

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 3, стр. 205-225
ТЕРМИЧЕСКОЕ ПОВЕДЕНИЕ ПОЛИ(АКРИЛОНИТРИЛ-СО-1-ВИНИЛИМИДАЗОЛА) В ПРОЦЕССЕ СТАБИЛИЗАЦИИ
Р. В. Томс a, b, А. Ю. Гервальд a, Н. И. Прокопов a, Н. И. Осипова b, c, А. В. Плуталова c, Е. В. Черникова b, c, *
a МИРЭА–Российский технологический университет,
Институт тонких химических технологий им. М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
c Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, д. 1, стр. 3, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 24.03.2022
После доработки 11.04.2022
Принята к публикации 20.04.2022
- EDN: HDVGPB
- DOI: 10.31857/S230811392270005X
Аннотация
Классической радикальной полимеризацией и полимеризацией с обратимой передачей цепи в ДМСО синтезированы сополимеры акрилонитрила и 1-винилимидазола. Показано, что реакционная способность 1-винилимидазола в сополимеризации ниже, чем у акрилонитрила (0.22 ± 0.02 и 0.88 ± 0.02 соответственно). Состав сополимера практически не изменяется на протяжении сополимеризации при мольном содержании 1-винилимидазола в мономерной смеси до 30%, что обеспечивает образование сополимеров с высокой композиционной однородностью. Механизм обратимой передачи цепи способствует низкой дисперсности сополимеров. Изучение процессов термостабилизации синтезированных сополимеров показывает, что молекулярно-массовое распределение играет незначительную роль в термическом поведении сополимеров акрилонитрила с 1-винилимидазолом в отличие от состава сополимера. Энергия активации реакции циклизации линейно возрастает с повышением содержания 1-винилимидазола в сополимере. Увеличение мольной доли 1-винилимидазола в сополимере приводит к более медленному развитию лестничной структуры. В целом 1-винилимидазол можно рассматривать как инертный в циклизации мономер. При постоянной температуре скорость образования лестничной структуры в атмосфере аргона и на воздухе резко различается. Конверсия нитрильных групп в группы –C=N–, равная 10%, достигается через 2.5 ч термообработки в аргоне или через 15 мин на воздухе. Существенная разница в скорости реакций циклизации в аргоне и на воздухе позволяет разработать новую стратегию получения формуемых из расплава прекурсоров углеродного волокна.
ВВЕДЕНИЕ
Области применения углеродного волокна интенсивно расширяются и в настоящее время не ограничиваются рынком аэрокосмической и оборонной промышленности, но включают в себя автомобилестроение, спортивные товары, ветроэнергетику, хранение сжатого газа и т.д. [1–6]. Следовательно, растет интерес к технологиям, позволяющим снизить затраты на производство углеродного волокна. Производство углеродного волокна представляет собой сложный процесс: синтез полимера, формование волокна, термоокислительную стабилизацию и карбонизацию [7]. В этой технологической цепочке получение прекурсора углеродных волокон на основе полиакрилонитрила методом формования из расплава вместо традиционного формования из раствора представляет заметный интерес [8]. Действительно, формование из расплава решает проблему утилизации органического растворителя, его регенерации и очистки [9]. Однако для сополимеров акрилонитрила (АН) оно ограничено способностью ПАН вступать в реакцию циклизации при нагревании [10]. Таким образом, сополимеры АН, перерабатываемые из расплава, должны иметь температуру течения ниже температуры начала циклизации. Это условие может быть достигнуто выбором соответствующего состава сополимера, его молекулярной массы и молекулярно-массового распределения и/или применением пластификатора.
Растущий интерес к сополимерам ПАН, перерабатываемым из расплава, стал заметным, начиная с 2000-х годов [11–16]. Первые попытки исследователей были направлены на использование уже известных сополимеров АН. С этой целью были изучены бинарные сополимеры акрилонитрила и метилакрилата (МА) и их тройные сополимеры с сомономером, содержащим кислотную группу [12, 17–26]. Закономерности их термостабилизации подробно описаны в работах [26–31]. Основная задача состоит в поиске способов увеличения времени “жизни” расплава сополимеров. Основные подходы к ее решению включают увеличение содержания МА в сополимере, снижение ММ сополимера и применение различных пластификаторов [21, 25, 26]. Однако типичное время “жизни” расплава для этого типа сополимеров при 200–225°С составляет около 30 мин, после чего вязкость быстро растет за счет образования лестничной структуры [23].
Получаемые волокна затем подвергаются термоокислительной стабилизации, однако она обычно протекает выше температуры течения полимера. Следовательно, перед проведением термоокислительной стабилизации необходима химическая модификация (сшивка) полученных волокон для предотвращения их плавления. Различные способы облучения сформованных волокон (электронно-лучевое, УФ, плазменное) приводят к образованию сшитого полимера, что предотвращает его течение в процессе термоокислительной стабилизации [32–35]. Однако использование облучения требует применения дополнительного оборудования. Другой подход основан на сополимеризации с фоточувствительным мономером, способным генерировать радикалы при УФ-облучении [36]. Такие мономеры обычно включают термостабильную бензофеноновую группу, которая запускает реакции сшивания под действием УФ-облучения [37]. В этом случае после формования волокон из расплава их облучают УФ-светом, а уже сшитый полимер подвергают термоокислительной стабилизации. В обоих случаях прочностные характеристики полученных волокон на основе ПАН и углеродных волокон были относительно невысокими [36].
Недавно было показано, что 1-винилимидазол (ВИМ) является перспективным мономером для получения расплавного ПАН. Сополимеры АН и ВИМ синтезировали радикальной сополимеризацией из смеси мономеров с мольной долей ВИМ fВИМ в диапазоне 10–25% в ДМФА в присутствии 1-додекантиола в качестве агента передачи цепи [38]. Сополимеры характеризуются широким ММР; их температура стеклования Tс уменьшается от 117 до 93°С с увеличением fВИМ от 13 до 19 мол. %. Однако при fВИМ = 19 мол. % авторы наблюдали вторую температуру стеклования при 160°C, обусловленную взаимодействиями полярных нитрильных групп [39]. При нагревании все сополимеры теряют массу при температуре выше ~310°C и характеризуются выходом коксового остатка, сравнимым с показателем для коммерческих волокон ПАН [40]. После нагревания на воздухе выше 210°С сополимеры теряют способность растворяться и меняют цвет на темно-коричневый за счет образования лестничной структуры. Однако сополимер, полученный из смеси с fВИМ = 18 мол. %, способен течь в атмосфере азота и при этом не подвергаться циклизации. Его степень кристалличности составляет ~ 35%, прочность на разрыв равна 1.4–1.6 сН/дтекс, удлинение при разрыве 7.1–11.2 [41]. Недавно были получены более подробные механические характеристики волокна на основе сополимера АН и ВИМ, синтезированного из fВИМ = 18 мол. % в ДМФА и сформованного из расплава, стабилизированного волокна и карбонизированного волокна [42]. Предел прочности при растяжении для исходного волокна из сополимера с М = (36–47) × 103 составляет от 300 до 491 МПа, а для стабилизированного волокна – от 750 до 975 МПа. Модуль упругости исходного волокна находится в пределах 90–110 ГПа, а для стабилизированного волокна варьируется от 128 до 158 в зависимости от ММ сополимера. Карбонизированное волокно имеет предел прочности при растяжении 1.43 ГПа и модуль упругости 156 ГПа. Эти характеристики относительно невысокие, однако углеродные волокна с такими характеристиками могут найти широкое применение в областях, не требующих высокой прочности или модуля упругости.
Дальнейшее увеличение долговечности расплава сополимеров АН и ВИМ может быть достигнуто добавлением пластификаторов [7, 43, 44]. B.L. Batchelor с сотрудниками [45] разработал новый пластификатор на основе олигомера АН и метил-2-(1Н-имидазол-1-ил)акрилата (20 мол. %) с Mn = (2–5) × 103, который оказал наиболее существенное влияние на свойства расплава. Например, температура стеклования сополимера АН и ВИМ снизилась на 40°C, а напряжение сдвига уменьшилось вдвое после добавления 8 мас. % олигомера. При этом температура начала циклизации изменилась всего на 5–10°С по сравнению с исходными значениями. Полный цикл от формования расплава пластифицированного сополимера АН и ВИМ в волокно до его последующих термоокислительной стабилизации и карбонизации привели к получению углеродного волокна с пределом прочности при растяжении 1.9 ГПа и модулем упругости 190 ГПа.
Приведенные результаты позволяют считать, что сополимеры АН и ВИМ действительно являются потенциальными кандидатами для создания расплавного ПАН. В упомянутых выше работах условия формования сополимеров выбирали, исходя из экспериментально установленных значений времени “жизни” расплава при разной температуре. Однако температурные режимы протекания циклизации при нагревании этих сополимеров в инертной атмосфере и на воздухе в литературе не описаны. Очевидно, что данная информация необходима для оптимизации условий формования волокна и его термостабилизации.
Сомономеры в макромолекуле ПАН не только нарушают последовательность звеньев АН, но и могут ускорять реакцию циклизации (виниловые кислоты или амиды), ингибировать ее (стирол) или играть роль инертного мономера (алкилакрилаты) [46]. Химическая структура ВИМ позволяет предположить, что ВИМ не способен ускорять циклизацию, однако его роль в процессах термоокислительной стабилизации остается неизвестной.
В настоящей работе для синтеза сополимеров акрилонитрила с 1-винилимидазолом различного состава в растворе ДМСО использовали классическую радикальную и ОПЦ-сополимеризацию. Применяя методы ДСК, ТГА и ИК-фурье-спектроскопии, впервые было проведено сравнительное исследование термического поведения полученных сополимеров в инертной атмосфере и на воздухе в зависимости от состава сополимера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Акрилонитрил (99%, “Aldrich”) и 1-винилимидазол (98%, “Fluka”) перед использованием перегоняли при атмосферном и пониженном давлении, соответственно. Персульфат калия (ПСК, 98%, “Aldrich”) применяли без дополнительной очистки. ОПЦ-агент дибензилтритиокарбонат (C6H5CH2–S–C(=S)–S–CH2C6H5, БТК) был синтезирован и охарактеризован, как описано ранее [47]. ДМСО (99%, “Fluka”), ДМФА (ВЭЖХ, “Fluka”) перед использованием перегоняли при пониженном давлении.
Радикальную сополимеризацию проводили при 55°С в атмосфере аргона в трехгорлой колбе емкостью 100 мл, снабженной верхнеприводным перемешивающим устройством с мешалкой якорного типа. В колбу помещали раствор, содержащий расчетное количество ПСК (6.5 × 10–3 моль/л) в ДМСО, и загружали смесь АН и ВИМ. Общая концентрация мономеров в ДМСО составляла 20 мас. %, а мольная доля ВИМ в мономерной смеси – 0, 5, 10, 15, 20, 25 и 30%. Реакционную смесь барботировали аргоном в течение 30 мин, колбу помещали в предварительно нагретую до 55°С баню. В заданное время отбирали пробы для определения конверсии, молекулярно-массовых характеристик и состава сополимеров. После полимеризации реакционные смеси быстро охлаждали до комнатной температуры, при необходимости разбавляли ДМСО и затем осаждали в избытке воды. Осадок отфильтровывали, промывали водой, затем сушили в вакууме до постоянной массы. Конверсию мономера определяли гравиметрически.
ОПЦ-сополимеризацию осуществляли при 55°С. Реакционные смеси готовили растворением необходимого количества ПСК (10–3 моль/л) и БТК (3 × 10–3 моль/л) в ДМСО. Затем добавляли мономеры, суммарная концентрация которых в ДМСО составляла 40 мас. %. Приготовленные растворы заливали в ампулы, дегазировали путем трехкратного повторения циклов замораживание–откачка–размораживание и запаивали. Ампулы погружали в термостат, предварительно нагретый до 55°С, на необходимое время. Выделение сополимеров проводили, как описано выше.
Для определения констант сополимеризации сополимеры синтезировали радикальной сополимеризацией в ДМСО при конверсии мономера ниже 5%.
Для анализа сополимеров методами ИК-НПВО-спектроскопии и ДСК образцы готовили путем растворения 8–10 мас. % сополимера в ДМСО. Растворы отливали на стеклянную подложку. Затем образцы помещали в вакуумную печь при 80°C на ночь. Полученные пленки промывали водой в течение 24 ч для удаления остаточного ДМСО, сушили до постоянной массы и отделяли от поверхности. Пленки со средней толщиной 20–50 мкм разрезали на куски размером 20 × 20 мм и анализировали.
Среднюю молекулярную массу и дисперсность (Ð = Mw/Mn) определяли методом ГПХ. Измерения проводили в ДМФА, содержащем 0.1 мас. % LiBr, при 50°C со скоростью потока 1.0 мл/мин на хроматографе GPC-120 “PolymerLabs” (“Hichrom Limited”, Великобритания), оборудованном рефрактометром и двумя колонками PLgel MIXED B в диапазоне М = (5 × 102)–(1 × 107). Молекулярную массу рассчитывали по узкодисперсным линейным стандартам ПММА с ММ в интервале от 800 до 2 × 106.
Состав сополимера анализировали с помощью спектроскопии ЯМР 1H и ИК-НПВО. Спектры ЯМР 1H растворов сополимеров в ДМСО-d6 регистрировали на импульсном Фурье-спектрометре “Bruker DPX-500” с частотой 500 МГц. Внутренним стандартом служил тетраметилсилан. ИК-Фурье-спектроскопию в режиме НПВО (кристалл алмаза) регистрировали на ИК-Фурье-спектрометре “Spectrum Two” (“PerkinElmer”) в диапазоне 4000–400 см–1. Для количественного анализа состава пленок синтезированных сополимеров получали калибровочную кривую, полученную из смесей мономеров известного мольного соотношения (мольную долю ВИМ изменяли в интервале 0.5–70 мол. %). Для расчетов использовали соотношение интенсивностей А характеристических полос валентных колебаний имидазольного кольца νC–N= = 665 см–1 (ВИМ) и нитрильной группы νCN = 2229 см–1 (АН) [48]. Для подтверждения достоверности калибровочных кривых брали сополимеры известного состава (определенного с помощью спектроскопии ЯМР 1Н).
Дифференциальную сканирующую калориметрию применяли для изучения термостабилизации сополимеров ПАН. Термограммы регистрировали на приборе “Netzsch DSC 204” (“Netzsch”, Германия) в атмосфере сухого газа (воздух, аргон) при расходе 100 мл/мин в диапазоне 30–500°С в динамическом режиме при скорости нагревания в интервале 5–20 град/мин в аргоне и 10 град/мин на воздухе.
Энергию активации реакций определяли методом Киссинджера [49]
(1)
$ - \frac{{{{E}_{a}}}}{R} = \frac{{d\left[ {{\text{ln}}\left( {\frac{\varphi }{{T_{p}^{2}}}} \right)} \right]}}{{d\left( {\frac{1}{{{{T}_{p}}}}} \right)}},$ИК-Фурье-спектры в режиме НПВО (кристалл алмаза) пленок, подвергнутых термообработке на воздухе (при 180, 200, 225 и 250°С) и в аргоне (при 225 и 250°С), регистрировали на ИК-фурье-спектрометре “Spectrum Two” (“Perkin-Elmer”) в диапазоне 4000–400 см–1.
Долю непрореагировавших нитрильных групп φCN [50] и индекс стабилизации Es находили по уравнениям [51]
(2)
${{\varphi }_{{{\text{CN}}}}} = \frac{{{{A}_{{2240~\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}{{{{A}_{{2240~\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}} + f{{A}_{{1590~\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}$(3)
${{E}_{s}} = \frac{{{{A}_{{1590\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}{{{{A}_{{2240~\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}$Здесь ${{A}_{{2240~\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}$ – интенсивность полосы, отнесенной к группе –C≡N, ${{A}_{{1590\,{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}$ – интенсивность полосы, отвечающей группе −C=N−, f – отношение молярных коэффициентов экстинкции, равное 0.29 [50].
Термическую стабильность сополимеров изучали методом ТГА. Анализ проводили на синхронном термоанализаторе STA 449 F3 Jupiter (“Netzsch”). Потерю массы образца сополимера массой 5–10 мг, помещенного в корундовый тигель, анализировали в режиме линейного нагрева (10 град/мин от 25 до 600°C) в токе аргона 50 мл/мин или на воздухе.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез сополимеров
Сополимеризацию АН и ВИМ проводили сначала в условиях классической радикальной сополимеризации при суммарной концентрации мономера 20 мас. % в диапазоне fВИМ = 0–30 мол. %. Сополимеризация протекает с достаточно высокой скоростью, и конверсия мономера 60–70% достигается в течение 2 ч.
Состав сополимеров анализировали методами ИК-фурье-спектроскопии и ЯМР-спектроскопии. Типичные спектры приведены на рис. 1. Для сополимеров АН и ВИМ валентные колебания СН имидазольного кольца наблюдаются при 3117 см–1 (ν=CH), валентные колебания СН2 основной цепи – при (νasCH) и 2865 см–1 (νsCH), валентные колебания нитрильной группы C≡N – при 2443 см–1 (νC≡N), валентные колебания групп C=C – при 1620 см–1 (νC=C), валентные колебания групп C=N имидазольного кольца – при ~1500 см–1 (νC=N), валентные колебания групп C–N имидазольного кольца – при ~1229, 910 и 665 см–1 (νC–N=), деформационные колебания –СН – при 1082 см–1. Полосы при 2443 и 665 см–1, относящиеся к АН и ВИМ, не перекрываются с другими полосами и поэтому подходят для аналитических целей определения состава сополимеров.
Рис. 1.
Спектры ИК-НПВО (a) и ЯМР 1H (б) сополимеров АН и ВИМ. Цветные рисунки можно посмотреть в электронной версии.

Повышение содержания ВИМ в смеси мономеров приводит к увеличению мольной доли ВИМ в сополимере (рис. 2а). Кроме того, средний состав сополимера остается постоянным на протяжении всей сополимеризации при содержании ВИМ в мономерной смеси до 20 мол. %. На рис. 2б приведена диаграмма состава сополимера. Константы сополимеризации определяли методами наименьших квадратов [52] (rВИМ = 0.22 ± 0.02 и rАН = 0.88 ± 0.02) и Файнемана–Росса [53] (rВИМ = 0.3 ± 0.2 и rАН = 0.88 ± 0.02). Оба метода дают схожие результаты и показывают, что АН имеет более высокую реакционную способность, чем ВИМ. Азеотропному составу при сополимеризации в растворе ДМСО соответствует состав смеси fВИМ = 0.13 и fАН = 0.87. Константы сополимеризации в ДМСО отличаются от значений, определенных в ДМФА (rАН = 0.12, rВИМ = 0.24) [48]. Для АН характерна разница между коэффициентами реакционной способности, определенными в разных растворителях; аналогичная тенденция наблюдалась при классической радикальной сополимеризации АН и метилакрилата [54].
Рис. 2.
а – Зависимость среднего состава сополимера 〈FВИМ〉 от конверсии мономера для сополимеров, образующихся при радикальной сополимеризации АН и ВИМ при fВИМ = 5 (1), 10 (2), 15 (3), 20 (4), 25 (5) и 30 мол. % (6); состав сополимеров определен методом ИК-НПВО-спектроскопии (темные точки) и методом ЯМР1Н (светлые). б – Зависимость мгновенного состава сополимера FВИМ от состава исходной мономерной смеси fВИМ.

Термическое поведение сополимеров АН при циклизации зависит от химической природы сомономера, его содержания и микроструктуры цепи [27, 31]. ПАН, синтезированный по радикальному механизму, является атактическим, и его макромолекулы имеют спиральную конформацию [39]. Формирование лестничной структуры за счет реакции циклизации вызывает избыточное напряжение в этой конформации и может привести к прекращению развития сопряженной структуры [55]. Включение другого мономера в цепь по-разному влияет на реакцию циклизации ПАН. Он может инициировать или ингибировать циклизацию или быть инертным мономером [56]. Поскольку роль ВИМ в циклизации ПАН неизвестна, представляет интерес оценить микроструктуру цепей на основе определенных значений констант сополимеризации.
Триадный состав при низкой конверсии мономера можно оценить из уравнений [57]
(4)
${{F}_{{{\text{АН--АН--АН}}}}} = {{\left( {1 - \frac{1}{{1 + \frac{{{{r}_{{{\text{АН}}}}}{{f}_{{{\text{АН}}}}}}}{{{{f}_{{{\text{ВИМ}}}}}}}}}} \right)}^{2}}$(5)
${{F}_{{{\text{АН--АН--ВИМ}}}}} = \frac{2}{{1 + \frac{{{{r}_{{{\text{АН}}}}}{{f}_{{{\text{АН}}}}}}}{{{{f}_{{{\text{ВИМ}}}}}}}}}\left( {1 - \frac{1}{{1 + \frac{{{{r}_{{{\text{АН}}}}}{{f}_{{{\text{АН}}}}}}}{{{{f}_{{{\text{ВИМ}}}}}}}}}} \right)$(6)
${{F}_{{{\text{АН--ВИМ--АН}}}}} = {{\left( {\frac{1}{{1 + \frac{{{{r}_{{{\text{АН}}}}}{{f}_{{{\text{АН}}}}}}}{{{{f}_{{{\text{ВИМ}}}}}}}}}} \right)}^{2}}$Мгновенная среднечисловая длина последовательности АН, 〈NАН〉n и ВИМ, 〈NВИМ〉n может быть оценена следующим образом:
(7)
${{\langle {{N}_{{{\text{АН}}}}}\rangle }_{n}} = \frac{1}{{1 - {{p}_{{{\text{AA}}}}}}}\quad {\text{и}}\quad {{\langle {{N}_{{{\text{ВИМ}}}}}\rangle }_{n}} = \frac{1}{{1 - {{p}_{{{\text{BB}}}}}}},$(8)
$\begin{gathered} {{p}_{{{\text{AA}}}}} = \frac{{{{r}_{{{\text{АН}}}}}{{f}_{{{\text{АН}}}}}}}{{{{f}_{{{\text{АН}}}}}\left( {{{r}_{{{\text{АН}}}}} - 1} \right) + 1}}\quad {\text{и}} \\ {{p}_{{{\text{BB}}}}} = \frac{{{{r}_{{{\text{ВИМ}}}}}{{f}_{{{\text{ВИМ}}}}}}}{{{{f}_{{{\text{ВИМ}}}}}\left( {{{r}_{{{\text{ВИМ}}}}} - 1} \right) + 1}} \\ \end{gathered} $Триадный состав и среднечисловая длина последовательности звеньев АН и ВИМ приведены в табл. 1. Согласно данным табл. 1, в триадном составе макромолекул преобладают триады FАН–АН–АН и FАН–АН–ВИМ. Среднечисловая длина последовательности ВИМ равна единице в области fВИМ до 30 мол. %. Таким образом, можно предположить, что последовательности звеньев АН в основном нарушены одиночными звеньями ВИМ. Очевидно, что снижение содержания АН в мономерной смеси приводит к падению 〈NАН〉n.
Таблица 1.
Теоретический триадный состав и среднечисловая длина последовательности звеньев АН и ВИМ в сополимере
| ВИМ, мол. % | FAН–AН–AН | FAН–AН–ВИМ | FAН–ВИМ–AН | 〈NAН〉n | 〈NВИМ〉n | $\frac{{\langle {{\sigma }^{2}}\rangle }}{{{{{\langle {{\sigma }^{2}}\rangle }}_{{{\text{max}}}}}}}$ × 103 |
|---|---|---|---|---|---|---|
| 5 | 0.89 | 0.11 | 0 | 17.0 | 1.0 | 0.10 |
| 10 | 0.79 | 0.20 | 0.01 | 9.0 | 1.0 | 0 |
| 15 | 0.69 | 0.28 | 0.03 | 6.0 | 1.0 | 0 |
| 20 | 0.61 | 0.34 | 0.05 | 4.5 | 1.0 | 0.04 |
| 25 | 0.53 | 0.40 | 0.07 | 3.6 | 1.1 | 0.11 |
| 30 | 0.45 | 0.44 | 0.11 | 3.0 | 1.1 | 0.27 |
Состав мономерной смеси постепенно изменяется с увеличением конверсии мономера p для мономеров с различной реакционной способностью. Это приводит к изменению состава сополимера и, следовательно, к возникновению конверсионной неоднородности по составу. Последнюю можно охарактеризовать дисперсией состава: $\langle {{\sigma }^{2}}\rangle = \langle F_{1}^{2}\rangle - {{\langle {{F}_{1}}~\rangle }^{2}},$ где $\langle {{F}_{1}}\rangle $ – средняя мольная доля мономера М1 в сополимере, а $\langle F_{1}^{2}\rangle $ определяется уравнением $\langle F_{1}^{2}\rangle = \frac{1}{p}\int_0^p {F_{1}^{2}dp} $. Для сравнения удобно использовать отношение текущего значения дисперсии к ее максимальному значению, полученному в случае смеси гомополимеров одного и того же среднего состава: ${{\langle {{\sigma }^{2}}\rangle }_{{{\text{max}}}}} = \langle {{F}_{1}}\rangle \langle {{F}_{2}}\rangle $. Соотношение текущих и максимальных значений дисперсии состава, рассчитанных при p = 70%, представлено в табл. 1. Эти значения являются достаточно низкими, что подтверждает высокую однородность состава сополимеров.
Сополимеры, образующиеся при конверсии мономера ниже 10–15%, имеют унимодальное ММР, типичное для радикальной полимеризации. Однако с увеличением продолжительности реакции ММР сополимеров уширяется. Повышение содержания ВИМ в мономерной смеси приводит к уширению ММР образующихся сополимеров (рис. 3а). Сополимеры характеризуются Mn = (100–160) × 103, дисперсностью ÐM = 1.8–2.0 при низкой конверсии мономера и Мn = (180–230) × 103, ÐM = 2.7–4.2 при высокой конверсии мономера. Столь значительное уширение ММР при радикальной полимеризации в растворе не свойственно для гомополимеризации АН. Следовательно, за этот процесс ответственны побочные реакции с радикалом ВИМ. Такое предположение согласуется с данными A. Chapiro [58] об образовании сшитого полимера в концентрированном растворе полимера в мономере, вероятно, за счет атаки радикала роста на двойные связи имидазольных колец в полимере. Кроме того, этот результат согласуется с данными ГПХ для сополимеров АН и ВИМ, синтезированных радикальной сополимеризацией в ДМФА, о которых сообщалось в работе [38].
Рис. 3.
Кривые ГПХ сополимеров АН и ВИМ, синтезированных через 3 ч реакции: а – классическая радикальная сополимеризация, fВИМ = 0 (1), 10 (2), 20 (3), 25 (4) и 30 мол. % (5); б – ОПЦ-сополимеризация, fВИМ = 10 (1), 15 (2), 20 (3), 25 (4) и 30 мол. % (5); в – ОПЦ-сополимеризация в присутствии 0.5 (1), 1 (2), 2 (3) и 5 мол. % уксусной кислоты по отношению к ВИМ (4), fВИМ = 20 мол. %.

Для синтеза сополимеров также использовали ОПЦ-полимеризацию, чтобы проверить возможное влияние ширины ММР сополимеров на термостабилизацию сополимеров АН. Известно, что ОПЦ-полимеризация ВИМ обычно приводит к низкой конверсии мономера [59]. Такой недостаток можно преодолеть при сополимеризации с более активным мономером АН, взятым в избытке. ОПЦ-сополимеризацию проводили в условиях, сходных с классической радикальной сополимеризацией, за исключением того, что использовали общую концентрацию мономера 40 мас. %, а концентрация ПСК составляла 10-3 моль/л, чтобы обеспечить достаточное количество “живых” цепей, образованных по механизму ОПЦ. Оказалось, что максимальная конверсия мономера (13–16%) достигается в течение 1 ч, а затем ОПЦ-сополимеризация быстро затухает. Увеличение содержания ВИМ в мономерной смеси ускоряет замедление реакции. При этом ОПЦ-гомополимеризация АН в ДМСО в присутствии БТК протекает без видимого замедления [60]. Таким образом, наблюдаемое явление вызвано участием в сополимеризации ВИМ. Причиной замедления могут быть побочные реакции, такие как перекрестный и квадратичный обрыв радикальных интермедиатов, образующихся при ОПЦ-полимеризации [61]. В то же время сополимеры, образованные по механизму ОПЦ, имеют более узкое ММР (рис. 3б). Они характеризуются Mn ~ (50–90) × 103 и дисперсностью ÐM = 1.56–1.68. Недавно было показано, что ОПЦ-гомополимеризация ВИМ в уксусной кислоте приводит к высокому выходу полимера с контролируемой ММ и узкой ММР [59]. В связи с этим ОПЦ-сополимеризацию АН и ВИМ при fВИМ = 20 мол. % провели в условиях, аналогичных рассмотренным выше, но при меньшей концентрации БТК и в присутствии различных количеств уксусной кислоты. Во всех случаях достигается высокая конверсия мономера (70–90%), а ММР имеет тенденцию к сужению (ÐM снижается от 3.2 до 1.2) с ростом содержания уксусной кислоты (рис. 3в).
Состав сополимеров оказался сходным с составом сополимеров, полученных классической радикальной сополимеризацией при том же составе мономерной смеси и конверсии. Таким образом, “классические” сополимеры и сополимеры на основе агентов ОПЦ различаются только шириной ММР и ММ.
Термическая стабилизация в аргоне
Динамический термический анализ. Термическое поведение сополимеров изучали методом ДСК в динамических условиях нагревания. В соответствии с химической структурой ВИМ можно ожидать, что его включение в макромолекулу может еще больше увеличить жесткость цепи. Действительно, температура стеклования Tс сополимера повышается с ростом мольной доли ВИМ в сополимере независимо от ММР и ММ сополимеров (табл. 2). Во всех случаях значения Tс выше, чем у ПАН (105°С), и ниже, чем у ПВИМ (176°С) [62, 63].
Таблица 2.
Анализ термограмм ДСК сополимеров АН–ВИМ, снятых в атмосфере аргона
| Система | fВИМ, мол. % | Механизм | Mn × 10–3 | Tс, °C | Tp, °C | −ΔH, Дж/г | −ΔH/ΔT, Дж/(г K) | Тепловой поток при Tp, Вт/г | Ea, кДж/моль |
|---|---|---|---|---|---|---|---|---|---|
| ВИМ-0 | 0 | Классическая полимеризация | 62.1 | 105 | 277 | 794 | 189 | 37.5 | 99 ± 8 |
| ВИМ-5 | 5 | Классическая полимеризация | 60.3 | 115 | 306 | 476 | 42.1 | 8.0 | 98 ± 4 |
| ВИМ-10 | 10 | Классическая полимеризация | 56.6 | 117 | 317 | 476 | 16.3 | 3.5 | 110 ± 1 |
| ОПЦ-полимеризация | 26.0 | 112 | 313 | 456 | 22.9 | 4.8 | – | ||
| ВИМ-15 | 15 | Классическая полимеризация | 55.4 | 114 | 325 | 483 | 11.3 | 2.1 | 118 ± 2 |
| ОПЦ-полимеризация | 23.5 | – | 322 | 453 | 10.7 | 2.3 | – | ||
| ВИМ-20 | 20 | Классическая полимеризация | 53.5 | 123 | 329 | 470 | 10.1 | 1.8 | 122 ± 6 |
| ОПЦ-полимеризация | 22.1 | 113 | 331 | 377 | 7.6 | 1.3 | – | ||
| ВИМ-25 | 25 | Классическая полимеризация | 51.6 | 125 | 336 | 380 | 6.3 | 1.4 | – |
| ОПЦ-полимеризация | 21.9 | 120 | 335 | 644 | 10.3 | 2.1 | – | ||
| ВИМ-30 | 30 | Классическая полимеризация | 49.0 | 130 | 338 | 330 | 5.7 | 1.3 | 134 ± 5 |
| ОПЦ-полимеризация | 18.0 | 129 | 341 | 603 | 10.4 | 2.0 | – |
В атмосфере аргона реакция циклизации ПАН протекает в узком интервале температур с интенсивным тепловыделением (рис. 4, кривые 1). Введение ВИМ в ПАН приводит к смещению термограмм на 30°С и более в высокотемпературную область и к падению интенсивности теплового потока (рис. 4а, кривые 2–7, рис. 3б, кривые 2–6). Энтальпия реакции циклизации ΔH и величина ΔH/ΔT, отражающая количество тепла, выделяющегося в ходе температурного интервала реакции циклизации, ниже чем для ПАН и незначительно уменьшаются с ростом содержания ВИМ в сополимере (табл. 2). Согласно расчетам (табл. 1), единичные звенья ВИМ разбивают последовательность звеньев АН и длина последних уменьшается с увеличением содержания ВИМ в цепи. Энергия активации реакции циклизации была определена по методу Киссинджера [49] на основании термограмм сополимеров, зарегистрированных в аргоне при различных скоростях нагревания (рис. 5а). Обработка данных по уравнению (1) приводит к линейным зависимостям $\ln (\varphi {\text{/}}T_{p}^{2})$ от 1/Tp (рис. 5б). Рассчитанные по этим данным значения энергии активации реакции циклизации суммированы в табл. 2.
Рис. 4.
Термограммы ДСК сополимеров АН и ВИМ, полученных классической радикальной (а) и ОПЦ-сополимеризацией (б). а: fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5), 25 (6) и 30 мол. % (7); б: fВИМ = 0 (1), 10 (2), 15 (3), 20 (4), 25 (5) и 30 мол. % (6). Скорость нагревания 10 град/мин, среда – аргон.
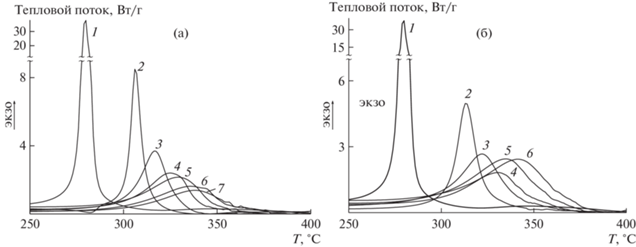
Рис. 5.
Термограммы ДСК сополимеров АН и ВИМ, полученные при fВИМ = 20 мол. % в среде аргона при скорости нагревания 5 (1), 10 (2), 20 (3) и 30 град/мин (4) (а), а также зависимость $\ln (\varphi {\text{/}}T_{p}^{2})$ от 1/Tp для сополимеров АН и ВИМ при fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5) и 30 мол. % (6) (б).
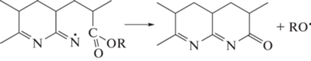
Энергия активации линейно возрастает с повышением содержания ВИМ в сополимере в пределах точности измерения (рис. 6). Значение энергии активации сополимера, полученного из мономерной смеси с fВИМ = 10 мол. %, аналогично значению энергии активации сополимеров АН и алкилакрилатов того же состава, синтезированных в аналогичных условиях [64]. Кроме того, введение ВИМ в ПАН вызывает такой же сдвиг термограмм в высокотемпературную область, как и для сополимеров АН и алкилакрилатов. Известно, что в последнем случае алкилакрилаты участвуют в реакции передачи цепи, приводящей к прекращению развития лестничной структуры и образованию нового полимерного радикала, способного инициировать циклизацию. Детальный механизм реакции или повторного инициирования циклизации неизвестен. Однако можно предположить, что радикал RO• может отрывать подвижный водород от макромолекулы, а углеродцентрированный радикал превращается в азотцентрированный радикал, способный повторно инициировать реакцию циклизации:


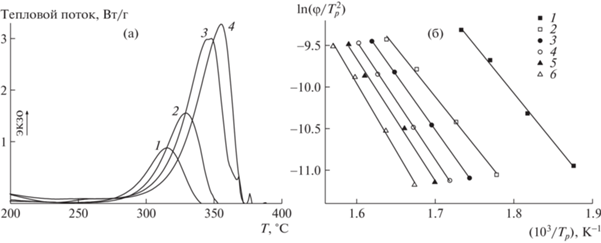
На рис. 7 представлены термограммы ТГА сополимеров АН и ВИМ, полученные в атмосфере аргона. Как видно, термостойкость сополимеров несколько повышается с увеличением содержания ВИМ в полимерной цепи. Между тем возрастание мольной доли ВИМ в сополимере приводит к большей потере массы при более высоких температурах. В интервале температур 20–600°С потеря массы чистого ПАН происходит в две стадии, а потеря массы сополимеров АН и ВИМ – в одну стадию. Реакция циклизации и деструкция цепей протекают в аналогичном интервале температур. Наблюдаемая потеря массы может быть вызвана образованием летучих продуктов (например, HCN, H2 и т.д.) и/или разрывом основной цепи, вызванным побочной реакцией передачи цепи через радикал –C=N• [65].
Рис. 7.
Зависимость потери массы от температуры в аргоне сополимеров АН и ВИМ, полученных из мономерных смесей разного состава. fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5) и 30 мол. % (6).

Изотермический анализ. Реакция циклизации сопровождается изменениями химической структуры макромолекул, которые можно проследить с помощью ИК-фурье-спектроскопии [66]. Пленки, приготовленные из сополимеров различного состава, прогревали в атмосфере аргона при 225 или 250°С в течение необходимого времени, а затем анализировали методом спектроскопии ИК-НПВО. На рис. 8 представлены спектры ИК-НПВО пленок сополимера, полученного при fВИМ = 10 мол. %, подвергнутых термообработке в атмосфере аргона при 225 и 250°С.
Рис. 8.
Спектры ИК-НПВО пленок образца ВИМ-10, подвергнутых термообработке в атмосфере аргона при 225 (а) и 250°С (б) при разном времени выдержки: 0 (1), 15 (2), 30 (3), 60 (4), 120 мин (5) (а) и 0 (1), 5 (2), 20 (3), 60 (4), 150 мин (5) (б).

Спектры ИК-НПВО сополимеров выявляют характерные полосы поглощения звеньев АН и ВИМ [48]. Валентные колебания нитрильной группы в звене АН наблюдаются при 2243 см–1 (νC≡N), групп СН имидазольного кольца – при 3110 см–1. Полосы при 667, 750, 826 и 907 см–1 можно отнести к внеплоскостным и плоскостным деформационным колебаниям кольца ВИМ. Полосы 1083, 1110, 1228 и 1290 см–1 соответствуют валентным и плоскостным деформационным колебаниям групп СН имидазольного кольца. Полоса 1495 см–1 может быть отнесена к валентным колебаниям групп С–С и С–N в ВИМ. Изгибные колебания групп СН основной цепи (δCHH) наблюдаются при 1450 см–1 и валентные колебания групп СН – при 2940 (νsC−H) и 2870 см–1 (νaC−H). В ходе циклизации происходит превращение группы −C≡N в группу –C=N-. Если это единственная реакция, протекающая при термообработке, то интенсивность полосы 2243 см–1 (νC≡N) должна уменьшиться, а затем появятся новые полосы при 1580 (ν−C=N−) и 1612 см–1 (νC=C), отвечающие образованию лестничной структуры [60]. При нагревании при 225°С интенсивность полосы 2243 см–1 несколько уменьшается (рис. 8а). Отчетливые полосы при 1580 и 1612 см–1 наблюдаются после 120 мин нагревания. Одновременно увеличивается интенсивность полос при 1412, 1363 и 1228 см–1.
Очевидные изменения видны при нагревании пленок сополимера при 250°С (рис. 8б). Наряду с уменьшением интенсивности полосы при 2243 см–1 данная полоса расщепляется на две полосы поглощения с максимумами при 2243 и 2200 см–1 (группа −C≡N, участвующая в сопряжении). Развитие лестничной структуры начинается при меньшем времени термообработки. Этому отвечает появление новых полос поглощения при 3350 (νNH), 1367 (δC−H, CH), 1237 и 1150 см–1 (νC−C, νC−N). При этом интенсивность колебаний групп СН основной цепи при 1450, 2940 и 2870 см–1 остается постоянной.
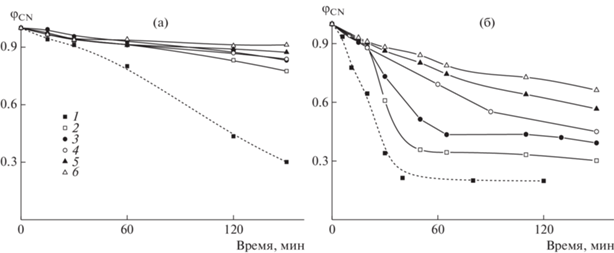
Доля непрореагировавших нитрильных групп φCN, рассчитанная по уравнению (2), зависит как от времени воздействия, так и от температуры. Зависимость φCN от времени термообработки при 225 и 250°С представлена на рис. 9. Низкая конверсия нитрильных групп наблюдается при 225°С независимо от содержания ВИМ в сополимере (рис. 9а). После выдержки пленок сополимера в течение 2 ч при 225°С они становятся светло-желтыми в отличие от пленки ПАН, которая становится темно-коричневой. Повышение температуры приводит к увеличению скорости циклизации (рис. 9б). Примечательно, что начальная скорость циклизации сополимеров ниже, чем у ПАН, и не зависит от содержания ВИМ. Однако через ~20 мин скорость циклизации увеличивается с уменьшением содержания ВИМ в сополимере. Этот результат может свидетельствовать об участии звеньев ВИМ в реакции циклизации.
Рис. 9.
Зависимости φCN от времени термообработки пленок сополимеров АН и ВИМ в аргоне при 225 (а) и 250°С (б). fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5) и 30 мол. % (6).
Индекс стабилизации Es (уравнение (3)) показывает превращение –C≡N в –C=N– и может рассматриваться как еще один количественный параметр реакции циклизации [51]. Как видно на рис. 10, при 225°С лестничная структура не формируется, а при 250°С она более развита. Увеличение содержания ВИМ в сополимере сокращает скорость образования лестничной структуры в сополимере. Такое поведение отличает сополимеры АН–ВИМ от сополимеров АН–акрилаты [64].
Рис. 10.
Зависимость Es от времени термической обработки пленок сополимеров АН и в аргоне при 225 (a) и 250°C (б). fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5) и 30 мол. % (6).

Механизм реакции передачи цепи на имидазольный цикл неизвестен и его изучение затруднено в связи с быстрой потерей растворимости полимера. Однако можно предположить, что радикал –C=N•, соседствующий со звеном ВИМ, атакует его и образует малоактивный радикал, который в дальнейшем может медленно реинициировать реакцию циклизации:

Данное предположение согласуется с лестничной структурой сополимера, предложенной в работе [42]. Предлагаемый механизм объясняет первоначальное замедление реакции циклизации и ее дальнейшее автоускорение. При этом не наблюдается дополнительной трансформации функциональных групп и не появляются новые полосы поглощения в спектрах ИК-НПВО. Кроме того, не предполагается никакой дополнительной деградации цепи, что согласуется с данными ТГА.
Таким образом, в атмосфере аргона развитие лестничной структуры в сополимерах АН и ВИМ происходит медленно, в отличие от ПАН. Этот эффект замедления регулируется содержанием ВИМ в сополимере.
Термическая стабилизация на воздухе
Динамический термический анализ. Термическое поведение сополимеров АН–ВИМ в условиях окислительной стабилизации отличается от поведения ПАН, а также от сополимеров АН–ВИМ в атмосфере аргона. На рис. 11а представлены термограммы ДСК сополимеров АН–ВИМ различного состава, зарегистрированные на воздухе. Сравнительные данные в аргоне и на воздухе для ПАН и сополимеров АН–ВИМ приведены на рис. 11б–11г. Термоокислительная стабилизация представляет собой сложный процесс: наряду с реакцией циклизации протекают реакции окисления и дегидратации, что приводит к появлению на термограммах ДСК суперпозиции нескольких экзотермических пиков разной интенсивности. Экзотермические процессы на воздухе начинаются при ~225°С для всех сополимеров АН–ВИМ. Интенсивность теплового потока снижается при увеличении содержания ВИМ в сополимере (табл. 3). Термограммы смещаются в высокотемпературную область при fВИМ ≤ 15 мол. %, после чего увеличение содержания ВИМ в сополимере по-прежнему влияет на тепловой поток и не влияет на положение термограмм на температурной шкале. В целом экзотермические реакции в ходе термоокислительной стабилизации начинаются при более низких температурах (более чем на 50°С раньше), чем экзотермические реакции в аргоне. Такое поведение не характерно для гомополимера АН. Интенсивный тепловой поток при термоокислительной стабилизации наблюдается при более высоких температурах, чем при термообработке в атмосфере аргона. Сравнение термограмм сополимеров АН–ВИМ, зарегистрированных в аргоне и на воздухе, показывает, что реакция циклизации протекает в одинаковом интервале температур при термообработке как в аргоне, так и на воздухе. Однако циклизации предшествуют другие экзотермические процессы.
Рис. 11.
Термограммы ДСК сополимеров АН и ВИМ, зарегистрированные на воздухе при скорости нагревания 10 град /мин (а), а также в аргоне (1) и на воздухе (2) (б–г). fВИМ = 10 (1), 15 (2), 20 (3), 30 мол. % (4) (а), а также 0 (б), 10 (в) и 20 мол. % (г).
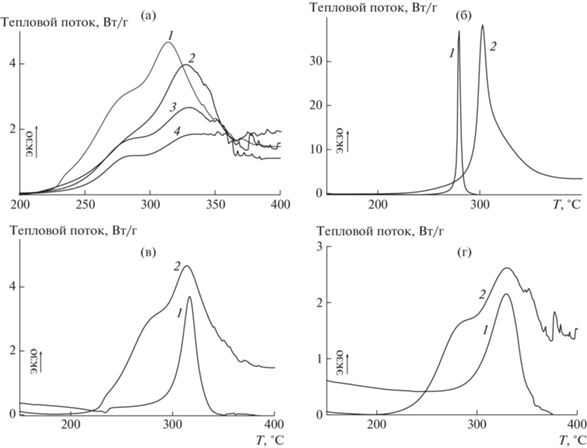
Таблица 3.
Анализ термограмм ДСК сополимеров АН, зарегистрированных на воздухе
| Образец | Tp, °C | −ΔH, Дж/г | Тепловой поток при Tр, Вт/г |
|---|---|---|---|
| ВИМ-0 | 297 и 325 | 5620 | 38.0 |
| ВИМ-10 | 277 и 314 | 1670 | 4.7 |
| ВИМ-15 | 279 и 331 | 1080 | 4.0 |
| ВИМ-20 | 281 и 328 | 1040 | 2.7 |
| ВИМ-30 | 281 и 332 | 1030 | 1.8 |
На рис. 12а представлены термограммы ТГА сополимеров АН и ВИМ, полученные в атмосфере воздуха. Как и в атмосфере аргона, увеличение мольной доли ВИМ в сополимере приводит к большей потере массы при более высоких температурах. В интервале температур 100–600°С потеря массы сополимеров АН и ВИМ происходит в три стадии. Первая незначительная потеря массы – в интервале температур 150–325°С. Вторая стадия (основной процесс) протекает в интервале 325–450°С. Чем выше содержание ВИМ в сополимере, тем более выражена потеря массы. Последняя стадия в исследованном интервале температур наступает после 450°С. Сравнение термограмм ТГА на воздухе и в аргоне показано на рис. 12б–12г. Очевидно, что деструкция сополимеров на воздухе начинается при более низких температурах, чем в аргоне.
Рис. 12.
Потеря массы при нагревании сополимеров АН и ВИМ на воздухе (а), а также в аргоне (1) и на воздухе (2) (б–г). (а) fВИМ = 0 (1), 10 (2), 20 (3), 30 мол. % (4), а также 10 (б), 20 (в) и 30 мол. % (г). Скорость нагревания 10 град/мин.

Изотермический анализ. Изменения химической структуры макромолекул при термоокислительной стабилизации также изучали с помощью ИК-фурье-спектроскопии. По данным ДСК мы ожидали обнаружить отличия в ИК-НПВО-спектрах сополимеров АН–ВИМ, подвергнутых термоокислительной стабилизации, за счет протекания дополнительных химических реакций по сравнению с термостабилизацией в аргоне. Спектры сополимеров, подвергнутых термоокислительной стабилизации при 200, 225 и 250°С при различном времени термообработки, представлены на рис. 13. Спектральные особенности формирования лестничной структуры наблюдаются уже при 200°С (рис. 13а). Скорость образования лестничной структуры увеличивается с повышением температуры. Основные различия в спектрах, зарегистрированных после термообработки в аргоне и на воздухе, проявляются в диапазонах 600–900 и 1000–1800 см–1. Новая полоса поглощения при ~1730 см–1 может быть отнесена к карбонильной (С=О) группе, образующейся при окислении имидазольного кольца радикалами НО•. Появление новой полосы при 805 см–1 связано с реакцией дегидратации (+О2; –Н2О) и образованием групп С=С в основной цепи. Вслед за появлением полосы при ~1730 см–1 в спектрах обнаруживается полоса при 805 см–1, а также наблюдается уширение полосы при 2243 см–1 и ее дальнейшее расщепление на две полосы. Возможные пути реакции приведены ниже.
Рис. 13.
Спектры ИК-НПВО пленок образца ВИМ-10, подвергнутых термоокислительной стабилизации при 200 (а), 225 (б) и 250оС (в) при разном времени выдержки: 0 (1), 30 (2), 60 (3), 120 (4), 240 мин (5) (а); 0 (1), 15 (2), 30 (3), 60 (4), 150 мин (5) (б); 0 (1), 10 (2), 40 (3), 80 (4), 120 мин (5) (в).

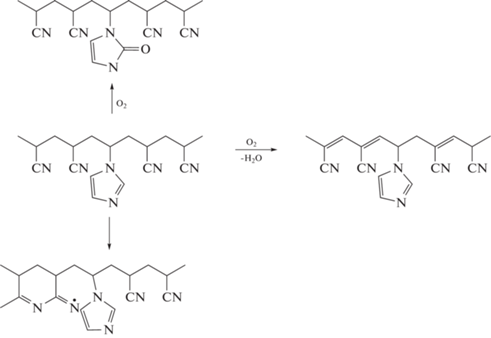
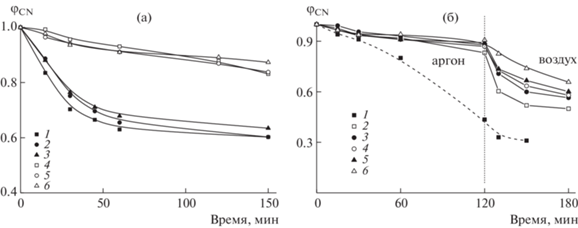
Таким образом, вначале термоокислительной стабилизации реакция окисления имидазольного кольца, вероятно, превалирует над реакцией циклизации и побочной реакцией радикала –C=N• с имидазольным кольцом. В результате лестничная структура развивается быстрее. Действительно, доля непрореагировавших нитрильных групп φCN уменьшается с повышением температуры термообработки (рис. 14а). Заметное количество нитрильных групп расходуется в ходе термоокислительной стабилизации уже при 180°C в отличие от термической обработки в аргоне. Скорость превращения нитрильных групп зависит как от температуры, так и от содержания ВИМ в сополимере. Конверсия групп −C≡N снижается в ряду ВИМ-5 ~ ВИМ-10 > ВИМ-15 ~ ВИМ-20 > ВИМ-30 (рис. 14б–14г). При этом скорость превращения нитрильных групп под действием термоокислительной стабилизации в ПАН аналогична ВИМ-30. Для наглядности сравнение конверсии нитрильных групп в аргоне и на воздухе дано на рис. 15а. Видно, что ~10%-ное превращение групп CN достигается после 2.5 ч термообработки при 225°С в аргоне или через ~15 мин на воздухе. Последовательная термообработка в аргоне и на воздухе позволяет регулировать скорость превращения нитрильных групп в сополимерах АН–ВИМ в отличие от ПАН (рис. 15б).
Рис. 14.
Зависимости φCN от времени термообработки пленок сополимера АН–ВИМ на воздухе. а: fВИМ = 10 мол. %, Т = 180 (1), 200 (2), 225 (3) и 250°С (4); б: T = 200°C, fВИМ = 10 (1), 15 (2), 20 (3) и 30 мол. % (4); в: T = 225°C, fВИМ = 10 (1), 15 (2), 20 (3) и 30 мол. % (4); (г) T = 250°C, fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5), 30 мол. % (6).

Рис. 15.
Зависимости φCN от времени термообработки при 225°С пленок сополимера АН–ВИМ. А – на воздухе (1–3) и в аргоне (4–6) при fВИМ = 10 (1, 4), 15 (2, 5) и 20 мол. % (3, 6); б: fВИМ = 0 (1), 5 (2), 10 (3), 15 (4), 20 (5), 30 мол. % (6) в атмосфере аргона, затем на воздухе.
Таким образом, скорость термостабилизации синтезированных сополимеров АН и ВИМ при заданной температуре может регулироваться газовой атмосферой. В инертной атмосфере (аргон) циклизация практически не происходит при температуре ниже 225°C. Окислительная стабилизация ускоряет формирование лестничной структуры. Полученные результаты могут быть положены в основу производства перерабатываемого в расплаве ПАН прекурсора. Предварительные эксперименты показывают образование волокна сополимера АН–ВИМ (fВИМ = 10 мол. %) при плавлении в капиллярном реометре в отсутствие воздуха (рис. 16). Этот эксперимент соответствует подходу, описанному в работе [38], где сополимер АН и ВИМ, содержащий 18 мол. % ВИМ, подвергали формованию из расплава в реометре в атмосфере азота.

ЗАКЛЮЧЕНИЕ
Результаты наших исследований показали, что 1-винилимидазол влияет на термическое поведение сополимеров акрилонитрила, синтезированных в ДМСО.
С учетом полученных значений констант сополимеризации можно считать, что ВИМ разрывает регулярную цепь ПАН, включаясь в нее единичными звеньями. Состав сополимера сохраняется постоянным на протяжении сополимеризации во всех исследованных системах. Это способствует образованию сополимеров с высокой композиционной однородностью, что позволяет аккуратно проследить влияние ВИМ на реакцию циклизации. В свою очередь механизм ОПЦ дополнительно обеспечивает низкую дисперсность сополимеров. Обнаружено, что молекулярно-массовое распределение в отличие от состава сополимера не оказывает заметного влияния на термическое поведение сополимеров акрилонитрила с 1-винилимидазолом.
В инертной атмосфере введение ВИМ в ПАН приводит к смещению экзотермических процессов, соответствующих реакции циклизации, в область более высоких температур. Увеличение содержания ВИМ в сополимере понижает интенсивность экзо-эффекта, как и в случае сополимеров АН–алкилакрилаты [64]. Кроме того, энергия активации циклизации для сополимеров АН–ВИМ и АН–алкилакрилаты одного состава одинакова. Таким образом, ВИМ можно рассматривать как инертный мономер, подобный алкилакрилатам. В отличие от алкилакрилатов конверсия нитрильных групп ниже для сополимеров АН–ВИМ. Мы предполагаем, что это связано с реакцией передачи цепи на имидазольное кольцо, следствием которой будет появление малоактивного радикала, медленно реинициирующего реакцию циклизации.
При проведении циклизации в условиях термоокислительной стабилизации появляются новые дополнительные источники радикальных частиц за счет окислительных реакций. В результате происходит ускорение формирования лестничной структуры. Она формируется на ~50°С раньше, чем при термообработке в аргоне. Такое поведение резко отличается от сополимеров АН с алкилакрилатами [64]. Существенная разница в скоростях реакций циклизации в аргоне и на воздухе для сополимеров АН–ВИМ позволяет разработать новую стратегию получения плавких прекурсоров углеродного волокна. Эта стратегия должна быть основана на формовании расплава сополимеров в инертной атмосфере с последующей термоокислительной стабилизацией.
Работа выполнена при финансовой поддержке Российского научного фонда (грант 17-79-30108-П).
Список литературы
Das T.K., Ghosh P., Das N.C. // Adv. Compos. Hybrid Mater. 2019. V. 2. № 2. P. 214.
Frank E., Hermanutz F., Buchmeiser M.R. // Macromol. Mater. Eng. 2012. V. 297. № 6. P. 493.
Huang X. // Materials. 2009. V. 2. № 4. P. 2369.
Burlyaev V.V., Burlyaeva E.V., Nikolaev A.I., Peshnev B.V. // Fine Chem. Technologies 2019. V. 14. № 1. P. 39.
Cai X., Zhang C., Zhang S., Fang Y., Zou D. // J. Mater. Chem. A. 2017. V. 5. № 6. P. 2444.
Dorey G. // J. Phys. D. 1987. V. 20. № 3. P. 245.
Morgan P. Carbon Fibers and their Composites. CRC Press, Taylor&Francis, 2005.
Gupta A.K., Paliwal D.K., Bajaj P. // J. Appl. Polym. Sci. 1998. V. 70. № 13. P. 2703.
Advances in Filament Yarn Spinning of Textiles and Polymers / Ed. by D. Zhang. Cambridge: Woodhead Publ. Ltd., 2014.
Grassie N., McGuchan R. // Eur. Polym. J. 1970. V. 6. № 9. P. 1277.
Rangarajan P., Yang J., Bhanu V., Godshall D., McGrath J., Wilkes G., Baird D. // J. Appl. Polym. Sci. 2002. V. 85. № 1. P. 69.
Godshall D., Rangarajan P., Baird D.G., Wilkes G.L., Bhanu V.A., McGrath J.E. // Polymer. 2003. V. 44. № 15. P. 4221.
Miller G.C., Yu J., Joseph R.M., Choudhury S.R., Mecham S.J., Baird D.G., Riffle J.S. // Polymer. 2017. V. 126. P. 87.
Lee J.H., Jin J.-U., Park S., Choi D., You N.-H., Chung Y., Yeo H. // J. Ind. Eng. Chem. 2018. V. 71. P. 112.
Udakhe J., Goud V. // J. Text. Assoc. 2011. V. 71. № 5. P. 233.
Naskar A.K., Walker R.A., Proulx S., Edie D.D., Ogale A.A. // Carbon. 2005. V. 43. № 5. P. 1065.
Penga W., Han N., Tang X., Liu H., Zhang X. // Adv. Mater. Research. 2011. Art. 332.
Han N., Zhang X., Yu W., Gao X. // Macromol. Res. 2010. V. 18. № 11. P. 1060.
Gao X., Han N., Zhang X., Yu W. // J. Mater. Sci. 2009. V. 44. № 21. P. 5877.
Bhanu V.A., Rangarajan P., Wiles K., Bortner M., Sankarpandian M., Godshall D., Glass T.E., Banthia A.K., Yang J., Wilkes G., Baird D., McGrath J.E. // Polymer. 2002. V. 43. № 18. P. 4841.
Rangarajan P., Yang J., Bhanu V., Godshall D., McGrath J., Wilkes G., Baird D. // J. Appl. Polym. Sci. 2002. V. 85. P. 69–83.
Bhanu V.A., Wiles K.B., Banthia A.K., Mansuri A., Sankarpandian M., Rangarajan P., Glass T.E., Baird D.G., Wilkes G.L., McGrath J.E. // Polymer. 2002. V. 43. № 18. P. 4841.
Rangarajan P., Bhanu V., Godshall D., Wilkes G., McGrath J., Baird D. // Polymer. 2002. V. 43. № 9. P. 2699.
Rwei S.-P., Way T.-F., Chiang W.-Y., Tseng J.-C. // Textile Res J. 2017. V. 88. № 13. P. 1479.
Lee J.H., Jin J.-U., Park S., Choi D., You N.-H., Chung Y., Ku B.-C., Yeo H. // J. Ind. Eng. Chem. 2018. V. 71. P. 112.
Chernikova E.V., Toms R.V., Gervald A.Yu., Proko-pov N.I. // Polymer Science C. 2020. V. 62. № 1. P. 17.
Bahrami S.H., Bajaj P., Sen K. // J. Appl. Polym. Sci. 2003. V. 88. № 3. P. 685.
Fu Z., Gui Y., Liu S., Wang Z., Liu B., Cao C., Zhang H. // J. Appl. Polym. Sci. 2014. V. 131. № 19. Art. 40834.
Ouyang Q., Cheng L., Wang H., Li K. // Polym. Degrad. Stab. 2008. V. 93. P. 1415.
Mailhot B., Gardette J.-L. // Polym. Degrad. Stab. 1994. V. 44. P. 223.
Moskowitz J.D., Wiggins J.S. // Polym. Degrad. Stab. 2016. V. 125. P. 76.
Paiva M.C., Kotasthane P., Edie D.D., Ogale A.A. // Carbon. 2003. V. 41. № 7. P. 1399.
Bullock R.E. // Fiber Sci. Technol. 1974. V. 7. № 2. P. 157.
Dietrich J., Hirt P., Herlinger H. // Eur. Polym. J. 1996. V. 32. № 5. P. 617.
Badawy S.M., Dessouki A.M. // J. Phys. Chem. B. 2003. V. 41. № 107. P. 11273.
Mukundan T., Bhanu V.A., Wiles K.B., Johnson H., Bortner M., Baird D.G., Naskar A.K., Ogale A.A., Edie D.D., McGrath J.E. // Polymer. 2006. V. 47. № 11. P. 4163.
Czech Z. // Int. J. Adhesion Adhesives. 2007. V. 27. P. 195.
Deng W., Lobovsky A., Iacono S.T., Wu T., Tomar N., Budy S.M., Long T., Hoffman W.P., Smith D.W. // Polymer. 2011. V. 52. № 3. P. 622.
Henrici-Olive G., Olive S. // Adv. Polym. Sci. 1979. V. 32. P. 123.
Gupta A.K., Paliwal D.K., Bajaj P. // J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1991. V. 31. P. 1.
Han N., Li H., Li C., Zhang X., Li W., Wang D. // Chem. J. Chineese Univ. 2015. V. 36. № 10. P. 2073.
Mahmood S.F., Batchelor B.L., Jung M., Park K., Voit W.E., Novak B.M., Yang D. // Carbon Lett. 2019. V. 29. № 6. P. 605.
Im Y.M., Choi H.M., Nathanael A.J., Jeong M.H., Lee S.O., Yun S.N., Oh T.H. // Fibers Polymers. 2020. V. 21. № 2. P. 376.
Im Y.M., Nathanael A.J., Jung M.H., Lee S.O., Oh T.H. // Fibers Polymers. 2022. V. 23. P. 321.
Batchelor B.L., Mahmood S.F., Jung M., Shin H., Kulikov O.V., Voit W., Novak B.M., Yang D.J. // Carbon. 2016. V. 98. P. 681.
Cho D.W., Hong S.C. // Polym. Degrad. Stab. 2019. V. 161. P. 191.
Chernikova E.V., Terpugova P.S., Garina E.S., Golu-bev V.B. // Polymer Science A. 2007. V. 49. № 2. 108.
Pekel N., Rzaev Z.M.O., Güven O. // Macromol. Chem. Phys. 2004. V. 205. P. 1088.
Kissinger H.E. // Anal. Chem. 1957. V. 29. № 11. P. 1702.
Collins G.L., Thomas N.W., Williams G.E. // Carbon. 1988. V. 26. № 5. P. 671.
Ouyang Q., Cheng L., Wang H., Li K. // Polym. Degrad. Stab. 2008. V. 93. № 8. P. 1415.
Езриелев А.И., Брохина Э.Л., Роскин Е.С. // Высокомолек. соед. А. 1969. Т. 11. № 8. С. 1670.
Fineman M., Ross S.D. // J. Polym. Sci. 1950. V. 5. № 2. P. 259.
Hao J., Liu Y., Lu C. // Polym. Degrad. Stab. 2018. V. 147. P. 89.
Hao J., An F., Lu C., Liu Y. // J. Macromol. Sci., Pure Appl. Chem. 2019. V. 56. P. 1012.
Bashir Z. // Carbon. 1991. V. 29. P. 1081.
Doremaele G.H.J., German A.L., Vries N.K., Vel-den G.P.M. // Macromolecules. 1990. V. 23. № 19. P. 4206.
Chapiro A. // Radiat. Phys. Chem. 1992. V. 40. № 2. P. 88.
Fan B., Wan J., McKay A.I., Qu Z., Thang S. // Polym. Chem. 2020. V. 11. P. 5649.
Chernikova E.V., Poteryaeva Z.A., Belyaev S.S., Ni-fant’ev I.E., Shlyakhtin A.V., Kostina Yu.V., Cherevan' A.S., Efimov M.N., Bondarenko G.N., Sivtsov E.V. // Polymer Science B. 2011. V. 53. № 7–8. P. 391.
Chernikova E.V., Golubev V.B., Filippov A.N., Garina E.S. // Polymer Science C. 2015. V. 57. № 1. P. 94.
Rahaman M.S.A., Ismail A.F., Mustafa A. // Polym. Degrad. Stab. 2007. V. 92. № 8. P. 1421.
Bayramgil N.P. // Polym. Degrad. Stab. 2008. V. 93. № 8. P. 1504.
Toms R.V., Balashov M.S., Gervald A.Yu., Prokopov N.I., Plutalova A.V., Chernikova E.V. // Polym. Int. 2021. Art. 6286.
Xue T.J., McKinney A., Wilkie C.A. // Polym. Degrad. Stab. 1997. V. 58. № 1–2. P. 193.
Wangxi Z., Jie L., Gang W. // Carbon. 2003. V. 41. P. 2805.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)