

Высокомолекулярные соединения (серия С), 2020, T. 62, № 1, стр. 3-19
Успехи в синтезе этилен-пропилен-диеновых эластомеров ионно-координационной полимеризацией на моноцентровых каталитических системах новых поколений
Н. М. Бравая a, Е. Е. Файнгольд a, *, Э. Р. Бадамшина a, Е. А. Сангинов a
a Институт проблем химической физики Российской академии наук
142432 Московская обл., Черноголовка, пр. ак. Семенова, 1, Россия
* E-mail: fine@cat.icp.ac.ru
Поступила в редакцию 06.06.2019
После доработки 12.11.2019
Принята к публикации 14.01.2020
Аннотация
Проанализированы работы, посвященные синтезу тройных сополимеров этилена, пропилена и несопряженных циклических диенов на моноцентровых каталитических системах нового поколения на основе металлоценовых комплексов, комплексов “напряженной геометрии”, моноциклопентадиенильных и пост-металлоценовых, хелатных, комплексов IVB-группы. Рассмотрены преимущества и недостатки этих каталитических систем по сравнению с традиционными ванадиевыми катализаторами, введенными в производство в 60-х годах ХХ века и используемыми до настоящего времени в промышленном синтезе каучуков. Значительная часть обзора посвящена разбору патентных публикаций, в особенности последних лет, принадлежащих основным производителям этилен-пропилен-диеновых каучуков в мире.
ВВЕДЕНИЕ
Этилен-пропилен-диеновые каучуки – тройные сополимеры этилена, пропилена и несопряженных диенов, в основном циклических, таких как 5-этилиден-2-норборнена (ЭНБ), дициклопентадиена (ДЦПД) и 5-винил-2-норборнена (ВНБ). Статистическое распределение сомономеров в цепи макромолекул определяет хорошие эластомерные характеристики СКЭПТ и широкий температурный диапазон их эксплуатации от –50 до +130°С. Основные цепи макромолекул сополимеров не содержат двойных связей, что обеспечивает высокую устойчивость к влиянию кислорода, озона, а также температурным воздействиям. Боковая двойная связь диена предоставляет возможности для вулканизации сополимеров, увеличивающей гибкость материала, или функционализации для приготовления различных полимерных компаундов, в том числе и с полярными каучуками. Кроме того, введение наполнителей и пластификаторов в СКЭПТ позволяет получать полимерные смеси с обширным спектром свойств. К настоящему времени СКЭПТ являются востребованными эластомерными материалами, используемыми во многих отраслях – это автомобилестроение, резинотехническая промышленность, производство строительных материалов, кабельная промышленность и другие. Согласно данным аналитического агентства “Global Information, Inc.”, объем мирового производства СКЭПТ в 2016 г. составил 1.3 млн тонн и достигнет 1.9 млн тонн к 2023 году [1].
Производство СКЭПТ основано исключительно на процессах каталитической координационной полимеризации. Ключевые характеристики СКЭПТ (микроструктура, кристалличность, температура стеклования, молекулярно-массовые и теплофизические характеристики), обусловливающие их эксплуатационные свойства, зависят от типа каталитической системы, которая, в свою очередь, определяет технологический процесс производства СКЭПТ: растворный, суспензионный или газофазный.
Ранние гомогенные каталитические системы, разработанные в начале 60-х годов ХХ века и используемые до настоящего времени, включают комбинацию соединения ванадия в качестве прекатализатора (VCl4, V(acac)3, чаще – VOCl3), и алюминийорганического соединения как сокатализатора (AlEt2Cl, AlEt3, Al2Et3Cl3) в небольшом избытке Al/V ∼ 10, а также хлорсодержащих промоторов, таких как этилтрихлорацетат, n-бутилперхлорокротонат, трихлортолуол и другие. Хотя природа активного центра ванадиевых каталитических систем до сих пор остается предметом дискуссий, в целом, роль компонентов каталитической системы сводится к следующему. Алюминийорганический сокатализатор алкилирует и активирует ванадиевый прекатализатор. Каталитически активные центры V(III) под действием сокатализатора восстанавливаются до неактивных, низко-валентных центров V(II). Процесс восстановления особенно выражен при повышенных значениях температуры (>60°C). Хлорсодержащие промоторы способны окислять (реактивировать) соединения V(II) с образованием хлорсодержащих V(III) центров, которые вновь реалкилируются алюминийорганическим соединением и включаются в полимеризационный цикл [2–4]. Введение хлорсодержащих промоторов в каталитическую систему на порядок увеличивает производительность ванадиевых каталитических систем.
К недостаткам ванадиевых катализаторов следует отнести невысокие активность (80–120 кгсопол мольV–1) и термическую стабильность даже при температуре 20–60°С. Следствием низкой активности является высокое остаточное содержание в полимере катализатора и хлорсодержащего промотора, которые обладают большой токсичностью и отрицательным влиянием на свойства сополимера при хранении (старении), а также высокое содержание непрореагировавшего диена. Наличие нежелательных примесей требует проведения дополнительных технологических операций по их удалению.
Несмотря на отмеченные недостатки, ванадиевые системы позволяют получать СКЭПТ c характеристиками, которые считаются ориентиром при выборе альтернативных каталитических систем. На ванадиевых каталитических системах получают аморфные высокоэластичные полимеры со статистическим распределением сомономеров в макромолекуле сополимера с высокой или сверхвысокой молекулярной массой. Коэффициенты полидисперсности (PDI) варьируются в диапазоне Ð = 2–10, но во многих случаях они имеют значения близкие к двум, что характерно для полимеров, формирующихся на моноцентровых каталитических системах. Вязкость по Муни (ML), определяемая как степень сопротивления вращению цилиндрического металлического ротора, погруженного в образец, помещенный в испытательную камеру вискозиметра Муни, является важной характеристикой эластомеров, обусловливающей их технологичность. СКЭПТ, получаемые на ванадиевых катализаторах, имеют значения ML от 30 до 100. При отсутствии соответствующего оборудования для оценки ML эластомера можно воспользоваться эмпирическим соотношением, установленным для большого числа синтетических и натуральных каучуков [5]:
где Mn и Mw – среднечисленная и среднемассовая ММ сополимера соответственно.Производство СКЭПТ на ванадиевых катализаторах осуществляется в растворных и суспензионных процессах при температуре 20–50°С. Реакционной средой в первом случае выступают легкие углеводороды, а во втором, как правило, жидкий пропилен. Недостатком первого процесса можно назвать ограничения на вязкость реакционной среды, формирующие невысокую степень конверсии, недостатком второго – более сложный процесс удаления остатков катализатора, окклюдированного образующимся полимером. Ванадиевые каталитические системы, в том числе и новых поколений, подробно рассмотрены в ряде обзорных публикаций (см., например, [6–11]).
Гетерогенные катализаторы TiCl4/MgCl2/AlEt3 с внутренними и внешними донорами проявляют высокую активность в процессах сополимеризации, в том числе и в синтезе СКЭПТ, позволяют осуществлять сополимеризацию при повышенных значениях температуры и получать конечный продукт в форме гранул, но не обеспечивают образования аморфных полимеров. Полицентровость этих каталитических систем определяет высокую степень полидисперсности образующихся сополимеров, их композиционную и фракционную неоднородность. Дополнительные сложности возникают с удалением непрореагировавшего диена из полимерного продукта.
Новые возможности в синтезе СКЭПТ открывают моноцентровые каталитические системы на основе металлоценовых, полусэндвичевых и пост-металлоценовых комплексов переходных металлов. По сравнению с ванадиевыми катализаторами, эти системы обладают рядом преимуществ: высокой активностью11; однотипностью активных центров, обеспечивающей однородность молекулярно-массовых характеристик, микроструктуры и композиционного состава каучуков; стабильностью каталитического действия при повышенных значениях температуры; возможностью целенаправленного создания СКЭПТ с заданными свойствами (степень внедрения и характер распределения сомономеров в цепи макромолекулы, введение разветвлений определенного типа и другие).
Металлоценовые катализаторы являются широким классом комплексов IVB-группы (Ti, Zr, Hf) и представляют собой сэндвичиевые соединения, где переходный металл связан π-связями с двумя циклопентадиенильными кольцами замещенных или незамещенных лигандов: циклопентадиенильного (Cp), инденильного (Ind), флуоренильного (Flu). Эти катализаторы можно условно разделить на два основных класса – немостиковые, в которых гапто-связанные лиганды свободно вращаются относительно переходного металла, и стереожесткие (ansa-комплексы), в которых гапто-связанные лиганды фиксированы ковалентными связями мостиковой группировки. Полусэндвичевые соединения переходных металлов аналогично металлоценам подразделяются на стереожесткие и немостиковые. Стереожесткие полусэндвичевые комплексы (“Сonstrained Geometry Catalysts” (CGC)) состоят из переходного металла, связанного с одним гапто- и одним гетероатом-содержащим лигандами, фиксироваными ковалентными связями мостиковой группировки. В немостиковых полусэндвичевых катализаторах гапто-связанный и гетероатом-содержащий лиганды не соединены между собой и свободно вращаются относительно переходного металла. Широким классом соединений выступают хелатные комплексы переходных металлов (пост-металлоценовые комплексы). Каталитические свойства комплексов этого типа легко модифицируются и “настраиваются” на получение конкретного полиолефина выбором органического лиганда, переходного металла, активатора при сравнительной простоте синтеза и доступности реагентов.
В настоящее время отсутствуют работы, где с единых позиций анализируются различные типы катализаторов для синтеза СКЭПТ. Известные обзоры по катализаторам для синтеза СКЭПТ, как правило, очень лаконичны [12, 13]. Согласно анализу патентной литературы, в последние годы наблюдается всплеск интереса основных мировых производителей эластомеров (“ARLANXEO”, “Mitsui Chemicals”, “Lion Copolymer” и другие) к моноцентровым каталитическим системам новых поколений, вызванный, вероятно, ужесточением требований к экологической безопасности производства и продуктов. Актуальность данного обзора определяется также тем, что в настоящий момент в России металлоценовые и пост-металлоценовые каталитические системы не применяются, а существующее малотоннажное производство каучуков основано исключительно на ванадиевых катализаторах, недостатки которых были указаны выше. С анализом производства СКЭПТ в мире и отдельных каталитических систем для их синтеза можно ознакомиться в работах российских авторов [14, 15].
При подготовке настоящего обзора была поставлена задача проследить хронологию и достижения в использовании металлоценовых, полусэндвичевых и пост-металлоценовых каталитических систем в синтезе СКЭПТ с акцентами на наиболее интересные с научной и прикладной точки зрения катализаторы.
МЕТАЛЛОЦЕНОВЫЕ КАТАЛИЗАТОРЫ ДЛЯ СИНТЕЗА ЭТИЛЕН-ПРОПИЛЕН-ДИЕНОВЫХ КАУЧУКОВ
Сополимеризация этилена и пропилена с линейными несопряженными диенами
Для несопряженных диенов возможно три способа их внедрения в цепь макромолекулы:
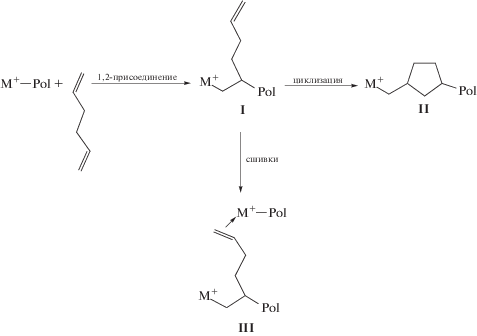
Так, 1,2-присоединение диена демонстрирует образование структуры I. Макромолекулы такого сополимера имеют разветвления с концевыми двойными связями, которые можно использовать для функционализации, вулканизации сополимера или вовлечения их в полимеризацию в качестве макромономера для формирования полиолефинов с длинноцепочечными разветвлениями. Также концевые двойные связи разветвлений способны принимать дальнейшее участие в полимеризационном процессе с представлением циклических структур (II) и сшитых полимеров (III). Структура II образуется в результате внутримолекулярной циклизации после 1,2-присоединения диена. Полиолефины с циклическими звеньями в цепи макромолекулы аналогично сополимерам с циклическими олефинами (например, норборненом) характеризуются повышенной температурой стеклования и высокой прозрачностью, применяются в качестве оптического материала [16, 17]. Только реакция циклизации является нежелательной при целевом синтезе СКЭПТ. Вовлечение второй двойной связи в процесс роста цепи на другом активном центре приводит к формированию сшитых полимеров (структура III), плохо поддающихся переработке и формованию. Однако небольшое содержание сшивок (≤2 мол. %) может повлечь увеличение теплостойкости полиолефинов и улучшение реологических свойств.
Образование тех или других структур определяется типом диена, каталитической системой, условиями проведения процесса сополимеризации, но прохождение реакций с формированием структур II и III доминирует в сополимеризации с несопряженными линейными диенами на металлоценовых каталитических системах и наиболее выражено в случае 1,5-гексадиена и 1,7-октадиена [18–30].
Реакция циклизации становится несущественной для 1,9-декадиена и более длинных диенов. В сополимеризации пропилена с 1,9-декадиеном на двух металлоценовых катализаторах – синдиоспецифическом Ph2CCpFluZrCl2 и изоспецифическом rac-Me2Si[(4-tBu,2-Me)Cp]2ZrCl2 c метилалюмоксаном (МАО) в качестве сокатализатора диен внедряется преимущественно в виде разветвлений с концевыми двойными связями [31]. Полученные сополимеры были использованы в качестве добавки для улучшения совместимости изотактического и синдиотактического полипропиленов. Мостиковые бис-Flu цирконоцены Me2SiFlu2ZrMe2 и EtFlu2ZrCl2 также почти не катализируют процессы циклизации и сшивания [32, 33]. Содержание чередующихся последовательностей (этилен/диен) в двойном сополимере, полученном на Me2SiFlu2ZrMe2 при 0°С, составляет 58 мол. % при концентрации диена 0.5 моль/л и 98 мол. % при концентрации 2.7 моль/л [33]. Диен входит в сополимер в виде линейных разветвлений без побочных реакций сшивки и циклизации. Сополимеры имеют высокие молекулярные массы Mw = (33–45) × 104, небольшие коэффициенты полидисперсности Ð = 1.6–1.8 и низкие значения температуры стеклования Tg от –55 до –66°C. Здесь следует отметить крайне низкую активность этих систем.
С целью получения линейных сополимеров с диенами и исключения реакций циклизации и сшивки используют также асимметричные диены с внутренней двойной связью. Сравнение сополимеризации этилена и пропилена с различными несопряженными линейными (1,7-октадиен, 1,9-декадиен) и разветвленными (2-метил-1,4-пентадиен, 2-метил-1,5-гексадиен, 7-метил-1,6-октадиен) диенами на каталитической системе rac-EtInd2ZrCl2/ММАО (модифицированный МАО; [AlMMAO]/[Zr] = 10 000, [олефин] : [диен] = 2 : 1) приведено в работе [34]. Система обеспечивает внедрение 1–2 мол. % диена в макромолекулу сополимера при содержании диена ∼30 мол. % в смеси сомономеров. Производительность катализатора в сополимеризации этилена с диенами лишь в 1.4–1.8 раза ниже, чем в гомополимеризации этилена (3000 кгПЭ (мольZr ч атм)–1). В сополимеризации диена с пропиленом активность катализатора приблизительно в 20 раз ниже, чем в гомополимеризации пропилена (1100 кгПП (мольZr ч атм)–1). Анализ микроструктуры сополимеров показал присутствие разветвлений с концевыми двойными связями и отсутствие циклических структур и сшивок. Показано, что образования сшивок удается полностью избежать и в случае сополимеризации 5,7-диметилокто-1,6-гептадиена с этиленом на каталитических системах Cp2ZrCl2/MAO, Me2SiCpNtBuTiCl2/MAO, rac-EtInd2ZrCl2/MAO [34–36] и пропиленом на каталитической системе rac-EtInd2ZrCl2/MAO [37].
В целом, все же можно отметить, что сополимеризация олефинов с линейными несопряженными диенами на металлоценовых каталитических системах значительно расширяет возможности создания новых полимерных гибридных структур, хотя в большинстве случаев характеристики получаемых СКЭПТ неудовлетворительны.
Сополимеризация этилена с пропиленом
Поскольку базовыми сомономерами в синтезе СКЭПТ считаются этилен и пропилен, то выбор каталитической системы определяется, в первую очередь, спецификой сополимеризации именно этих сомономеров. Для получения СКЭПТ предпочтительнее каталитические системы, формирующие статистические сополимеры. Распределение сомономеров в цепи макромолекулы создается произведением констант сополимеризации rЕ = kЕЕ/kЕP и rP = kPP/kPE, где константы kEE, kEP, kPP и kPE – относительные вероятности гомо- (EE или PP) или гетеро-присоединений (EP или PE) сомономеров [38, 39]. Для ванадиевых каталитических систем в зависимости от их состава и условий сополимеризации эти параметры варьируются в пределах rE = 3–20, rP = 0.01–0.07, rErP = 0.1–1.0, но в большинстве случаев – rErP < 1 [40–46]. Другим важным свойством конкретной каталитической системы является способность формировать высокомолекулярный сополимер.
Таким образом, можно отметить определенные тенденции в сополимеризации для некоторых классов металлоценовых комплексов, хотя однозначной корреляции между симметрией прекатализатора и константами сополимеризации нет. Немостиковые бис-Cp и бис-Ind цирконоцены симметрии C2V при активации МАО ведут сополимеризацию с выраженным предпочтением в отношении этилена [47, 48] (rE на три–четыре порядка больше rP). При этом бис-Cp прекатализаторы приводят к образованию низкомолекулярных сополимеров, но с относительно высокой активностью. Мостиковые незамещенные бис-Ind металлоцены симметрии С2 с умеренной активностью формируют сополимеры достаточно высокой молекулярной массы, а rErP = 0.5–1.0 [46–49]. На каталитической системе rac-EtInd2ZrCl2/МАО (Т = 40°C) в сополимеризации этилена с пропиленом значения констант составляют rE = 6.1, rP = 0.1 [47], rE = 8.0, rP = 0.1 [49]. Константы сополимеризации объемных сомономеров, таких как норборнен и ЭНБ существенно ниже, чем пропилена. В сополимеризации этилена с норборненом rE = 3.0, r = 0.03, а в сополимеризации с ЭНБ rE = 10.5, rЭНБ = 0.004 [50]. В работе показано, что на этом катализаторе при мольном соотношении ЭНБ : этилен = 22.5 удается получить сополимер, содержащий до 43 мол. % ЭНБ.
Высокоактивные изоспецифические цирконоцены симметрии С2 проявляют склонность к образованию блочных сополимеров (rErP = 2–3) [46, 49, 51] и способствуют созданию полимерных продуктов значительно меньшей ММ по сравнению с гомополимеризацией соответствующих мономеров. Так, каталитические системы rac-Me2Si(2-Me,4-Ph-1-Ind)2ZrCl2/MAO и rac-Me2Si(2-Me,4-Naph-1-Ind)2ZrCl2/MAO формируют полиэтилен и полипропилен высокой (Mw > 30 × 104) и очень высокой (Mw ≥ 100 × 104) молекулярной массы [52]. Однако при сополимеризации образуются сополимеры значительно меньшей ММ [51, 53–59]. Здесь более перспективными являются гафноцены симметрии С2, несмотря на их невысокую активность. Каталитическая система rac-EtInd2HfCl2/MAO проявляет незначительную тенденцию к снижению молекулярной массы сополимера с увеличением содержания пропилена в смеси Mw = от 55 × 104 (Ð = 2.3) при E/P = 3.46 до 39.3 × 104 при E/P = 0.97 [46]. Сополимеры, полученные на этой каталитической системе, имеют приемлемые значения молекулярных масс, а по теплофизическим характеристикам не уступают сополимерам, полученным на гомогенных ванадиевых катализаторах.
Мостиковые Cp–Flu цирконоцены симметрии Сs и C1 проявляют высокую активность (до 20 000–50 000 кгсопол (мольZr ч атм)–1) и способствуют образованию высокомолекулярных сополимеров (Mw до 106 г моль–1), при этом значения rE лишь в несколько раз больше, чем rP, а rErP$ \ll $ 1 [54, 55, 60, 61].
Металлоценовые каталитические системы для синтеза СКЭПТ с циклическими диенами
В настоящее время ЭНБ и другие производные норборнена, ВНБ и ДЦПД являются основными диенами для синтеза СКЭПТ. Двойные связи диенов различаются по реакционной способности. Норборненовая двойная связь, будучи высоко реакционноспособной, участвует в сополимеризации с образованием основной насыщенной цепи сополимера. Другая, менее реакционноспособная двойная связь, как правило, остается незатронутой и может быть использована для вулканизации полученного каучука серой, пероксидами или для функционализации.
Следует отметить, что эти диены значительно (в 3–4 раза) дороже этилена и пропилена. Низкая конверсия диена требует использования высокой концентрации мономера и последующей его рекуперации из реактора. Таким образом, поиск систем, проявляющих повышенную реакционную способность по отношению к диену, и условий проведения процесса являются важной задачей. В частности, в работе [50] показано, что прекатализаторы rac-EtInd2ZrCl2 и rac-Me2SiInd2ZrCl2 при активации МАО (Т = 40°С) обеспечивают близкую к 100% конверсию ЭНБ в сополимеризации с этиленом при мольном соотношении сомономеров в смеси ∼30 : 70. Значительное снижение конверсии до 24% можно было наблюдать только при высоком содержании ЭНБ ∼90 мол. %. Цирконоцен rac-Me2CInd2ZrCl2 в этих условиях обеспечивает также близкое к 100% вхождение ЭНБ в сополимер. Введение объемного заместителя в инденильную систему цирконоцена (rac-Me2C(tBuInd)2ZrCl2) влечет снижение конверсии до 17% при прочих равных условиях.
Замечено, что эффективность внедрения ЭНБ или ВНБ в цепь макромолекулы по сравнению с пропиленом значительно ниже, в основном, из-за стерического фактора, а увеличение концентрации диена сопровождается снижением ММ сополимеров. Так, каталитическая система Сp2ZrCl2/MAO (AlMAO/Zr = 1500 моль/моль, 35°С) ведет сополимеризацию этилена с ВНБ, обеспечивая вхождение диена в сополимер на уровне 14 мол. % (41 мас. %) [62]. Активность системы с увеличением концентрации диена снижается более чем на порядок. Уменьшаются и значения средневязкостных молекулярных масс сополимеров. Значения констант сополимеризации составляют rE = 18.6, rВНБ = 0.3. Система Ph2CCpFluZrCl2/MAO приводит к образованию сополимеров этилена с изолированными звеньями ВНБ в цепи [63] с содержанием ВНБ до 18 мол. % (49 мас. %). Активность этой каталитической системы в сополимеризации достигает 5700–6500 кгсопол (мольZr ч атм)–1, но существенно снижается с увеличением концентрации диена, которое, в свою очередь, сопровождается значительным уменьшением молекулярной массы сополимеров Mw = с 87.7 × 104 до 8.6 × 104.
Сопоставление каталитических свойств гомогенных каталитических систем VCl4/AlEt2Cl (Al/V = 5) и rac-EtInd2ZrCl2/MAO (Al/Zr = 2500) в тройной сополимеризации этилен–пропилен–ЭНБ [64] показало, что металлоценовая система примерно в 15 раз активнее. Внедрение ЭНБ происходит также более эффективно, что позволяет работать с меньшими значениями концентрации диена. Аналогичные эффекты можно было наблюдать при сопоставлении каталитических свойств систем rac-EtInd2ZrCl2/MAO (Al/Zr = 1000, 3000) и VOCl3/Al2Et3Cl3 (Al/V = 10, 15) в тройной сополимеризации этилен–пропилен–ЭНБ [65]. Циркониевая система производительнее ванадиевой и обеспечивает бόльшее содержание диена в сополимере. Однако молекулярные массы сополимеров, получаемых на циркониевом катализаторе, существенно ниже, чем на ванадиевом. Вместе c тем, сополимеры с содержанием ∼50 мол. % этилена аморфны, тогда как ванадиевые сополимеры имеют кристаллизующиеся этиленовые домены.
Интересно сопоставить данные по синтезу СКЭПТ на металоценовых катализаторах, приведенные в патенте [66], поскольку протокол проведения процесса был одинаковый для металлоценов различного состава и симметрии. Активаторами служили МАО, перфторфенилборан B(C6F5)3, перфторфенилбораты (Ph3CB(C6F5) и Me2NHPhB(C6F5)4), т.е. соединения, традиционно используемые для активации металлоценовых комплексов [67–69]. Мостиковые незамещенные бис-Ind цирконоцены симметрии C2 – rac-Me2SiInd2ZrCl2 и rac-EtInd2ZrCl2 при активации МАО проявляют очень высокую активность (до 26 000 и 34 700 кгсопол (мольZr ч атм)–1) в тройной сополимеризации с ЭНБ, обеспечивая внедрение диена до 7 и 5 мас. % и высокие молекулярные массы сополимеров 16.4 × 104 и 14.2 × 104 соответственно [66]. Однако ML сополимеров имеет низкие значения. Соответствующие гафноцены показывают значительно меньшую активность, которая тем не менее выше активности ванадиевых систем (210 и 1160 кгсопол (мольHf ч атм)–1), но формируют СКЭПТ c высокой молекулярной массой (Mw = 16.2 × 104 и 51.4 × 104) с содержанием 4 и 12 мас. % диена. Вязкость по Муни (125°С) для сополимера, полученного на rac-EtInd2HfCl2, равна 70. Активности систем в тройной сополимеризации с ДЦПД при прочих равных условиях в 2–3 раза ниже, чем с ЭНБ. При этом различия характеристик тройных сополимеров, полученных с ЭНБ и ДЦПД, незначительны. Хорошую комбинацию свойств показали каталитические системы на основе анса-цирконоцена Cs-симметрии – Ph2CCpFluZrCl2. Их активность в тройной сополимеризации с ЭНБ и ДЦПД достигает 10 800 кгсопол (мольZr ч атм)–1, содержание диена в сополимере составляет ∼7 мас. %, Mw ∼ 23 × 104, Ð = 2.0, ML ∼ 20 (Т = 125°С). Из данных, приведенных в патенте, можно заключить, что в тройной сополимеризации с ЭНБ стерические характеристики цирконоценов проявляются в бόльшей степени, чем в сополимеризации этилена с пропиленом. Так, цирконоцен, имеющий метильные заместители во втором положении инденильной системы, при том же составе смеси сомономеров проявляет очень низкую активность и не обеспечивает внедрение диена. Не обнаружено внедрение диена и при использовании Me2SiFlu2ZrCl2. В работе [67] для структурно сходного цирконоцена с этиленовым мостиком EtFlu2ZrCl2 также отмечено отсутствие внедрения ЭНБ в макромолекулу в тройной сополимеризации этилен–пропилен–ЭНБ при активации МАО, хотя показано, что катализатор образует комплекс с ЭНБ.
Перспективность металлоценовых каталитических систем симметрии Cs в синтезе СКЭПТ продемонстрирована в недавно опубликованных патентах фирмы “Mitsui Chemicals” [71–73]:
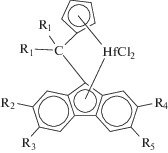
(R1 = 4-MePh, 4-MeOPh, 4-Me2NPh, 4-N–morpholinylPh; R2–5 = Alk).
Предметом изобретений является серия гафноценов c различными заместителями в метиленовой мостиковой группировке и алкильными заместителями в периферийных положениях флуоренильного лиганда. Комплексы предварительно алкилируют триизобутилалюминием (Al/Hf = 700–2000 моль/моль), который частично расходуется на очистку реакционной среды от примесей влаги, и активируют CPh3B(C6F5)4 (B/Hf = 4, 10 моль/моль). Сополимеризацию проводят при 80–120°С (суспензионный и растворный процессы). Катализаторы обеспечивают внедрение ЭНБ на уровне 7–11 мас. %. Сополимеры имеют высокие значения молекулярной массы (Mw = (1–2) × 106) и низкие PDI (Đ = 2.5–3.0). Наибольшую активность (10–20 тоннполимера (мольHf ч атм)–1) продемонстрировали гафноцены с R1 = 4-MePh, 4-MeOPh, 4-Me2NPh и R2 = = R3 = R4 = R5 = Me. По активности, рабочему температурному диапазону, расходу реагентов, а также по характеристикам получаемых сополимеров эти каталитические системы значительно превосходят традиционные ванадиевые катализаторы.
Другим перспективным типом металлоценовых катализаторов для синтеза СКЭПТ являются новые Cp–Ind-комплексы С1-симметрии, разработанные компанией “Lotte Chemical” [74]:
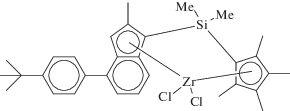
Комплексы имеют замещенный или незамещенный Cp-лиганд, связанный различными мостиковыми группировками с инденильным лигандом, имеющим метильный заместитель во втором положении и замещенный арильный фрагмент (фенил, нафтил, дифенил и т.д.) в четвертом положении. Наилучшие свойства показал цирконоцен. Активность системы при активации МАО в тройной сополимеризации этилен–пропилен–ЭНБ составляет 155–190 тонн (мольZr ч)–1 при 80°С. Получены каучуки с высокими значениями Mw = (16.3–20.9) × 104 и PDI (Đ = 5.6–9.3). Содержание ЭНБ в тройных сополимерах составляет 7.1–8.5 мас. %.
В подавляющем большинстве публикаций для активации металлоценовых прекатализаторов в синтезе СКЭПТ используют МАО. Однако МАО является дорогостоящим компонентом при необходимости использовать его в больших мольных избытках к активируемому прекатализатору (тысячи и десятки тыс. моль/моль). Кроме того, следует отметить нестабильность МАО при хранении, что влечет потерю активирующих свойств и невоспроизводимости характеристик получаемых полимеров. Новые эффективные активаторы металлоценовых катализаторов для синтеза этилен-пропиленовых и этилен-пропилен-диеновых каучуков на основе изобутилалюмоксанов (–O–Al(iBu)–)n и арилоксидов изобутилалюминия Al(iBu)3 – nOArn предложены в работах [75–79]. Под действием этих активаторов прекатализаторы легко переводятся в каталитически активное состояние при низких мольных соотношениях Al : Zr (200–300) и проявляют активности, сопоставимые с МАО. С применением каталитических систем на основе rac-Et(2-MeInd)2ZrMe2 и изобутилалюмоксанов разного состава и строения [77], получены СКЭПТ с содержанием ЭНБ до 5 мол. % (18 мас. %) и Mw от 4.06 × 104 до 79.0 × 104. Близкие по свойствам сополимеры формируются и при использовании арилоксидов изобутилалюминия [76, 78]. В отличие от традиционных систем, разработанные каталитические системы не приводят к образованию последовательностей PPP и PEP в сополимерах, что определяет их хорошие эластомерные свойства. Приведенные данные позволяют рассматривать изобутилалюмоксаны и арилоксиды изобутилалюминия в качестве перспективных альтернативных активаторов металлоценовых систем для синтеза СКЭПТ.
Новый эффективный активатор на основе смеси борарильных соединений LiB(C6F5)4 и B(C6F5)3 предложен исследователями компании “UniroyalChemicalComp.” [66]. Этот активатор обеспечивает стабильную кинетику процессов сополимеризации даже при повышенных значениях температуры. Для примера, система на основе цирконоцена Ph2CCpFluZrCl2 и комбинированного активатора проявляет стабильное каталитическое действие в ходе процесса сополимеризации при температуре 40–70°С, характеризуется высокой активностью и способностью к внедрению диенов (ЭНБ+ВНБ) на уровне 4–7 мас. % и формирует сополимеры с Mw = (70–90) × 104. В отличие от каталитических систем с МАО, металлоценовые комплексы с таким активатором образуют СКЭПТ с высокой вязкостью по Муни (ML = = 40–90; 125°С).
КОМПЛЕКСЫ “НАПРЯЖЕННОЙ” ГЕОМЕТРИИ
Отдельное место в синтезе СКЭПТ занимают мостиковые моноциклопентадиенильные комплексы, так называемые комплексы “напряженной” геометрии (CGC). Комплекс содержит η5-циклопентадиенильный фрагмент, подвесную донорную группировку (D) и мостиковую группу (Br) между циклопентадиенильной частью и донором:
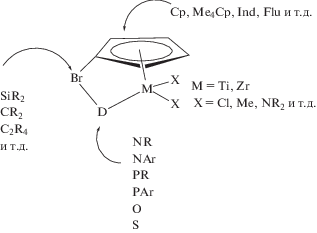
Мостиковая группа уменьшает угол Cp–M–D на ⁓20°–30° по сравнению с Cp–M–Cp, что делает переходный металл (М) в активном центре более доступным и облегчает внедрение сомономеров. Донорная группировка соединена σ-связью с переходным металлом и обеспечивает высокую стабильность катализаторам этого типа при повышенных значениях температуры (до 160–180°С). Вариации любой из частей лиганда CGC, а также типа переходного металла влияют на каталитические свойства комплексов, такие как активность, способность к внедрению сомономера(ов), ММ образующихся полимеров. Дизайн катализаторов основан на идеях J.E. Bercaw и J. Okuda, реализованных в синтезе мостиковых моноциклопентадиениламидных комплексов скандия, железа и титана [80, 81]. Разработка, синтез и применение CGC в процессах сополимеризации олефинов подробно рассмотрены в обзорах [13, 82, 83]:
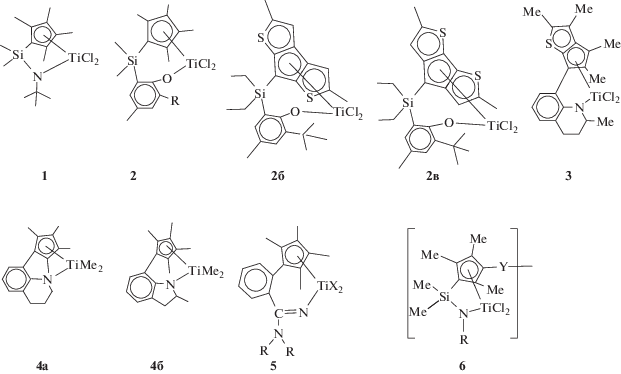 ,
,
где 2a – R = iPr, tBu, Adm; 5 – R = iPr, Cy; 6 – R = = tBu, Cy, Y = –C6H4–, –CH2–C6H4–.
Комплексы CGC (комплекс 1) были коммерциализированы DOW в середине 90-х годов для гомогенных полимеризационных процессов получения линейного полиэтилена низкой плотности [83–87]. Эти комплексы с замещенным Cp-кольцом оказались также эффективны для синтеза СКЭПТ [86, 87], что позволило DOW выпустить целую линейку эластомеров (Nordel IP) c разным составом сополимеров и кристалличностью. Компанией “Exxon” показана также возможность использования подобной каталитической системы для получения сополимеров с длинными разветвлениями на основе этилена, октена-1 и ВНБ [88]. Сравнение каталитического действия Me2SiCp*NtBuTiCl2 (комплекс 1) в тройной сополимеризации этилен–пропилен–ЭНБ в присутствии различных активаторов приведено в патенте [66]. Активность системы при использовании МАО в ∼2 раза ниже, чем с борсодержащими активаторами, ниже и молекулярные массы сополимеров. Однако активация комплексов Ph3СB(C6F5)4, Me2NHPhB(C6F5)4 и B(C6F5)3 приводит к быстрой дезактивации каталитической системы (в течение нескольких минут). Использование комбинированного активатора на основе LiB(C6F5)4 и B(C6F5)3 позволяет получать высокоактивную стабильную каталитическую систему и формировать СКЭПТ с хорошими свойствами (Mw = 50.9 × 104, Đ = 2.3, E : P = 68 : 32, ЭНБ = = 4.2 мас. %, Тg = –40°С; ML = 89, 125°С).
В конце 1990–начале 2000-х гг. фирмой “Sumitomo” был разработан новый тип CGC, получивший обобщенное название “PHENICS” (Phenoxy-Induced Complex of Sumitomo). Ключевым элементом в катализаторах такого типа было наличие боковой донорной фенокси-группировки с объемным заместителем в орто-положении. Комплексы 2: R = iPr, tBu, Adm – наиболее эффективные представители катализаторов этого типа, обеспечивающие полимеризацию при повышенных значениях температуры (до 180°С). В полимеризации пропилена в присутствии комбинированного активатора AliBu3–Ph3CB(C6F5)4 катализаторы типа 2a обеспечивают высокую активность (до 1500 кгПП/(ммоль Ti ч)), формируют синдио-обогащенный полипропилен очень высокой молекулярной массы до 2 × 106, Đ = 1.5–1.6 [82]. Комплексы 2 с сопряженными гетероциклами являются высокоэффективными высокотемпературными катализаторами в синтезе сополимеров этилена с гексеном-1 c активностью 27 200 при 80°С и 6000 кгсопол (мольTi ч)–1 при 180°С, формируя при этих температурах сополимеры с Mw = (23.4 и 6.1) × 104, Đ ∼ 2 и содержанием гексена в сополимере до 10–30 мол. % [89, 90]. Прекатализатор 2a является перспективным и для синтеза СКЭПТ [91]. При активации CPh3B(C6F5)4 в присутствии ТИБА при 60°С и мольном соотношении сомономеров E : P : ЭНБ = 0.76 : 2.08 : 0.016 он обеспечивает активность 53200 кгсопол (мольTi ч)–1 и образует сополимер мольного состава 66.7/30.8/2.5.
В последние годы возрос интерес к катализаторам этого типа со стороны ведущих производителей СКЭПТ. Титановый комплекс компании “Lotte Chemical”, содержащий сопряженный с тиофеновым кольцом Cp-лиганд (комплекс 3), эффективно активируется традиционными активаторами и с активностями 2500–5200 кг (мольTi ч)–1 проводит тройную сополимеризацию этилен–пропилен–диен (ЭНБ или ВНБ) при высоких значениях температуры (80–180°С) [92]. С участием этого катализатора получены каучуки, содержащие 3–7 мас. % диена, с высокими молекулярными массами (Mw = (18.5–32.0) × 104, Đ = = 2.9–3.9) и ML = 110–168 (1+8, 125°C).
Бинарные каталитические системы на основе смеси тетрагидрохинолинил- и тригидроиндолинил-циклопентадинильных комплексов титана (комплексы 4) разработаны “LG Chem” [93–96]. При использовании Me2NHPhB(C6F5)4 в качестве активатора они обеспечивают высокую активность (до 18 300–49 000 кгполимер/(моль Ti ч)) и работают при температуре до 160°С в проточном реакторе. Утверждается, что эти системы позволяют в широких пределах варьировать состав образующихся сополимеров (E : P : ЭНБ = (50–70) : (24–48) : (5–12) мас. %.), получать сополимеры с высокой молекулярной массой (Mw = (15–30) × 104, Đ = 2.7–3.2) и ML = 30–80.
Разновидностью комплексов напряженной геометрии считаются некоторые производные широко известных полусэндвичевых k1-амидинатных титановых катализаторов компании “ARLANXEO” (“Lanxess”), в которых циклопентадиенильное кольцо соединено через фенильный заместитель с амидиновым лигандом (комплекс 5; X = Cl, Me) [97]. Титановые комплексы такого типа эффективно активируются МАО и ведут полимеризацию этилен–пропилен–ЭНБ при повышенных значениях температуры (90°С) в проточном режиме. Эффективность комплексов в синтезе СКЭПТ сопоставима с полусэндвичевыми k1-амидинатными титановыми катализаторами (см. далее в разделе “Полусэндвичевые комплексы титана”). Эти комплексы обеспечивают получение СКЭПТ с высокой молекулярной массой (Mw = = (36–56) × 104, Đ = 2.2–2.6), содержанием этилена 38–42 мас. %, пропилена 56–59 мас. % и умеренным содержанием циклического диена (до 2.5 мас. %). Такой тип комплексов используется в технологии получения СКЭПТ компании “ARLANXEO” (“Keltan ACE”).
Новые эффективные CGC на основе биметаллических комплексов (комплекс 6) разработаны “Kumho Polychem” – одним из крупнейших производителей СКЭПТ в мире [98]. Катализаторы с высокой активностью в широком диапазоне температуры (до 150°С) способны вести сополимеризацию этилена с пропиленом и тройную сополимеризацию этилен–пропилен–ЭНБ. На этих катализаторах получают каучуки с высокими и легко регулируемыми молекулярными массами (Mw = (22–40) × 104, Đ = 2.5–5.1). Соединения проявляют высокую способность к внедрению пропилена и диенов. Содержание пропилена в сополимерах достигает 50 мас. %, а ЭНБ до 20 мас. %.
ПОЛУСЭНДВИЧЕВЫЕ КОМПЛЕКСЫ ТИТАНА
Перспективным классом катализаторов для синтеза СКЭПТ являются полусэндвичевые титаноцены, сoдержащие донорные лиганды, прочно соединенные с металлом σ-связью и π-донорным взаимодействием титана с O- или N-атомом. В качестве донорных выступают арилоксидные [99, 100], фосфинимидные [101–106], кетимидные [13, 101, 107], гуанидинатные [13, 107, 108], иминоимидазолидинатные [13, 102–105, 107] и k1-координированные амидинатные [13, 97, 107, 108–121] лиганды. Этот подкласс комплексов позволяет получать каучуки с широким набором свойств и лежит в основе современных технологий промышленного производства СКЭПТ.
Разработка и изучение свойств арилоксидных полусэндвичевых титановых комплексов началась с их открытия в 1998 г. группой Nomura [122, 123]. Комплексы, активированные МАО или Ph3CB(C6F5)4/AliBu3, проявляют высокую активность в полимеризации этилена и демонстрируют высокую способность к внедрению α-олефина в сополимеризации этилен–бутен-1 и этилен–гексен-1 [122–124]. Они также эффективно ведут сополимеризацию этилена с циклическими олефинами различного строения, позволяя получать сополимеры с высоким содержанием сомономера (до 35 и 60 мол. % в случае циклогексена и норборнена соответственно) [125–127].
Результаты по тройной сополимеризации этилен–α-олефин–диен на этих катализаторах, представленные патентами компаний “Asahi Kasei Chem.” [99] и “SK Innovation” [100], свидетельствуют об их большом потенциале для синтеза СКЭПТ:
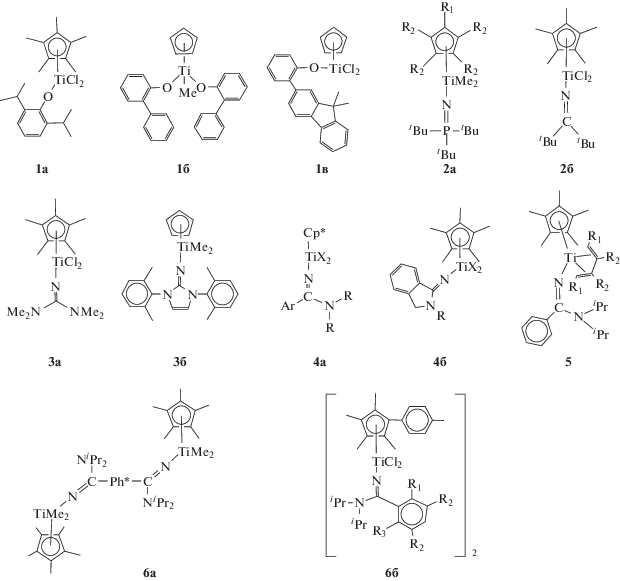
Здесь 2a – R1 = H, C6F5, Me; R2 = Me. 4a – Cp* = Cp, Me5Cp, (C6F5)Cp, nBuCp, (TMS)Cp, Ind; Ar = Ph, 2-MePh, 2,6-F2Ph, 2,6-Cl2Ph, 9-An, 4-Me2NPh, 4-CF3Ph, 4-tBuPh; R = iPr, Cy; X = Cl, Me. 4б – R = = 2,6-Me2Ph, Alk, CycloAlk; X = Cl, Me. 5 – R1 = H, Ph; R2 = H, Me. 6a – Ph* = p-C6H4, m-C6H4, C6H4–CH2–C6H4–. 6б – R1 = H, F, iPr; R2 = H, CF3; R3 = H, F.
Так, титановый комплекс 1a, активированный МАО, демонстрирует высокую эффективность в проведении тройной сополимеризации этилен–гексен–1–1,5-гексадиен [99]. Циклизации диена на катализаторах этого типа не происходит. Молекулярные массы сополимеров составляют (21–30) × 104 (Đ = 2), содержание гексена-1 достигает 39 мол. % (49 мас. %), а 1,5-гексадиена – 4 мол. % (9 мас. %).
Новые полусэндвичевые титаноцены, содержащие арильную группу в орто-положении арилоксилиганда, заявлены в патенте “SK Innovation” [100]. На примере комплексов 1б и 1в продемонстрирована способность этих соединений с высокой активностью (14 000 и 15 000 кгсопол (мольTi ч атм)–1 соответственно) проводить сополимеризацию этилен–пропилен–ЭНБ в диапазоне температуры 80–120°С. Молекулярные массы получаемых сополимеров Mw = (6–60) × 104 (Đ = 2–3), а содержание ЭНБ достигает 15 мас. %. Конверсия ЭНБ составляет 90–94%, что существенно превышает конверсию на ванадиевых каталитических системах (∼40% для системы VCl3/ (C2H5)3Al2Cl3).
Фосфинимидные и кетимидные полусэндвичевые титановые катализаторы были разработаны компанией “Nova Chemical” для процессов полимеризации этилена [128–130] и сополимеризации этилена с α-олефинами [130]. Первый патент на процесс получения СКЭПТ в условиях суспензионной (65–80°С) и растворной (100–140°С) сополимеризации этилен–пропилен–ЭНБ с применением таких катализаторов получен в 2001 году [101]. Наиболее эффективными структурами для синтеза СКЭПТ оказались соединения, содержащие объемные трет-бутильные группы в составе фосфинимидного лиганда (комплексы 2). Достоинствами катализаторов являются простота синтеза комплексов, способность к активации Ph3CB(C6F5)4. Наибольшую активность при 65°С показали комплексы типа 2a (R1 = = R2 = H и R1 = C6F5, R2 = H: 17 000–44 000 кгполимер (мольTi ч атм)–1), тогда как активность кетимидного комплекса 2б существенно ниже (3600–14 800 кгполимер (мольTi ч атм)–1). Сополимеры, образующиеся на этих системах имеют высокую молекулярную массу (до 56 × 104). Активация комплексов МАО дает менее активные системы (4400 кгполимер (мольTi ч атм)–1), но обеспечивает Mw сополимеров до 76 × 104. На комплексах этих типов возможно получение сополимеров с содержанием диена до ∼10 мас. %.
В 2007 году компанией “DSM Elastomers” (позднее “Lanxess”, а в настоящее время – “ARLANXEO”) было объявлено о заключении лицензионного соглашения с компанией “NOVA Chemical” и приобретении исключительных прав на производство СКЭПТ с применением каталитической технологии, основанной на фосфинимидных и кетимидных катализаторах [131]. Новая технология предстала под названием “Keltan Advanced Catalyst Elastomer” (“Keltan ACETM”).
Особый коммерческий интерес представляют СКЭПТ, получаемые с участием ВНБ, в том числе в комбинации с ЭНБ. Более реакционноспособная двойная связь ВНБ по сравнению с ЭНБ определяет возможность контролируемого образования сшивок и облегчает процессы вулканизации при малом содержании диена в сополимере, тем самым улучшая экструзионные свойства сополимеров и позволяя использовать значительно меньшее количество пероксидов [2, 132]. Тройные сополимеры с ВНБ имеют повышенную стойкость при высокотемпературном старении на воздухе или в присутствии полярных жидкостей, проявляют улучшенные характеристики при отрицательных значениях температуры. Фосфинимидные комплексы оказались перспективными катализаторами для синтеза таких сополимеров [102–106]. Содержание ВНБ в сополимере достигает 4.0 мас. %. Комплексы с объемными заместителями в составе фосфинимидного лиганда (трет-бутильными и изо-пропильными) проявили наибольшую активность в сополимеризации этилена, пропилена и смеси диенов ЭНБ+ВНБ при активации модифицированным МАО так же, как и в сополимеризации с ЭНБ при активации комплексов боратными активаторами. Катализаторы включены в технологию получения СКЭПТ “Keltan ACETM”.
В 2002 году W.P. Kretschmer с соавторами был разработан новый тип полусэндвичевых титановых комплексов, содержащих гуанидинатные и иминоимидазолидинатные лиганды, эффективные в гомополимеризации этилена, пропилена и сополимеризации этилена с α-олефинами (пропилен, пентен-1, гексен-1), стиролом, изопреном [132–134]. Катализаторы проявляют высокую активность, эффективны при высоких значениях температуры процесса (вплоть до 150°С) и позволяют получать полимеры с высокими молекулярными массами (Mw = (1–10) × 104). Содержание α-олефина в сополимерах достигает 50 мас. %. Комплексы 3 показали также высокую эффективность в синтезе СКЭПТ с ЭНБ и ВНБ (53 000–89 000 кгсопол (мольTi ч)–1, содержание диенов достигает 5 мас. %) [102–105, 108] и используются в технологии “Keltan ACETM”.
Развитие работ “DSM Elastomers” по исследованию влияния состава лиганда, содержащего –С=N– группу, на каталитические свойства полусэндвичевых комплексов титана для синтеза СКЭПТ привело к открытию новых эффективных катализаторов, содержащих k1-амидинатный лиганд –N=C(Ar)NR2 [2, 13, 97, 107–121]. k1-Амидинатные комплексы типа 4 проявляют высокую активность (до 1157 тоннполимер (мольTi ч)–1 в тройной сополимеризации олефинов и диенов и позволяют получать сополимеры с высоким содержанием диенов (до 15 мас. % ЭНБ, 5 мас. % ВНБ и 20 мас. % ДЦПД). При этом, как и для комплексов 2, в процессе сополимеризации с ВНБ на этих системах подавляется образование сшивок [13]. k1-Амидинатные катализаторы стабильны в процессах сополимеризации при температуре до 120°С и формируют сополимеры с высокими молекулярными массами (Mw = (6– 200) × 104). Строение лигандов в этих комплексах существенно влияет на их каталитические свойства и свойства сополимера. Разнообразие синтезированных в настоящее время комплексов позволяет получать СКЭПТ разного состава и с широким набором свойств [108–121]. Наиболее активные катализаторы содержат перметилированный циклопентадиенильный лиганд и 2,6-дигалогенидфенильный или изоиндолиновый заместитель в амидинатной группе. Вместе с тем, наибольшее внедрение пропиленового и диеновых сомономеров наблюдается для наименее активных каталитических систем. Интересным типом k1-амидинатных катализаторов являются комплексы с иминоизоиндолиновой группой [111, 118, 121], которые позволяют получать СКЭПТ с высоким содержанием пропилена (до 70 мас. %) и ЭНБ (до 15 мас. %) и сверхвысокой молекулярной массой (70–200) × 104. Характеристическая вязкость [η], измеренная при температуре 135°С, достигает 50 дл г–1. k1-Амидинатные катализаторы эффективно активируются борарильными активаторами и МАО при больших мольных избытках по отношению к титану (Al/Ti = 103–104). В последнем случае для увеличения эффективности системы добавляют 2,6-дитретбутил-4-метилфенол (BHT), связывающий ТМА, содержащийся в МАО.
Одним из перспективных подходов можно назвать использование комплексов титана, стабилизированных диеном (комплекс 5), что, в частности, позволяет исключить применение дорогостоящего МАО [114, 115, 120]. Эти комплексы эффективно активируются CPh3B(С6F5)4 и B(C6F5)3 (B/Ti = 2) в присутствии небольших мольных избытков изо-бутилалюмоксана (Al/Ti = 100) в комбинации с BHT (BHT/Ti = 2). Активности этих каталитических систем и их способность к внедрению диеновых сомономеров сопоставимы со структурно сходным дихлоридным катализатором, активированным МАО. Представленные результаты указывают на перспективность способа удешевления k1-амидинатных титановых катализаторов путем создания диен-содержащих аналогов.
Использование лигандов с двумя амидиновыми группами позволяет получать биметаллические k1-амидинатные титановые комплексы (комплекс 6a) [112, 113]. Соединения показали высокую активность в сополимеризации этилен–пропилен–ЭНБ+ВНБ. Биметаллические титановые катализаторы в сравнении с близкими по строению монометаллическими комплексами (комплекс 4a) обладают более высокой способностью к внедрению пропилена в сополимер (48–51 мас. %), но низкой – по отношению к диенам (0.4–0.7 мас. %). Высокое сродство к пропилену объясняется меньшей донорной способностью амидинатных лигандов биметаллических комплексов [112]. Низкая способность к внедрению диенов определяется стерическим фактором соединений.
Другой тип полусэндвичевых амидинатных биметаллических титановых каталитических комплексов (комплексы типа 6б) исследуется в компании “LG” [117]. Соединение двух титановых каталитических центров осуществляется мостиковой бисфениленовой группой между тетраметилциклопентадиенильными гаптосвязанными лигандами. Комплексы проявляют умеренную активность в процессе сополимеризации, но обеспечивают высокую способность к внедрению пропилена (60–65 мас. %) и ЭНБ (5.8–8.1 мас. %). СКЭПТ, формирующиеся на этих системах, имеют высокие значения Mw = (44.5–56.0) × 104 и обладают прекрасными механическими свойствами.
Перспективность данного класса комплексов, огромное разнообразие лигандов и простота синтеза позволяют использовать возможности методов комбинаторной химии для рационального дизайна новых эффективных каталитических систем. Такие исследования активно проводят в компании “ARLANXEO” по установлению новых оптимальных структур полусэндвичевых k1-амидинатных титановых комплексов для синтеза СКЭПТ [2, 118, 121, 135, 136]. В изобретениях [118, 121] описан способ приготовления катализаторов из η5-LTiCl3 и амидинов, депротонированных одним эквивалентом MeMgCl, с последующим применением полученных соединений in situ в процессах сополимеризации. Изучено влияние большого числа гапто-связанных лигандов и амидинатных лигандов на каталитические свойства полусэндвичевых титановых комплексов. Примененные подходы существенно расширили число эффективных структур катализаторов и марок получаемых на них СКЭПТ. Возможность дальнейшей разнообразной модификации амидинов и создания новых комбинаций полусэндвичевых титановых комплексов свидетельствует о большом потенциале разработок в данной области. k1-Амидинатные катализаторы также включены в технологию “Keltan ACE™”, на базе которой в настоящее время организовано крупномасштабное производство СКЭПТ на двух заводах в Нидерландах и Китае, объемом 95 и 160 тыс. тонн/год соответственно [2, 107, 137].
ПОСТ-МЕТАЛЛОЦЕНОВЫЕ КАТАЛИЗАТОРЫ
Бис-(фенил)феноксидные катализаторы
Бис-(фенил)феноксидные комплексы циркония и гафния, оказавшиеся эффективными пост-металлоценовыми катализаторами полимеризации олефинов [138–140], были обнаружены компанией “Symyx Technologies” методами комбинаторной химии, разработанными ранее для скрининга катализаторов полимеризации олефинов [141, 142]:
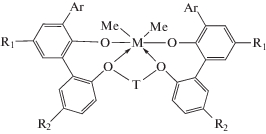
(M = Zr, Hf; R1 = Alk; R2 = H, Cl, F, tBu; T = Alk, CycloAlk).
Последние 10 лет катализаторы этого класса активно разрабатывает компания “Dow Elastomers” для синтеза каучуков [143–148]. Этот тип комплексов обладает уникальными каталитическими характеристиками, превосходящими многие известные классы катализаторов для производства СКЭПТ. Достоинствами бис-(фенил)феноксидных катализаторов являются способность к эффективной активации низкими мольными количествами активатора (AlМАО/M = 10–200, Bборат/M = = 0–5 моль/моль), высокая активность в процессах сополимеризации при 120–180°С. Преимуществом высокотемпературного процесса, осуществляемого в режиме растворной полимеризации (полимер растворим в реакционной среде), считается формирование аморфных сополимеров с широким ММР. Особенностью этих систем является также их способность образовывать сополимеры с высокими молекулярными массами (Mw = (10–100) × 104) и низким содержанием гель-фракции [85]. С применением таких катализаторов получены каучуки с высоким содержанием пропилена (до 50 мас. %) и диеновых сомономеров разного типа (ЭНБ, ВНБ, ДЦПД и другие, до 16 мас. %). Структурными элементами бис-(фенил)феноксидных комплексов, определяющих их свойства, можно назвать тип переходного металла (Zr, Hf), тип мостиковой эфирной группы (T = Alk, CycloAlk), арильного лиганда (карбозолил, октагидроантраценил, дитретбутилфенил) и тип пара-заместителей в фенильных фрагментах (R1 = Alk; R2 = H, F, Cl, tBu). Этот класс катализаторов включен в основу разработанной в 2015 г. технологии “Advanced Molecular Catalyst” (AMC) для производства СКЭПТ [85]. Высокоэффективная технология AMC пришла на смену NordelTM (“DOW Elastomers”), основанной на катализаторах CGC. В 2018 году в г. Плакимине (США) был запущен новый завод мощностью 200 тыс. тонн СКЭПТ в год, работающий на новой AMC-технологии [149, 150].
Фенокси-иминные катализаторы
Фенокси-иминные комплексы переходных металлов – известный класс пост-металлоценовых катализаторов, развиваемый в компании “Mitsui” под руководством Т. Fujita и применяемый для полимеризации и олигомеризации этилена, полимеризации пропилена и сополимеризации этилена с α- и циклическими олефинами [151]. В сополимеризации этилена и пропилена эффективность катализаторов существенно зависит от типа переходного металла и строения фенокси-иминного лиганда. Здесь проявляют активность и способность к внедрению пропилена лучше других циркониевые комплексы. Структурными элементами фенокси-иминных лигандов, определяющими каталитические свойства соединений, являются тип алициклических или ароматических заместителей при имине и заместители в орто-положении фенокси-группы. Наиболее высокую эффективность (активность и способность к внедрению пропилена) показывают соединения, содержащие фенильный заместитель с пара-электронодонорным заместителем при имине и незамещенный фенильный заместитель в орто-положении фенокси-группы. Влияние фенильного заместителя в орто-положении фенокси-группы на свойства катализатора объясняется комбинацией электронного фактора и пространственной доступностью формируемого каталитически активного центра [152]. Предполагается, что благодаря свободному вращению фенильной группы обеспечивается возможность более эффективной координации α-олефина на переходном металле и протекания процесса сополимеризации.
Информация о возможности целенаправленного синтеза этилен–α-олефин–диеновых каучуков на фенокси-иминных катализаторах представлена в патенте [153]:
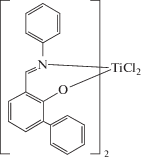
Продемонстрирован синтез этилен–пропилен–ЭНБ каучуков с высокой активностью (9000–27 000 кг (мольTi ч)–1) и при высоких значениях температуры реакции (80–120°С). Конверсия ЭНБ в тройной сополимеризации достигает 99%. Согласно данным патента на фенокси-иминных комплексах можно получать СКЭПТ с содержанием α-олефина до 50 мол. % и несопряженных диенов до 49 мол. %. Характеристическая вязкость [η] в декалине при 135°С составляет 0.02–10 дл г–1. С применением этого катализатора получены каучуки, характеризующиеся низкотемпературной гибкостью. Представленные результаты указывают на большой потенциал фенокси-иминных комплексов в качестве катализаторов для синтеза СКЭПТ.
ЗАКЛЮЧЕНИЕ
Несмотря на то что этилен-пропилен-диеновые каучуки выпускаются промышленностью уже более 50 лет, анализ литературных данных показывает, что возможностей усовершенствовать технологии синтеза СКЭПТ и расширить их ассортимент остается существенно много. Такой потенциал обеспечивается разработкой каталитических систем новых поколений на основе комплексов IVB-группы, альтернативных малоактивным, нестабильным и токсичным ванадиевым катализаторам. В пользу этого, в частности, свидетельствует высокая исследовательская активность мировых лидеров в производстве СКЭПТ по разработке новых классов катализаторов за последние 10–15 лет. Основными преимуществами современных систем по сравнению с ванадиевыми являются более высокая активность, экологическая безопасность, возможность целевого дизайна эластомеров и реализации инноваций в цепочке “катализатор–процесс–продукт”. Применение этих каталитических систем уже позволило вывести на рынок различные марки новых СКЭПТ с широким, легко варьируемым диапазоном состава, структур, молекулярно-массовых характеристик и свойств (например, NordelTM IP компании “Dow Elastomers” или Keltan ACETM компании “ARLANXEO”, VistalonTM производителя “Exxon” или Mitsui EPTTM производителя “Mitsui” и другие), что обусловливает актуальность и перспективность рассмотренных в настоящем обзоре каталитических систем для синтеза СКЭПТ и дальнейшего развития исследований в этом направлении.
Работа выполнена при финансовой поддержке фонда Министерства образования и науки (Госзадание № 01201055317).
Список литературы
Global Ethylene Propylene Diene Monomer (EPDM) Industry Production, Sales and Consumption Status and Prospects Professional Market Research Report 2018–2023, https://www.giiresearch.com/report/qyr606224-global-ethylene-propylene-diene-monomer-epdm.html
van Duin M., van Doremaele G., van der Aar N. // KGK. 2017. V. 11–12. P. 14.
Transition Metal Catalyzed Polymerizations: Ziegler-Natta and Metathesis Polymerizations / Ed. by R.P. Quirk, R.E. Hoff. Cambridge: Cambridge University Press, 1988.
Ma Y., Reardon D., Gambarotta S., Yap. G. // Organometallics. 1999. V. 18. № 15. P. 2773.
Mark. H.F. // Encyclopedia of Polymer Science and Technology. New York: Wiley, 2004. V. 6.
Kramer O., Good W.R. // J. Appl. Polym. Sci. 1972. V. 16. № 10. P. 2677.
Davis S.C., von Hellens W., Zahalka H.A., Richter K.-P. // Polymer Material Encyclopedia / Ed. by J.C. Salamone. Boca Raton: CRC Press Inc., 1996. V. 3. P. 2264.
Karpeles R., Grossi A.V. // Handbook of Elastomers: 2nd Ed. / Ed. by A.K. Bhowmick, H.L. Stephens. New York: Marcel Dekker, 2001. P. 845.
Ver Strate G. // Encyclopedia of Polymer Science and Engineering. New York: Wiley, 1986. V. 6. P. 522.
Nomura K., Zhang S. // Chem. Rev. 2011. V. 111. № 3. P. 2342.
Noordermeer J.W.M. // Ethylene-Propylene Polymers. Kirk-Othmer Encyclopedia of Chemical Technology. New York: Wiley, 2002.
Li X., Hou Z. // Coord. Chem. Rev. 2008. V. 252. № 15–17. P. 1842.
Baier M.C., Zuideveld M.A., Mecking S. // Angew. Chem. Int. Edit. 2014. V. 53. № 1. P. 2.
Ильин В.М., Резова А.К. // Каучук и резина. 2016. V. 1. № 1. P. 38.
Арутюнов И.А., Иванисько О.Л., Костоглодов П.В., Максимов Д.А., Кочнев А.И., Казаков Ю.М. // Каучук и резина. 2010. V. 5. № 5. P. 22.
Tritto I., Boggioni L., Ferro D.R. // Coord. Chem. Rev. 2006. V. 250. № 1–2. P. 212.
Boggioni L., Tritto I. // Adv. Polym. Sci. 2013. V. 258. P. 117.
Resconi L., Waymouth R.M. // J. Am. Chem. Soc. 1990. V. 112. № 12. P. 4953.
Coates G.W., Waymouth R.M. // J. Am. Chem. Soc. 1991. V. 113. № 16. P. 6270.
Resconi L., Coates G.W., Mogstad A., Waymouth R.M. // J. Macromol. Sci. Chem. 1991. V. A28. № 11–12. P. 1225.
Coates G.W., Waymouth R.M. // J. Am. Chem. Soc. 1993. V. 115. № 1. P. 91.
Bergemann Ch., Cropp R., Luft G. // J. Mol. Catal. 1997. V. 116. № 3. P. 317.
Pietikäinen P., Väänänen T., Seppälä J.V. // Eur. Polym. J. 1999. V. 35. № 6. P. 1047.
Naga N., Shiono T., Ikeda T. // Macromolecules. 1999. V. 32. № 5. P. 1348.
Pietikäinen P., Seppälä J.V., Ahjopalo L., Pietilä L.-O. // Eur. Polym. J. 2000. V. 36. № 1. P. 183.
Kim I., Shin Y.S., Lee J.K. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 9. P. 1590.
Kokko E., Pietikäinen P., Koivunen J., Seppälä J.V. // J. Polym. Sci., Polym. Chem. 2001. V. 39. № 21. P. 3805.
Kim I., Shin Y.S., Lee J.-K., Cho N.J., Lee J.-O., Won M.-S. // Polymer. 2001. V. 42. № 23. P. 9393.
Naga N., Imanishi Y. // Macromol. Chem. Phys. 2002. V. 203. № 4. P. 771.
Naga N., Okada S., Imanishi Y. // Polymer. 2004. V. 45. № 1. P. 117.
Tynys A., Eilertsen J.L., Seppдlд J.V., Rytter E. // Polymer. 2007. V. 48. № 10. P. 2793.
Yu Z., Marques M., Rausch N., Chien J.C.W. // J. Polym. Sci., Polym. Chem. 1995. V. 33. № 16. P. 2795.
Uozumi T., Tian G., Ahn C.-H., Tsubaki S., Sano T., Soga K. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 10. P. 1844.
Lee D.-H., Yoon K.-B., Park J.-R., Lee B.-H. // Eur. Polym. J. 1997. V. 33. № 4. P. 447.
Santos J.M., Ribeiro M.R., Portela M.F., Cramail H., Deffieux A. // Macromol. Chem. Phys. 2001. V. 202. № 15. P. 3043.
Santos J.M., Ribeiro M.R., Portela M.F., Cramail H., Deffieux A., Antinollo A., Otero A., Prashar S. // Macromol. Chem. Phys. 2002. V. 203. № 1. P. 139.
Hackmann M., Repo T., Jany G., Rieger B. // Macromol. Chem. Phys. 1998. V. 199. № 8. P. 1511.
Odian G. // Principles of Polymerization. New York: Wiley, 1991.
Ham G.E. // Copolymerization. New York: Interscience Publ., 1964.
Tosi C., Valvassori A., Ciampelli F. // Eur. Polym. J. 1969. V. 5. № 4. P. 575.
Фирсов A.П., Мешкова И.Н., Кострова Н.Д., Чирков Н.M. // Высокомолек. соед. 1966. Т. 8. № 11. С. 1860.
Cozewith C., Ver Strate G. // Macromolecules. 1971. V. 4. № 4. P. 482.
Bushick R.I. // J. Polym. Sci. A. 1965. V. 3. № 5. P. 2047.
Natta G., Mazzanti G., Valvasorri A., Sartory G., Barbagallo A. // J. Polym. Sci. 1961. V. 51. № 156. P. 429.
Ткачева Е.Н., Софронова О.В., Михеева В.А., Борейко Н.П., Галанин Д.А. // Вестн. Казанского технол. ун-та. 2010. № 5. P. 115.
Starck P., Lehtinen C., Lofgren B. // Angew. Makromol. Chem. 1997. V. 249. № 1. P. 115.
Lehtinen C., Lofgren B. // Eur. Polym. J. 1997. V. 33. № 1. P. 115.
Chien J.C.W., He D.J. // J. Polym. Sci., Polym. Chem. 1991. V. 29. № 11. P. 1585.
Lu L., Niu H., Dong J.-Y., Zhao X., Hu X. // J. Appl. Polym. Sci. 2010. V. 118. № 6. P. 3218.
Meshkova I.N., Shchegolikhin A.N., Raspopov L.N., Kiseleva E.V., Kuznetsov S.P., Grinev V.G., Novokshonova L.A. // Additives and Polymers. Analysis and Applications / Ed. by A. Berlin, S.Z. Rogozina, G.E. Zaikov. New York: Imprint Apple Acad. Press, 2015.
Friederichs N., Wang B., Budzelaar P.H.M., Coussens B.B. // J. Mol. Catal. A. 2005. V. 242. № 1– 2. P. 91.
Resconi L., Cavallo L., Fait A., Piemontesi F. // Chem. Rev. 2000. V. 100. № 4. P. 1253.
Galimberty M., Piemontesi F., Fusco O. // Metallocene-Based Polyolefins. Preparation, Properties and Technology / Ed. by J. Scheirs, W. Kaminsky. New York: Wiley, 1999. P. 309.
Weingarten U. // Ph.D. Thesis. Hamburg: University of Hamburg, 2000.
Arrowsmith D., Kaminsky W., Schauwienold A.-M. // J. Mol. Catal. A. 2000. V. 160. № 1. P. 97.
Bergstra M.F., Weikert G., Meier G.B. // Macromol. Reaction. Eng. 2009. V. 3. № 7. P. 433.
Ishihara T., Ban H.T., Hagihara H., Shiono T. // J. Polym. Sci., Polym. Chem. 2007. V. 45. № 24. P. 5731.
Forlini F., Fan Z.-Q., Tritto I., Locatelli P., Sacchi M.C. // Macromol. Chem. Phys. 1997. V. 198. № 8. P. 2397.
Stagnaro P., Boragno L., Losio S., Canetti M., Alfonso G.C., Galimberti M., Piemontesi F., Sacchi M.C. // Macromolecules. 2011. V. 44. № 10. P. 3712.
Arndt M., Kaminsky W., Schauweinold A.-M., Weingarten U. // Macromol. Chem. Phys. 1998. V. 199. № 6. P. 1135.
Fan W., Leclerc M.R., Waymouth R.M. // J. Am. Chem. Soc. 2001. V. 123. № 39. P. 9555.
Marathe S., Sivaram S. // Macromolecules. 1994. V. 27. № 5. P. 1083.
Lasarov H., Pakkanen T.T. // Macromol. Chem. Phys. 2000. V. 201. № 14. P. 1780.
Dolatkhani M., Cramail H., Deffieux A. // Macromol. Chem. Phys. 1996. V. 197. № 8. P. 2481.
Sobhanmanesh K., Hajizadeh A. // Iran. Pol. J. 2004. V. 13. № 4. P. 257.
Gillis D.J. Karpeles R. Pat. US6225426B1 USA. 2001.
Chen E.Y., Marks T.J. // Chem. Rev. 2000. V. 100. № 4. P. 1391.
Bochmann M. // Organometallics. 2010. V. 29. № 21. P. 4711.
Zijlstra H.S., Harder S. // Eur. J. Inorg. Chem. 2015. № 1. P. 19.
Yu Z., Marques M., Rausch M.D., Chien. J.C.W. // J. Polym. Sci., Polym. Chem. 1995. V. 33. № 16. P. 2795.
Endo K., Hiwara M., Matsuura S., Mizobuchi Yu., Yamamura Yu., Noguchi Yu., Ishii Yu., Sakai T., Shishido K., Ichino K., Inomata K., Takeuchi F., Ide K. Pat. US2016/0347894A1 USA. 2016.
Ichino K., Kikuchi Yo., Tohi Ya., Matsugi T., Yanagimoto Ya., Arino M., Shishido K., Hosoya M. Pat. US2016/0355622A1 USA. 2016.
Ichino K., Yamaguchi T., Aita Yu., Noguchi Yu. Pat. US2018/0072877A1 USA. 2018.
Pat. KR101792956B1 South Korea. 2017.
Бравая Н.М., Панин А.Н. Пат. РФ 2477289 Россия. 2013.
Файнгольд Е.Е., Бравая Н.М., Панин А.Н., Саратовских С.Л., Бабкина О.Н. Пат. РФ2588496 Россия. 2016.
Bravaya N.M., Panin A.N., Faingol’d E.E., Saratovskikh S.L., Babkina O.N., Zharkov I.V., Perepelitsina E.O. // Polym. Bull. 2016. V. 73. № 2. P. 473.
Faingol’d E.E., Bravaya N.M., Panin A.N., Babkina O.N., Saratovskikh S.L., Privalov V.I. // J. Appl. Polym. Sci. 2016. V. 133. № 14. P. 43276.
Faingol’d E.E., Zharkov I.V., Bravaya N.M., Panin A.N., Saratovskikh S.L., Babkina O.N., Shilov G.V. // J. Organomet. Chem. 2018. V. 871. P. 86.
Shapiro P.J., Bunel E., Schaefer W.P., Bercaw J.E. // Organometallics. 1990. V. 9. № 3. P. 867.
Okuda. J. // Chem. Ber. 1990. V. 123. № 8. P. 1649.
Braunschweig H., Breitling F.M. // Coord. Chem. Rev. 2006. V. 250. № 21–22. P. 2691.
Klosin J., Fontaine P.P., Figueroa R. // Accounts Chem. Res. 2015. V. 48. № 7. P. 2004.
Stevens J.C. // Stud. Surf. Sci. Catal. 1996. V. 101. P. 11.
Thakur V., Shan C.L.P., Li G., Han T., Doelder J.D. // Plast. Rubber Comp. 2019. V. 48. № 1. P. 32.
Stevens J.C., Timmers F.J., Wilson D.R., Schmidt G.F., Nickias P.N., Rosen R.K., Knight G.W., Lai S.-Ya. Pat. EP0416815A2 EU. 1991.
Canich J.A.M. Pat. EP0420436A1 EU. 1991.
Canich J.A.M., Licciardi G.F. Pat. US5057475 USA. 1991.
Nabika M., Katayama H., Watanabe T., Kawamura-Kuribayashi H., Yanagi K., Imai A. // Organometallics. 2009. V. 28. № 13. P. 3785.
Senda T., Hanaoka H., Okado Y., Oda Y., Tsurugi H., Mashima K. // Organometallics. 2009. V. 28. № 24. P. 6915.
Katayama H., Nabika M., Imai A., Miyashita A., Watanabe T., Johohji H., Oda Yo., Hanaoka H. Pat. US6329478B1 USA. 2001.
Pat. KR101501853B1 South Korea. 2013.
Park S.H., Yoon S.C., Kim S.K., Ko J.S., Park S.E., Choi S.Y. Pat. US9428600B2 USA. 2016.
Kim S.K., Park S.H., Yoon S.C., Ko J.S., Choi S.Y. Pat. US9493593B2 USA. 2016.
Yoon S.-C., Park S.-H., Ko J.-S., Choi S.-Y. Pat. US9410008B2 USA. 2016.
Park S.H., Yoon S.C., Kim S.K., Ko J.S., Park S.E., Choi S.Y. Pat. US9650460B2 USA. 2017.
Berthoud A., Norambuena V.Q., van Doremaele G., Zuideveld M.A., Mountford P., Scott R.T.W. Pat. US2016/0122455A1 USA. 2016.
Pat. KR101216691B1 South Korea. 2012.
Hasebe K. Pat. JP2005314504A. 2004.
Byoungtak Y., Youngwook K., Myungahn O., Jongsok H., Dongcheol S. Pat.WO2010/079906. 2010.
Brown S.J., Wang Q. Pat. EP1162214A1 EU. 2001.
Windmuller P.J.H., van Doremaele G.H.J. Pat. WO2005/005496A2. 2005.
Ijpeij E.G., Arts H.J., van Doremaele G.H.J., Beijer F.H., van Der Burgt F., Zuideveld M.A. Pat. WO2005/014601A2. 2005.
Windmuller P., Doremaele G. Pat. US2006/0205900A1 USA. 2006.
Windmuller P., Doremaele G. Pat. US7829645B2 USA. 2010.
Van Doremaele G.H.J., Kranenburg M., Dullaert K. Pat. US2010/0105851A1 USA. 2010.
Van Doremaele G., van Duin M., Valla M., Berthoud A. // J. Polym. Sci., Polym. Chem. 2017. V. 55. № 18. P. 2877.
Van Doremaele G.H.J., Zuideveld M.A., Leblanc A., Norambuena V.F.Q. Pat. EP 2319874A1 EU. 2009.
Van Doremaele G.H.J., Zuideveld M.A., Norambuena V.F.Q., Leblanc A. Pat. WO2011054927A1. 2011.
Ijpeij E., Arts H., van Doremaele G., Windmuller P., van der Burgt F., Zuideveld M.A. Pat. US7956140B2 USA. 2011.
Van Doremaele G.H.J., Berthoud A., Norambuena V.Q., Rupnicki L., Karbaum P. Pat. WO2015113957A1. 2015.
Collins R.A., Russell A.F., Scott R.T.W., Bernardo R., van Doremaele G.H.J., Berthoud A., Mountford P. // Organometallics. 2017. V. 36. № 11. P. 2167.
Berthoud A., van Doremaele G., Norambuena V.Q., Scott R., Zuideveld M.A., Arts H.J. Pat. EP2816050B1 EU. 2013.
Van Doremaele G.H.J., Zuideveld M.A., Mountford P., Heath A., Scott R.T.W. Pat. WO2011076775A1. 2011.
Van Doremaele G.H.J., Zuideveld M.A. Pat. WO2011076772A1. 2011.
Van Doremaele G.H.J., Kranenburg M., Dullaert K. Pat. US2010/0105851A1 USA. 2010.
Pat. KR101828001B1 South Korea. 2015.
Karbaum P., Scott R.T.W., van De Moosdijk J. Pat. US2017/0240663A1 USA. 2017.
Berthoud A., Van Doremaele G.H.J., Scott R.T.W., Perez F., Bernardo R. Pat. WO2017/029141A1. 2017.
Scott R.T.W., Zuideveld M.A., Mountford P. Pat. WO2014180913A1. 2014.
Scott R.T.W., Bernardo R., van de Moosdijk J., Berthoud A. Pat. EP3272761A1. 2018.
Nomura K., Naga N., Miki M., Yanagiand K., Imai A. // Organometallics. 1998. V. 17. № 11. P. 2152.
Nomura K., Naga N., Mikiand M., Yanagi K. // Macromolecules. 1998. V. 31. № 22. P. 7588.
Nomura K., Liu J., Padmanabhan S., Kitiyanan B. // J. Mol. Catal. A. 2007. V. 267. № 1–2. P. 1.
Zhao W., Nomura K. // Catalysts. 2016. V. 6. № 11. P. 175.
Nomura K., Wang W., Fujiki M., Liu J. // Chem. Commun. 2006. № 25. P. 2659.
Wang W., Tanaka T., Tsubota M., Fujiki M., Yamanaka S., Nomura K. // Adv. Synth. Catal. 2005. V. 347. № 2–3. P. 433.
Stephan D.W., Stewart J.C., Guerin F., Spence R.E.V.H., Xu W., Harrison D.G. // Organometallics. 1999. V. 18. № 7. P. 1116.
Stephan D.W., Guerin F., Spence R.E.V.H., Koch L., Gao X., Brown S.J., Swabey J.W., Wang Q., Xu W., Zoricak P., Harrison. D.G. // Organometallics. 1999. V. 18. № 11. P. 2046.
Mcmeeking J., Gao X., Spence R.E.V.H., Brown S.J., Jeremic D. Pat. WO9914250A1 USA. 1999.
Пpecc-peлиз: https://www.pressreleasefinder.com/pr/DSMELPR005/en/
Dees M., van Duin M., Twigg C., Dikland H. // Rubber World. 2009. V. 240. P. 13.
Kretschmer W.P., Dijkhuis C., Meetsma A., Hessen B., Teuben J.H. // Chem. Commun. 2002. V. 6. № 6. P. 608.
Kretschmer W. Pat. US2004/0192541A1 USA. 2004.
Bernardo R. Fundamental Investigation and Screening of Iminato Based Titanium Catalysts / Ed. by Ph.D. Thesis. Napoli: Universita degli Studi di Napoli Federico II, 2013.
Valla M., Bernardo R., Coussens B., Berthoud A. // Proceedings of XVIII Netherland’s Catalysis and Chemistry Conference, March 6–8 2017, Noordwijkerhout.
Интернет ресурс: http://keltan.com.br/en/about-keltan/keltan-ace/
Boussie T.R., Bruemmer O., Diamond G., Goh C., Lapointe A.M., Leclerc M.K., Shoemaker J.A. Pat. WO03/091262A1. 2003.
Boussie T., Bruemmer O., Diamond G., LaPointe A., Leclerc M., Micklatcher C., Sun P., Bei X. Pat. US2006/0025548A1 USA. 2006.
Boussie T., Diamond G., LaPointe A., Leclerc M., Micklatcher C., Sun P., Bei X. Pat. US2006/0052554A1 USA. 2006.
Boussie T.R., Diamond G.M., Goh C., Hall K.A., LaPointe A.M., Leclerc M., Lund C., Murphy V., Shoemaker J.A.W., Tracht U., Turner H., Zhang J., Uno T., Rosen R.K., Stevens J.C. // J. Am. Chem. Soc. 2003. V. 125. № 14. P. 4306.
Boussie T.R., Diamond G.M., Goh C., Hall K.A., LaPointe A.M., Leclerc M., Murphy V., Shoemaker J.A.W., Turner H., Rosen R.K., Stevens J.C., Alfano F., Busico V., Cipullo R., Talarico G. // Angew. Chem. Int. Edit. 2006. V. 45. № 10. P. 3278.
Konze W.V., Vanderlende D.D. Pat. WO2007/136506A2. 2007.
Boone H.W., Iverson C.N., Konze W.V., Vanderlende D.D. Pat. WO2007/136494A2. 2007.
Konze W.V., Stevens J.C., Vanderlende D.D. Pat. WO2007/136495A2. 2007.
Klosin J., Thomas P.J., Iverson C.N., Aboelella N.W., Frazier K.A. Pat. US9029487B2 USA. 2015.
LiPiShan C., Karjala T.W., Smith M.L., Spencer L.P., Klosin J. Pat. US 9422383 B2 USA. 2016.
Spencer L.P., Kirschner J.M. Pat. US9534070B2 USA. 2017.
Пpecc-peлиз https://www.icis.com/resources/news/ 2018/02/01/10189241/us-dowdupont-to-start-up-ldpe-epdm-plants-in-q1/
Пресс-релиз компании Dow Elastomers; https://www.dow.com/elastomers/news/2017/ 20171009a.htm#.W5jAF7hI5ec
Makio H., Terao H., Iwashita A., Fujita T. // Chem. Rev. 2011. V. 111. № 3. P. 2363.
Terao H., Iwashita A., Matsukawa N., Ishii S., Mitani M., Tanaka H., Nakano T., Fujita T. // ACS Catal. 2011. V. 1. № 4. P. 254.
Matsuura S., Murakami H., Hasada Ya., Saito J., Fujita T., Matsui S. Pat. US2005/0176890A1 USA. 2005.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)