 28, Россия
b Московский государственный университет им. М.В. Ломоносова. Химический факультет
119899 Москва, Ленинские горы, Россия
c Санкт-Петербургский государственный университет.
Ресурсный центр “Магнитно-резонансные методы исследования”
198504 Петергоф, Санкт-Петербург, Университетский пр., 26, Россия
* E-mail: shulevich@vstu.ru
Поступила в редакцию 24.06.2019
После доработки 12.07.2019
Принята к публикации 26.07.2019
Аннотация
Методом спектроскопии ЯМР изучены кинетические особенности радикальной полимеризации триметилметакрилоилоксиэтиламмоний метилсульфата в мицеллярных растворах противоположно заряженного поверхностно-активного вещества – додецилсульфата натрия. Показано, что матричная полимеризация по zip-механизму протекает только в разбавленных растворах ПАВ, содержащих сферические мицеллы, при мольном соотношении [мономер]/[ПАВ] ≤ 1 и существенном возрастании скорости полимеризации. Увеличение концентрации ПАВ приводит к изменению механизма матричной полимеризации, сопровождающемуся снижением скорости полимеризации, что, вероятно, обусловлено изменением формы мицелл. Молекулярная масса полученных полиэлектролитов зависит от мольного соотношения компонентов и минимальна при эквимольном соотношении [мономер]/[ПАВ].
ВВЕДЕНИЕ
Свободнорадикальная полимеризация является наиболее распространенным способом получения полимеров, в том числе и полиэлектролитов. Различные методы ее осуществления и большое разнообразие мономеров, инициаторов, ингибиторов и агентов передачи цепи позволяют получать полиэлектролиты различной молекулярной массы, состава и строения. Существенным недостатком свободнорадикальной полимеризации является неконтролируемость молекулярно-массовых характеристик, к тому же продукты синтеза отличаются высокой полидисперсностью.
Все известные способы контролируемого синтеза полимеров основываются на переводе растущей макромолекулярной цепи в структурно организованное состояние. В большинстве каталитических систем, осуществляющих контролируемую полимеризацию, присоединению мономера к растущей цепи предшествует стадия образования комплекса между активным центром и молекулой мономера.
В отечественной литературе первое упоминание о контролируемой полимеризации связано с работами Каргина, которые были посвящены полимеризации комплексно-связанных и организованных мономеров [1].
Современная химия высокомолекулярных соединений предлагает такие методы контролируемого синтеза полимеров как радикальная полимеризация с переносом атома, радикальная полимеризация с обратимой реакцией передачи цепи и другие аналогичные способы [2, 3], дающие возможность синтезировать полиэлектролиты с узким молекулярно-массовым распределением, но невысокой молекулярной массой. Несмотря на несомненные преимущества данных подходов, на практике методы контролируемой радикальной полимеризации зачастую остаются нереализованными.
Перспективным подходом, позволяющим контролировать одновременно и молекулярную массу, и полидисперсность полимеров может оказаться матричная полимеризация, поскольку в матричном синтезе молекулярно-массовые характеристики дочернего полимера определяются используемой матрицей [4, 5].
Основные принципы матричной полимеризации развиты с использованием виниловых мономеров, способных образовывать с матрицей водородные связи или ионные пары. В качестве матрицы обычно используют структурно и химически комплементарные макромолекулы, такие как поли(мет)акриловую кислоту [6], полистиролсульфонат натрия [7], полиэтиленгликоль [8] и другие. В этом случае продукт полимеризации представляет собой стехиометричный полиэлектролитный комплекс, который, как правило, нерастворим в известных органических растворителях, что существенно затрудняет процесс выделения дочернего полимера. Возможным вариантом решения проблемы является использование в качестве матрицы мицелл ПАВ.
Теоретическое обоснование предложенной идеи базируется на современных представлениях о строении мицелл ПАВ. Известно, что мицеллы ПАВ способны удерживать на своей поверхности противоионы. При этом если простые неорганические противоионы слабо ассоциируют с мицеллой, то степень связывания органических противоионов с мицеллой существенно выше [9, 10]. Поэтому, в смеси мицелл ПАВ и органических противоионов, содержащих двойную связь, возможно образование комплекса, в котором мицелла ПАВ будет выступать в качестве матрицы, на поверхности которой формируется слой мономера, обеспечивая высокую локальную концентрацию ионов мономера и их ориентацию в пространстве, тем самым способствуя проявлению структурного и кинетического матричного эффекта.
Успешным примером реализации полимеризации по такому типу являются работы по мицеллярной полимеризации поверхностно-активных мономеров, в том числе гексадецилтриметиламмоний метакрилата, додециламмоний (мет)акрилата, гексадецилтриметиламмоний 4-винилбензоата и его гомологов, додецил- и цетиламмоний 2-акриламидо-2-метилпропан сульфоната, N-акрилоил-11-аминоундекановой кислоты и ее производных [11–20]. Такие мономеры содержат мицеллообразующий ионогенный фрагмент, а противоин – двойную связь. В результате полимеризации контрионов, сконцентрированных вблизи поверхности мицеллы за счет электростатических и других взаимодействий, формируется стехиометрический комплекс полиэлектролит–ПАВ, представляющий практический и научный интерес [21–23].
Принципиальное отличие предлагаемого в данной работе подхода заключается в том, что мономер вводят в мицеллярный раствор противоположно заряженного ПАВ, что позволяет варьировать мольное соотношение мономер : ПАВ в широких пределах и получать в качестве продукта реакции как нерастворимые (стехиометричные), так и водорастворимые (нестехиометричные) комплексы полиэлектролит–ПАВ.
Несмотря на кажущуюся простоту предлагаемого подхода, возможность его практической реализации (a priori) неоднозначна.
Ионогенные мономеры, также как и ПАВ, являются амфифильными и поверхностно-активными. Значит, скорее всего, мономер может встраиваться в мицеллу с образованием смешанных мицелл. В случае очень сильного взаимодействия двух противоположно заряженных компонентов могут образоваться нерастворимые органические соли, как это наблюдалось при смешении противоположно заряженных ПАВ [9]. Для систем, в которых одно из ПАВ имеет относительно короткий алкильный фрагмент, характерно формирование множества жидкокристаллических фаз с большим количеством фазовых переходов [9]. Понятно, что и в первом, и во втором случае полимеризация, если она и возможна, не будет являться матричной.
Ранее в работах [24–27] методом ЭПР-спектроскопии было детально изучено взаимодействие ионогенных мономеров с мицеллами противоположно заряженных ПАВ. Показано, что определяющее влияние на место локализации молекул мономера в мицеллах ПАВ оказывает гидрофобность мономера. Так, мономер триметилметакрилоилоксиэтиламмоний метилсульфат (М), не содержащий гидрофобных заместителей, сорбируется на поверхности мицелл додецилсульфата натрия (ДДС), замещая их собственные противоионы, причем высокая концентрация компонентов (вплоть до 2.5 моль/л мономера и 1.5 моль/л ДДС) не препятствует связыванию. Введение в структуру мономера объемных гидрофобных заместителей (на примере стиролсульфоната натрия) приводит к встраиванию мономера в мицеллы додецилтриметиламмоний бромида, поэтому двойные связи, по которым должна идти полимеризация, оказываются экранированы от воздействия инициатора или разнесены на большие расстояния друг от друга. Полимеризация этого мономера по матричному механизму, по-видимому, невозможна.
По совокупности результатов, полученных при изучении взаимодействия ионогенных мономеров с мицеллами противоложно заряженных ПАВ, в настоящей работе были определены наиболее перспективные условия для реализации матричного механизма полимеризации триметилметакрилоилоксиэтиламмоний метилсульфата в мицеллярных растворах ДДС: 0.5 ≤ ≤ [М]/[ДДС] ≤ 2 и 70 мМ ≤ [ДДС] ≤ 250 мМ. Однако прямым доказательством реализации матричного механизма полимеризации являются только так называемые матричные эффекты, выявляемые в процессе изучения кинетики полимеризации и молекулярно-массовых характеристик полученных полиэлектролитов.
Цель данной работы – изучение кинетических закономерностей полимеризации триметилметакрилоилоксиэтиламмоний метилсульфата в мицеллярных растворах ДДС и молекулярно-массовых характеристик полученных полимеров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Триметилметакрилоилоксиэтиламмоний метилсульфат получали алкилированием диметиламиноэтилметакрилата диметилсульфатом (“Sigma-Aldrich”, Германия) в ацетоне. Синтезированный мономер перекристаллизовывали из ацетона, сушили под вакуумом 1.5–2.0 мм рт.ст. и температуре 20 ± 0.5°С до постоянной массы. Чистоту мономера (структура (I)) подтверждали элементным анализом: С 41.30, Н 6.20, N 5.20, S 11.92 мас. %, что полностью соответствует теории (С 40.29, Н 6.75, N 5.22, S 11.94 мас. %):

Водорастворимый инициатор радикальной полимеризации 2,2'-азо-бис-(2-метилпропионамидин)дигидрохлорид (I) (“Wako Pure Chemical Corporation”, Япония), структурная формула (II) которого приведена ниже, дополнительно не очищали:

Додецилсульфат натрия (“Amresco”, США) использовали без дополнительной очистки. Критическая концентрация мицеллообразования, определенная методом солюбилизации маслорастворимого красителя Oil Yellow OB по стандартной методике, соответствовала литературным значениям и составила 8 мМ [9].
Для изучения кинетики полимеризации готовили исходные растворы 1.0 моль/л мономера, 0.5 моль/л раствора ДДС и 0.01 моль/л раствора I, которые применяли для приготовления растворов мономера в воде и в мицеллярных растворах ДДС в требуемой концентрации и мольных соотношениях.
В качестве растворителя в экспериментах по ЯМР использовали дейтерированную воду (“Cambridge Isotope Laboratories”, США), во всех остальных экспериментах – бидистилированную воду.
Кинетику полимеризации изучали методом спектроскопии ЯМР. Для чего приготовленный раствор помещали в чистую и сухую ампулу для ЯМР, продували аргоном в течение 5–7 мин, закрывали крышкой и устанавливали в ЯМР-спектрометр, предварительно термостатированный на 60°С. Момент погружения ампулы в спектрометр фиксировали и принимали за начальное время реакции. Спектры ЯМР 1Н записывали через каждые 20 с как сумму четырех сканирований (рис. 1).

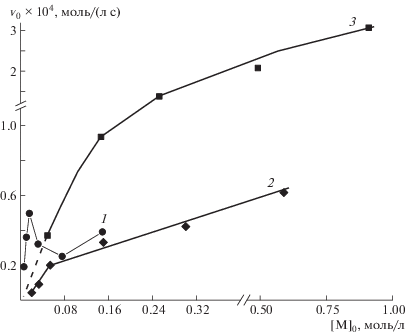
Рис. 2.
Концентрационная зависимость начальной скорости полимеризации; [I] = 0.5 мМ, Т = 60°С. [ДДС] = 15 (1) и 150 мМ (2), а также без ПАВ (3; начальный участок кривой представлен для [M] < 50 мМ пунктирной линией, поскольку рассчитан экстраполяцией).
За кинетикой полимеризации следили, фиксируя интенсивность сигналов от протонов винильной группы (обозначены как а, b на структурной формуле (I)) и протонов метильных заместителей в четвертичном атоме азота (c). Отношение интенсивностей групп (Inta/Intc и Intb/Intc) в начальный момент времени принимали за единицу. Уменьшение отношения интенсивностей сигналов относительно единицы использовали для расчета значений конверсии.
Конверсию мономера (X, %) рассчитывали отдельно по группам а и b по формуле
где Int – нормированная интенсивность сигнала группы а или b (Inta/Intc или Intb/Intc).Используя полученные значения X и начальную концентрацию мономера [M]0, находили текущую концентрацию мономера [M] по выражению
Зависимость [M] = f(t) строили отдельно по группам а и b, скорость полимеризации определяли как тангенс угла наклона начального прямолинейного участка прямой (5% < X < 15%). Результирующую скорость полимеризации рассчитывали как среднее арифметическое из двух значений скорости.
Так как использованный мономер характеризуется высокой реакционной способностью и склонен к спонтанной полимеризации (в 2 моль/л растворе мономера в отсутствие инициатора при 60°С конверсия мономера составляла 10% за ∼1.5 ч, концентрация мономера не превышала 1 моль/л).
Порядок реакции полимеризации по инициатору и мономеру, а также энергию активации реакции находили по стандартным методикам при эквимольном соотношении мономера и ПАВ ([М]/[ПАВ]). Принимая во внимание склонность ДДС к формированию цилиндрических мицелл и возможное влияние размеров и формы мицелл ПАВ на скорость полимеризации [28], порядок полимеризации по инициатору и энергию активации определяли при двух значениях концентрации, соответствующих существованию сферических ([ДДС] = 15 мМ) и цилиндрических ([ДДС] = 150 мМ) мицелл соответственно [29–31].
При определении порядка реакции по мономеру зависимости, полученные при фиксированных значениях концентрации ПАВ и инициатора, носят сложный характер, и не спрямляются в логарифмических координатах. Поэтому и в этом случае фиксировали эквимольное соотношение [М]/[ПАВ].
Для получения образцов полиэлектролитов и дальнейшего нахождения их молекулярно-массовых характеристик полимеризацию осуществляли в мицеллярных растворах ДДС различной концентрации ампульным методом в атмосфере аргона. Мольное соотношение мономера и ПАВ ([М]/[ПАВ]) составляло 0.5, 1.0, 2.0 и 5.0. Синтез проводили при [I] = 0.5 мМ и температуре 60 ± 0.5°С.
Непосредственным продуктом полимеризации являются комплексы полиэлектролит–ПАВ, которые в зависимости от концентрации и соотношения исходных реагентов могут быть как растворимыми, так и нерастворимыми в воде [32]. Возможность выделения индивидуального полиэлектролита из комплекса основана на подавлении электростатических взаимодействий при увеличении ионной силы раствора и разрушении комплексов полиэлектролит–ПАВ в больших ионных силах. Методики выделения полиэлектролитов зависели от фазового состояния продукта полимеризации.
Для выделения полиэлектролита из геля (водорастворимого комплекса) небольшое количество геля растворяли в 3.5 моль/л растворе NaCl. Выделившийся из комплекса и высалившийся ДДС отфильтровывали, а раствор полиэлектролита диализовали для удаления соли. Контроль за отмыванием раствора полимера от соли осуществляли, измеряя концентрацию ионов Cl– в диализате с помощью иономера “Мультитест–ИПЛ-513”. Очищенный полиэлектролит лиофильно сушили.
Для выделения полиэлектролита из нерастворимого продукта полимеризации небольшое количество сухого осадка также растворяли в 3.5 моль/л растворе NaCl. Под действием высокой ионной силы комплекс переходит в диспергированное состояние, при этом раствор представляет собой смесь взвешенного мелкодисперсного ДДС, стабилизированного раствором полимера. Для полного выделения ДДС в раствор добавляли BaCl2, который связывает ДДС в нерастворимую соль – додецилсульфат бария. Далее процесс выделения полиэлектролита был аналогичен описанному выше. Высушенные полиэлектролиты хранили в эксикаторе над Р2О5.
По данным элементного анализа выделенные полиэлектролиты не содержали серы, что подтверждает полное удаление ПАВ из образцов в процессе их выделения и очистки.
Молекулярно-массовые характеристики всех синтезированных полиэлектролитов находили методом статического лазерного рассеяния света с помощью широкоуглового фотометра рассеянного лазерного света “PhotoCor Complex” (“PhotoCor”, США) с He–Ne-лазером мощностью 10 мВт и длиной волны 632 нм в качестве источника света. Растворы перед измерениями тщательно обеспыливали путем фильтрования через мембранные фильтры Millipore с размером пор 0.45 мкм. Инкремент показателя преломления растворов dn/dc определяли с помощью дифференциального рефрактометра “Optilab T-rEX” (“Waytt Technology”, США) с He–Ne-лазером мощностью 2 мВт и длиной волны 632 нм в качестве источника света. При измерениях dn/dc в качестве раствора сравнения использовали растворитель, приведенный в диализное равновесие с раствором.
Для исследования молекулярно-массовых характеристик полиэлектролитов готовили их растворы, используя водный раствор 1 моль/л NaCl. Затем растворы разбавляли через сутки после приготовления, а все измерения выполняли через 2–3 ч после разбавления.
Измерения интенсивности рассеянного света осуществляли при 25°С в диапазоне углов рассеяния от 30° до 140° с шагом в 10° для серии растворов из пяти значений концентрации. Для определения среднемассовой молекулярной массы Mw полученные результаты обрабатывали методом двойной экстраполяции Зимма.
Элементный анализ проводили в лаборатории центра коллективного пользования “Физико-химические методы исследования” Волгоградского государственного технического университета на CHNOS-элементном анализаторе фирмы (“Vario EL Cube”, Германия).
Исследования методом спектроскопии ЯМР выполняли в ресурсном центре “Магнитно-резонансные методы исследования” Научного парка Санкт-Петербургского государственного университета, используя спектрометр ЯМР “Bruker Avance III 500” (рабочая частота 500 МГц на ЯМР 1H).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Кинетические особенности полимеризации мономера в присутствии противоположно заряженного ПАВ
В общем случае под матричной полимеризацией понимают процесс, в котором на макромолекулах, предварительно введенных в реакционную систему, образуются другие макромолекулы (дочерний полимер), структурно и химически комплементарные исходным [4].
О матричном механизме полимеризации судят по так называемым “матричным эффектам” – кинетический эффект, выражающийся в изменении скорости полимеризации; молекулярный эффект, заключающийся во влиянии матрицы на молекулярную массу и молекулярно-массовое распределение дочернего полимера; эффект регулярности молекулярной структуры, когда дочерний полимер приобретает структуру, комплементарную структуре используемой матрицы [33].
Кроме того, различают два типа матричной полимеризации: первый тип (zip) соответствует сильному специфическому взаимодействию компонентов, сопровождающемуся адсорбцией мономера на матрице; второй тип (pick-up) характерен для случая, когда мономер и матрица не взаимодействуют или слабо взаимодействуют друг с другом, но, начиная с некоторой степени полимеризации, матрица “перехватывает” растущие полимерные цепи за счет электростатического или иного кооперативного связывания.
Для идентификации того или иного типа полимеризации используют кинетические данные, представленные в координатах “начальная скорость полимеризации–отношение мономер : матрица” [34]. При матричной полимеризации по zip-механизму в точке эквимольности на зависимости виден излом, связанный с тем, что помимо полимеризации на матрице, протекающей, как правило, с большей скоростью за счет локального концентрирования мономеров, в системе начинает проходить параллельный процесс свободнорадикальной полимеризации в растворе. Ни при свободнорадикальной полимеризации, ни при матричной полимеризации по pick-up-механизму изломов на зависимости не наблюдается вплоть до значительных избытков (концентрации) мономера.
На рис. 2 показана концентрационная зависимость начальной скорости полимеризации в мицеллярных растворах ПАВ разной концентрации (кривые 1 и 2), а также для сравнения представлены соответствующие данные для полимеризации М в воде (кривая 3). Так как для каждой из кривых 1 и 2 на рис. 2 концентрация ПАВ, т.е. матрицы, была постоянной, то рост концентрации мономера соответствовал росту отношения мономер : матрица, и такое представление кинетических данных можно считать качественно эквивалентным координатам “начальная скорость полимеризации – отношение мономер : матрица”, при этом для кривой 1 точка эквимольности соответствует концентрации мономера 15 мМ, а для кривой 2 – 150 мМ.
Полученные данные являются, по меньшей мере, неожиданными, так как за исключением составов [М]/[ПАВ] ≤ 2 при [ПАВ] = 15 мМ, введение ДДС приводит к заметному снижению скорости полимеризации. Более того, концентрация ПАВ, влияющая на форму мицелл, также имеет принципиальное значение. Для более концентрированного раствора ДДС (кривая 2) ход зависимости в целом аналогичен кривой 3 (без ПАВ), однако можно отметить наличие незначительного излома в области состава [М]/[ПАВ] = 0.5. При минимальном содержании ДДС в растворе зависимость начальной скорости от соотношения мономер : ПАВ имеет сложный характер с явным максимумом в точке эквимольности, соответствующей [М] = 15 мМ (кривая 1). Рост скорости полимеризации с ростом концентрации мономера вплоть до стехиометрии понятен и считается очевидным признаком матричной полимеризации. Действительно, эффективное связывание мономера с мицеллами ДДС [25] влечет его локальное концентрирование и, как следствие, рост скорости полимеризации вплоть до стехиометрии, когда мицеллы ПАВ оказываются полностью “насыщены” ионами мономера. Повышенный порядок реакции по мономеру, согласно работам [28, 35], также свидетельствует об ассоциации мономера на мицеллах ПАВ (таблица 1).
Таблица 1.
Кинетические параметры полимеризации мономера триметилметакрилоилоксиэтиламмония метилсульфата в воде и в мицеллярных растворах ДДС
| Кинетический параметр | Условия эксперимента | ||||
|---|---|---|---|---|---|
| [I], мМ | [М], мМ | [ДДС], мМ | [М]/[ДДС] | t, °C | |
| Ea, кДж/(моль К) | |||||
| 96 | 0.50 | 150 | 0 | – | 50–80 |
| 20 | 0.50 | 15 | 15 | 1 | 50–80 |
| 43 | 0.50 | 150 | 150 | 1 | 50–80 |
| ni | |||||
| 0.5 | 0.08–1.0 | 150 | 0 | – | 60 |
| 0.33 | 0.1–1.0 | 15 | 15 | 1 | 60 |
| 0.29 | 0.08–0.8 | 150 | 150 | 1 | 60 |
| nm | |||||
| 1.0 | 0.50 | 49–910 | 0 | – | 60 |
| 1.52 | 0.50 | 6–22 | 6–22 | 1 | 60 |
| – | 0.50 | 49–200 | 49–200 | 1 | 60 |
Однако резкое снижение скорости полимеризации при увеличении концентрации мономера сверх стехиометрии и в целом сравнительно низкая скорость полимеризации в присутствии ДДС, особенно в 150 мМ растворе ДДС, являются неожиданными и нуждаются в объяснении.
Концентрационные зависимости приведенной скорости полимеризации показаны на рис. 3. Видно, что при концентрации ДДС 15 мМ, когда матрицей служат сферические мицеллы ПАВ, скорость полимеризации растет вплоть до стехиометричного соотношения, а в точке эквимольности компонентов в ∼4.5 раза превышает скорость полимеризации в воде. Незначительное увеличение приведенной скорости наблюдается и для [ДДС] = 150 мМ на начальном участке кривой (кривая 2) вплоть до [М]/[ПАВ] = 0.5. Увеличение концентрации мономера сверх эквимольного соотношения влечет уменьшение приведенной скорости полимеризации, особенно резкое для полимеризации в присутствии сферических мицелл ДДС. При этом аналогичный эффект – заметное уменьшение приведенной скорости полимеризации с ростом концентрации мономера – можно видеть и в отсутствие ПАВ (рис. 3б).
Рис. 3.
Концентрационные зависимости приведенной скорости полимеризации (а) и полимеризации мономера в воде (б). а: [ДДС] = 15 (1) и 150 мМ (2); кривые 1' и 2' – теоретические зависимости, рассчитанные по правилу аддитивности в условиях избытка мономера. [I] = 0.5 мМ, Т = 60°С.
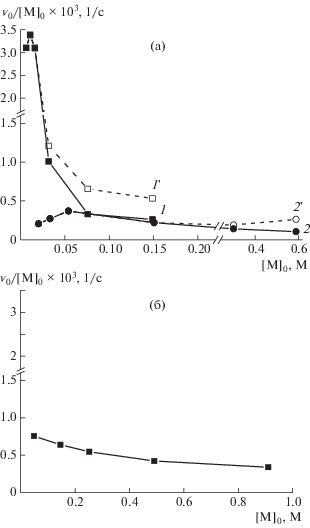
В работе [31] приведена аналогичная зависимость для полимеризации структурно схожего мономера, противоионом которого вместо метилсульфат-аниона был хлорид-анион. Показано, что такое снижение приведенной скорости с ростом концентрации мономера характерно для полимеризации ионогенных мономеров, способных к образованию флуктуационных ассоциатов [28]. Было высказано предположение, что увеличение концентрации мономера в растворе вызывает рост ассоциатов, что сопровождается уменьшением степени диссоциации мономера. Результатом чего является увеличение вероятности столкновения растущих цепей, и, как следствие, увеличение константы обрыва цепи, что и влечет уменьшение приведенной скорости полимеризации таких мономеров с ростом их концентрации [36].
В присутствии ПАВ ([ДДС] > ККМ), в условиях избытка мономера, в реакционной среде появляется мономер, не связанный с мицеллами ПАВ, причем его доля возрастает с увеличением мольного соотношения [М]/[ПАВ]. Инициирование полимеризации водорастворимым инициатором в такой системе приводит к тому, что рост полимерных цепей может осуществляться как на поверхности мицелл ПАВ за счет мономера, адсорбированного на них, так и в растворе за счет мономера, находящегося в “свободном” состоянии. Рост этих двух типов полимерных цепей может осуществляться независимо друг от друга, по крайней мере, на начальных этапах полимеризации, а экспериментально определяемая скорость полимеризации, будет являться результирующей для двух процессов: полимеризации на мицеллах и полимеризации в растворе. На рис. 3а в области мольных соотношений компонентов, соответствующих избытку мономера, представлены зависимости (кривые 1', 2'), рассчитанные по правилу аддитивности11 с учетом того, что с ростом концентрации скорость полимеризации “свободного” мономера падает (рис. 3б). Видно, что экспериментально определенные значения скорости полимеризации (кривые 1, 2) уменьшаются несколько сильнее, чем теоретически рассчитанные, свидетельствуя о том, что присутствие мицелл ПАВ дополнительно замедляет процесс полимеризации. Предположительно, такое снижение также связано со склонностью изучаемого мономера к самоассоциации в растворах. Ранее методом ЭПР-спектроскопии было показано, что локальная подвижность ионов ДДС в мицеллах меняется с ростом концентрации мономера в растворе вплоть до пятикратного его избытка, причем как в разбавленных растворах ПАВ (15 мМ) [24], так и в более концентрированных растворах (вплоть до 0.5 моль/л) [25]. Это означает, что присутствие мицелл ДДС причем как сферических, так и цилиндрических приводит не только к сорбции мономера на мицеллу, прежде всего, за счет электростатических взаимодействий, но и вызывает формирование дополнительных слоев мономеров, ассоциированных вокруг мицеллы за счет π–π-взаимодействий двойных связей. Заметим, что склонность непредельных соединений к ассоциации за счет π–π-взаимодействий хорошо известна из литературы [37] и находит подтверждение в ряде экспериментальных работ [38, 39]. Можно полагать, что именно такая дополнительная самоассоциация мономера вокруг мицелл приводит к снижению скорости либо за счет неблагоприятной для протекания полимеризации локализации двойных связей, либо за счет рекомбинации растущих цепей, о чем было упомянуто выше.
Однако остается вопрос о причинах значительного снижения скорости полимеризации как экспериментально определенной, так и приведенной для полимеризации в 150 мМ растворе ДДС. Согласно литературным данным [28], полимеризация мицеллообразующих мономеров замедляется при переходе от сферических к цилиндрическим мицеллам. Одна из причин такого снижения может заключаться в том, что при изменении формы мицеллы заметно уменьшается площадь, приходящаяся на полярную группу ДДС, которой по стерическим причинам может быть недостаточно для электростатического связывания мономера с каждым ионом ПАВ. Как следствие, мицелла “насыщается” электростатически связанными с ней ионами мономера при составах смеси ниже эквимольных, а слой мономера на поверхности цилиндрических мицелл оказывается менее плотным, чем для сферических мицелл. В результате, максимум скорости наблюдается при составах, не достигающих стехиометрии (рис. 3а), а сама скорость матричной полимеризации заметно падает вследствие снижения локальной концентрации мономера в слое противоионов.
При всей условности и неоднозначности такого предположения, несомненно требующего дополнительных экспериментальных подтверждений, оно объясняет как уменьшение состава смеси, отвечающего максимуму приведенной скорости (рис. 3а), так и заметное снижение максимальной скорости для полимеризации в 150 мМ растворе ДДС.
Кроме того, представляется вероятным и переход от полимеризации по zip-механизму к полимеризации по pick-up-механизму (или смешанному) матричной полимеризации при изменении формы мицеллы, так как тип полимеризации очень чувствителен к эффективности связывания мономера с матрицей, и повышение собственной ионной силы раствора наряду с изменением формы мицелл может оказаться критичным для эффективности связывания, а соответственно, типа матричной полимеризации. Изменение механизма полимеризации при переходе от сильно разбавленного раствора ДДС, содержащего сферические мицеллы ПАВ, к более концентрированному, содержащему цилиндрические мицеллы, подтверждается и ходом зависимости скорости полимеризации от концентрации мономера при фиксированной концентрации ПАВ (рис. 2), не имеющей четкого излома.
Полученные экспериментальные данные, позволяют рассчитать порядки реакции полимеризации по инициатору ni и мономеру nm, а также энергию активации процесса полимеризации Еа (таблица 1).
Из данных таблицы 1 видно, что полимеризация М в воде подчиняется общим закономерностям свободнорадикальной полимеризации и характеризуются порядками по мономеру (nm = 1) и инициатору (ni = 0.5) и типичным значением энергии активации 96 кДж/(моль К).
Введение ПАВ оказывает заметное влияние на кинетические параметры полимеризации, определенные в широком интервале концентрации мономера при фиксированном соотношении [М]/[ДДС] = 1. При относительно низких значениях концентрации ПАВ, отвечающих образованию сферических мицелл, nm возрастает и составляет 1.52, что свидетельствует об ассоциации мономера [28] и является характерным для матричной полимеризации [5], в то время как ni уменьшается до 0.33, а энергия активации уменьшается практически в 5 раз и составляет 20 кДж/(моль К). При повышении концентрации ДДС энергия активации возрастает в ∼2 раза, до 43 кДж/(моль К), однако оставаясь заметно ниже энергии активации свободнорадикальной полимеризации в воде, что не противоречит высказанным выше предложениям. При этом наблюдается дальнейшее, но незначительное снижение порядка полимеризации по инициатору, что характерно для полимеризации с обрывом цепи на первичных радикалах [40]. К сожалению, определить nm для более высоких значений концентрации ДДС, соответствующих образованию цилиндрических мицелл, в аналогичных условиях, т.е. при эквимольном соотношении компонентов, не удалось, так как в эксперименте скорость полимеризации изменяется незначительно с ростом концентрации мономера.
Таким образом, исследование кинетических особенностей полимеризации триметилметакрилоилоксиэтиламмоний метилсульфата в мицеллярных растворах ДДС показало, что полимеризация протекает по матричному механизму, причем zip-механизм матричной полимеризации, характеризующийся резким ускорением процесса в недостатке мономера и достигающий максимальной скорости в точке эквимольности, реализуется только при низкой концентрации ДДС при [М]/[ДДС] ≤ 1. В то же время, характер кинетических зависимостей и значения кинетических параметров, полученных для больших значений концентрации ПАВ, позволяют утверждать, что и в этом случае полимеризация протекает по матричному механизму, предположительно pick-up-механизму или смешанному механизму, по крайней мере, при [М]/[ДДС] ≤ 1.
Молекулярно-массовые характеристики полиэлектролитов
Еще одним доказательством матричного характера полимеризации являются молекулярно-массовые характеристики полимеров, полученных в мицеллярных растворах ДДС (рис. 4).
Рис. 4.
Зависимость Мw полиэлектролитов, полученных полимеризацией М в мицеллярных растворах ДДС, от мольного соотношения [М]/[ДДС]; растворитель 1 моль/л NaCl, Т = 25°С. [ДДС] = 100 (1), 200 (2), 300 мМ (3).

Данные рис. 4 наглядно демонстрируют, что среднемассовая молекулярная масса полиэлектролитов зависит экстремально от мольного соотношения компонентов в реакционной среде с характерным минимумом вблизи точки эквимольности (0.9–1.4) × 106 в зависимости от концентрации мономера. Увеличение соотношения мономер : ПАВ приводит к росту ММ, причем, чем выше концентрация мономера, тем более заметен рост ММ. Предположительно, такой эффект связан с прогрессирующим вкладом свободнорадикальной полимеризации мономера, не связанного с мицеллами ПАВ и накапливающегося в растворе по мере роста содержания мономера сверх эквимольного.
ЗАКЛЮЧЕНИЕ
Изучение кинетических особенностей полимеризации триметилметакрилоилоксиэтиламмоний метилсульфата в мицеллярных растворах противоположно заряженного додецилсульфата натрия показало, что полимеризация носит матричный характер, причем матричная полимеризация по zip- механизму, сопровождающаяся резким увеличением скорости, протекает только при мольном соотношении [М]/[ДДС], не превышающем стехиометрии, и в разбавленных растворах, содержащих сферические мицеллы ПАВ. Вместе с тем, и кинетические параметры полимеризации, и молекулярно-массовые характеристики полученных полиэлектролитов свидетельствуют о матричном характере процесса в недостатке мономера и при больших значениях концентрации ДДС.
Работа выполнена в рамках соглашения о совместных научных исследованиях между Волгоградским государственным техническим университетом и Санкт-Петербургским государственным университетом (№ С-119/7) и проектной части Госзадания (№ 4.3230.2017/4.6).
Список литературы
Каргин В.А., Кабанов В.А. // Докл. АН СССР. 1966. Т. 67. № 1. С. 124.
Hadjichristidis N., Iatrou H., Pitsikalis M., Mays J. // Prog. Polym. Sci. 2006. V. 31. № 12. P. 1068.
Yakimansky A.V. // J. Polym. Sci. C. 2005. V. 47. № 1. P. 1.
Papisov I.M. // J. Polym. Sci. B. 1997. V. 39. № 3–4. P. 122.
Polowinski S. // Prog. Polym. Sci. 2002. V. 27. № 3. P. 537.
Кабанов В.А., Петровская В.А., Каргин В.А. // Высокомолек. соед. А. 1968. Т. 10. № 4. С. 925.
Каргин В.А., Кабанов В.А., Каргина О.В. // Докл. АН СССР. 1965. Т. 161. № 1. С. 1131.
Паписов И.М., Кабанов В.А., Осада Е., Лескано Брито М., Реймонт Ж., Гвоздецкий А.Н. // Высокомолек. соед. А. 1972. Т. 16. № 11. С. 2462.
Холмберг К., Йёнссон Б., Кронберг Б., Линдман Б. Поверхностно-активные вещества и полимеры в водных растворах / Пер. с англ. под ред. Б.Д. Сумма. М.: БИНОМ. Лаборатория знаний, 2007.
Bilibin A.Yu., Shcherbinina T.M., Girbasova N.V., Lebedev V.T., Kulvelis Yu.V., Molchanov V.S., Zorin I.M. // Design. Monomers Polym. 2016. V. 19. № 5. P. 369.
Lerebours B., Perly B., Pileni M.P. // Prog. Colloid Polym. Sci. 1989. V. 79. P. 239.
Hartmann P.C., Dieudonné Ph., Sanderson R.D. // J. Coll. Int. Sci. 2005. V. 284. P. 289.
Gerber M.J., Walker L.M. // Langmuir. 2006. V. 22. P. 941.
Kline S.R. // Langmuir. 1999. V. 15. P. 2726.
Kline S.R. // J. Appl. Crystallogr. 2000. V. 33. P. 618.
Kuntz D.M., Walker L.M. // J. Phys. Chem. B. 2007. V. 111. № 23. P. 6417.
Walker L.M., Kuntz D.M. // Curr. Opin. Coll. Int. Sci. 2007. V. 12. P. 101.
Bilibin A.Yu, Shcherbinina T.M., Kondratenko Yu.A., Zorina N.A., Zorin I.M. // Coll. Polym. Sci. 2015. V. 293. № 4. P. 1215.
Zorin I.M., Makarov I.A., Ushkova T.S., Melnikov A.B., Antonov E.A., Bilibin A.Y. // Spec. Iss.: Modern Trends in Polymer Science. 2010. V. 296. № 1. P. 407.
Roy S., Dey J. // J. Coll. Int. Sci. 2007. V. 307. № 1. P. 229.
Summers M., Eastoe J. // Adv. Coll. Int. Sci. 2003. V. 100–102. P. 137.
Thünemann A.F. // Prog. Polym. Sci. 2002. V. 27. № 8. P. 1473.
Zorin I., Scherbinina T., Fetin P., Makarov I., Bilibin A. // Talanta. 2014. V. 130. P. 177.
Motyakin M.V., Shulevich Yu.V., Zakharova Yu.A., Vasserman A.M., Navrotskii A.V., Novakov I.A. // Colloid J. 2009. T. 71. № 5. P. 672.
Shulevich Yu.V., Motyakin M.V., Zakharova Yu.A., Dukhanina E.G., Wasserman A.M., Navrotskii A.V., Novakov I.A. // Colloid J. 2015. V. 77. № 1. P. 108.
Shulevich Yu.V., Zakharova Yu.A., Motyakin M.V., Ionova I.S., Dukhanina E.G., Navrotskii A.V., Novakov I.A. // Colloid J. 2018. V. 80. № 6. P. 751.
Shulevich Yu.V., Ilyin S.O., Dukhanina E.G., Ozerin A.S., Tutaev D.S., Navrotskii A.V., Kulichikhin V.G., Novakov I.A. // Russ. Chem. Bull. 2016. V. 65. № 5. P. 1161.
Егоров В.В., Зайцев С.Ю., Зубов В.П. // Высокомолек. соед. A. 1991. Т. 33. № 8. С. 1587.
Miura M., Kodama M. // Bull. Chem. Soc. Jpn. 1972. V. 45. P. 428.
Kubota Y., Kodama M., Miura M. // Bull. Chem. Soc. Jpn. 1973. V. 46. P. 100.
Zhao J., Fung B.M. // Langmuir. 1993. № 9. P. 1228.
Shulevich Yu.V., Kovaleva O.Yu., Navrotskii A.V., Zakharova Yu.A., Zezin A.B., Novakov I.A. // J. Polym. Sci. C. 2007. V. 49. № 12. P. 1286.
Polymer Science: a Comprehensive Reference / Eds. by K. Matyjaszewski, M. Moller. Amsterdam: Elsevier, 2012
Tan Y.Y., Challa G. // Pure Appl. Chem. 1981. V. 53. P. 627.
Zorin I.M., Podolskaya E.P., Bilibin A.Yu. // Eur. Polym. J. 2019. V. 110. P. 355.
Егоров В.В., Батракова Е.В., Зубов В.П. // Высокомолек. соед. A. 1990. Т. 32. № 5. С. 927.
Кабанов В.А. Избранные труды / Под ред. А.Б. Зезина. М.: Наука, 2010.
Казанцев О.А., Ширшин К.В., Барута Д.С., Сивохин А.П., Каморин Д.М., Квашенников А.И. // Клеи. Герметики. Технологии. 2011. № 5. С. 19.
Казанцев О.А., Сивохин А.П., Барута Д.С., Ширшин К.В., Каморин Д.М., Квашенников А.И. // Клеи. Герметики. Технологии. 2011. № 3. С. 30.
Гладышев Г.П., Попов В.А. Радикальная полимеризация при глубоких степенях превращения. М.: Наука, 1974.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)