

Высокомолекулярные соединения (серия С), 2020, T. 62, № 1, стр. 20-54
Волокнообразующие сополимеры акрилонитрила: от синтеза к свойствам прекурсоров углеродного волокна и перспективам промышленного производства
Е. В. Черникова a, b, *, Р. В. Томс c, А. Ю. Гервальд c, Н. И. Прокопов c
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
c МИРЭА – Российский технологический университет
Институт тонких химических технологий им. М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 13.01.2020
После доработки 20.01.2020
Принята к публикации 05.02.2020
Аннотация
Обобщены последние достижения в области синтеза сополимеров акрилонитрила. Проанализированы новые экологичные способы получения сополимеров акрилонитрила в ионных жидкостях и в сверхкритических средах. Обсуждаются возможности разных вариантов контролируемой радикальной и анионной полимеризации в управлении структурой и свойствами сополимеров акрилонитрила. Рассмотрены способы получения сополимеров акрилонитрила, которые можно использовать при формовании волокон из расплава. Проведен патентный поиск и обобщена информация о новых способах и рецептурах синтеза сополимеров акрилонитрила.
ВВЕДЕНИЕ
В последние десятилетия значительно возрос интерес к армированным композиционным материалам разного назначения на основе углеродных волокон. В целом в период с 2005 по 2015 гг. мировое потребление углеродных волокон выросло более чем в два раза [1]. Особый интерес среди них вызывают высокопрочные волокна с прочностью на разрыв более 6 ГПа и модулем упругости выше 300 ГПа, основным производителем которых является японская компания “Toray” [2]. Следует отметить, что эти характеристики заметно ниже предсказанных теоретических значений, что, естественно, способствует появлению поисковых работ в области получения как прекурсоров, так и конечных углеродных волокон. Другой не менее важной задачей, является создание более экономичных и экологичных способов получения углеродных волокон, которые могут обладать не столь высокими механическими характеристиками.
Производство углеродных волокон – сложная многопараметровая задача, включающая синтез полимера (при необходимости), формование волокна (получение прекурсора) и последующие процессы термоокислительной стабилизации, карбонизации и графитизации. Каждая из этих стадий вносит вклад в свойства конечного продукта.
Среди известных прекурсоров наибольшее практическое значение имеют полимеры на основе акрилонитрила (АН), что обусловлено их высокой удельной прочностью и жесткостью в сочетании с малой массой и низкой стоимостью, а также высоким выходом углерода при карбонизации.
Впервые акриловые волокна текстильного назначения на основе сополимеров акрилонитрила были запатентованы в середине 1940-х гг. компанией “DuPont” и запущены в промышленное производство в 1950-х гг. [3]. Этот метод был основан на суспензионной полимеризации акрилонитрила и формовании волокна из раствора в ДМФА. В середине 1950-х, используя тот же метод синтеза ПАН, компанией “Monsanto” была разработана технология формования из ДМАА [4], а затем фирма “Cyanamid” запатентовала способ формования суспензионного ПАН из водного раствора NaSCN [5]. Последний вариант стимулировал разработку “Courtaulds” растворного метода синтеза ПАН и формования в водном растворе NaSCN [6]. Последующим производителям требовалось вносить новые элементы в технологии, чтобы не нарушать права на патенты предшественников. В итоге в 1970-х гг. были созданы все основные технологии получения ПАН-прекурсора, включавшие растворную (ДМСО, ДМФА, ДМАА, этилен- и пропиленкарбонат, ZnCl2 (водный), NaSCN (водный), HNO3) и гетерофазную (преимущественно, суспензионную и осадительную) полимеризации.
Альтернативным вариантом получения ПАН-прекурсора является формование не из раствора, а из расплава. Это накладывает определенные ограничения на молекулярные характеристики ПАН и на способы его получения. Кроме того, формование из расплава более экологично и экономично, поскольку не требует использования органических растворителей и существенно повышает концентрацию полимера при формовании волокна.
Использование (со)полимеров АН для производства углеродных волокон для конструкционного применения связано с работами японского ученого A. Shindo, который впервые разработал и запатентовал карбонизацию ПАН по периодическому способу без вытяжки с получением углеродного волокна с прочностью менее 2 ГПа [7]. Ориентация на гражданское применение углеродных волокон в последующем послужила серьезной причиной успеха японских производителей, которые совместными усилиями развивали технологии создания ПАН-прекурсора для углеродных волокон на основе собственных способов получения акриловых волокон. В США большее внимание уделялось углеродным волокнам из вискозы, поэтому американские производители изначально не разрабатывали свои технологии, а применяли готовый ПАН-прекурсор фирм “Courtaulds”, “Exlan” или “Toray”. Великобритания фокусировала разработки главным образом на военное применение, а не в гражданские сегменты экономики, что впоследствии послужило причиной низкой конкурентоспособности производимых углеродных волокон по сравнению с японскими. В России производство акриловых волокон было создано в Саратове в 1960-х годах на основе технологии “Courtaulds”. К настоящему времени несмотря на то, что все основные способы получения сополимеров АН полимеризацией в растворе или суспензии позволяют получить углеродные волокна близкого качества, абсолютным лидером по объемам и качеству производимых волокон является компания “Toray”.
Конечные свойства углеродных волокон зависят от эффективности предыдущих стадий, включая синтез (со)полимера [8–10]. Именно на стадии синтеза задается молекулярная структура цепи, его композиционная неоднородность и ММР, которые влияют на морфологию и структуру сформованного волокна и его поведение при термостабилизации [11–13]. Из-за низкой растворимости и кристалличности, которые обусловлены сильными взаимодействиями нитрильных групп макромолекул, гомополимер ПАН редко используют для получения углеродного волокна [14, 15]. Вместо этого применяют двойные и тройные сополимеры с массовым содержанием сомономеров не выше 15% [16–19]. Выбор сомономеров вызван их способностью влиять на циклизацию ПАН, приводящую к образованию лестничной структуры полимера. В общем случае циклизация инициируется различными дефектами в структуре цепи или остаточным инициатором и протекает по радикальному механизму, что сопровождается интенсивным экзотермическим эффектом [20]:
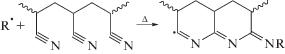
Однако в присутствии мономеров, содержащих кислотные или амидные группы, возможна реализация ионного механизма; циклизация начинается при более низкой температуре, протекает в более широком интервале температур, и ее суммарный экзо-эффект понижается [21]:
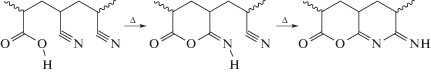
На практике используют либо двойные сополимеры с нейтральным (инертным) сомономером, например с метилакрилатом, либо тройные – с добавлением мономера, ускоряющего циклизацию [22–25]. При этом каждый год появляются десятки исследований, в которых авторы применяют новые мономеры с целью улучшения свойств ПАН-прекурсора или стремятся повысить степень стереорегулярности ПАН [26–29]. Однако предпринимаются также попытки разработать двойные сополимеры, в которых сомономер содержит одновременно сложноэфирную и кислотную или амидную группы, например метиловый эфир 3-аминокарбонил-3-бутеновой кислоты [30]:
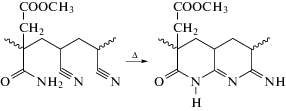
Благодаря взаимодействиям нитрильных групп вдоль цепи макромолекула ПАН имеет спиральную конформацию [31]. При циклизации образуется последовательность сопряженных циклов пиридина, и по достижении критической длины этой последовательности (по теоретическим оценкам составляет пять–семь звеньев) в цепи возникают стерические затруднения, препятствующие дальнейшей циклизации [32]. В результате в циклической лестничной структуре образуются дефекты [17, 33]. Понижение количества таких дефектов и, как следствие, повышение физико-механических свойств углеродных волокон является важной задачей. Один из способов ее решения – равномерное расположение нейтральных (не влияющих на циклизацию) и ускоряющих (инициирующих циклизацию) звеньев сомономеров вдоль цепи. Предполагают, что более высокая степень циклизации позволяет полимерам выдерживать высокотемпературную карбонизацию и обеспечивает более высокие прочностные характеристики углеродного волокна [34–36]. Первая попытка регулирования распределения сомономеров в цепи ПАН была предпринята J.S. Tsai с сотрудниками в 1991 г. [37] в классической радикальной растворной сополимеризации АН с метилакрилатом (МА), итаконовой кислотой и 2-этилгексилакрилатом, вводимыми в реактор с регулируемой скоростью. Было показано, что замедленное введение сомономеров приводит к уменьшению степени кристалличности и размеров кристаллитов, при этом нейтральные сомономеры (МА и 2-этилгексилакрилат) повышали, а кислотный сомономер (итаконовая кислота) понижали начальную температуру циклизации.
Попытки улучшить свойства ПАН-прекурсора способствовали активным фундаментальным исследованиям закономерностей гомо- и сополимеризации в растворе, суспензии, эмульсии и в осадительной полимеризации [38–43]. К настоящему моменту времени установлены основные кинетические закономерности (со)полимеризации АН в разных средах; так, в обзоре [44] обобщены данные по растворной полимеризации АН. Определены константы сополимеризации АН с большим числом сомономеров, включая виниловые кислоты, алкилакрилаты и т.д. [18, 45–47]. Данные об относительной активности мономеров в сополимеризации и состав мономерной смеси позволяют оценить среднюю длину блоков АН, которая оказывает заметное влияние на свойства прекурсора, например, при термоокислительной стабилизации [48]. С появлением современных методов контролируемой радикальной полимеризации оказалось возможным регулировать не только ММР, но и молекулярную структуру полимеров [49].
К настоящему времени накоплены многочисленные экспериментальные данные о способах получения (со)полимеров акрилонитрила, о влиянии их химического состава на способ и условия формования, а также на процессы термоокислительной стабилизации. Однако, на наш взгляд, последние достижения в области синтеза сополимеров АН с точки зрения возможности управления структурой и свойствами прекурсоров углеродных волокон и перспективы их промышленного применения требуют обобщения. Обсуждению этих вопросов и посвящен настоящий обзор.
“ЗЕЛЕНАЯ” ХИМИЯ В СИНТЕЗЕ ПОЛИМЕРОВ АКРИЛОНИТРИЛА
Особенностью полимеризации АН является нерастворимость полимера в своем мономере: уже при степени конверсии около 10% ПАН высаживается из раствора мономера [50]. Этот факт привел к интенсивному развитию и изучению как гомогенной (в растворе), так и гетерогенной (в массе, эмульсии, суспензии и осадительной) радикальной полимеризации АН. Но в последнее время усиливается внимание к экологическим проблемам окружающей среды, что приводит к поиску альтернативных сред для проведения процессов полимеризации. Наиболее широкое распространение среди так называемых “зеленых” сред получили сверхкритический CO2 и ионные жидкости. Первый является экологичной, относительно недорогой и пожаробезопасной средой [51–53]. Ионные жидкости, в свою очередь, в широком интервале температур находятся в жидком состоянии, обладают высокой удельной электропроводностью, хорошей растворяющей способностью, нелетучестью, негорючестью, невзрывоопасны, нетоксичны и возможно их многократное применение [54]. Рассмотрим их использование в синтезе ПАН и его сополимеров.
Полимеризация в сверхкритическом CO2
Критическими условиями для диоксида углерода являются температура Ткр = 31°С и давление Ркр = 7.4 МПа [55]. В сверхкритическом состоянии он обладает плотностью, характерной для жидкости, а диффузией и вязкостью, типичными для газа. Например, вязкость сверхкритического CO2 примерно на порядок ниже, чем у органических растворителей, а коэффициент самодиффузии того же порядка, что у газообразного СО2 и на один–два порядка выше, чем у молекул органических растворителей [56]. Благодаря этим свойствам сверхкритический СО2 легко растворяет мономеры и пластифицирует полимеры. При фиксированном давлении, отвечающем критическому значению Ркр, фазовое состояние системы сверхкритический СО2–мономер может различаться в зависимости от значения критической температуры последнего. В результате полимеризация может протекать в условиях сверхкритики, двухфазного или однофазного жидкого или газообразного состояния системы [56, 57].
Главное преимущество использования сверхкритического СО2 – отсутствие стадий сушки готового продукта и удаления растворителя. Основной проблемой для широкого распространения CO2 в качестве растворителя для синтеза полимеров является то, что в сверхкритическом состоянии он является плохим растворителем для многих полимеров, в том числе и для ПАН [58]. В связи с этим полимеризация АН в среде CO2 протекает гетерофазно. В основном ПАН в сверхкритическом CO2 получают дисперсионной или осадительной полимеризацией; в таких системах концентрация мономера обычно находится в диапазоне 10–15 мас. % [59–61].
Первая информация о полимеризации АН в сверхкритическом CO2 появилась в 2000 г. [59]. Авторы провели осадительную и дисперсионную полимеризацию АН; в последнем случае в качестве стабилизатора выступал полифторалкилакрилат, растворимый в сверхкритическом СО2, или его блок-сополимер со стиролом:
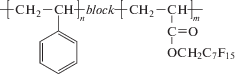
Осадительная и дисперсионная полимеризация с использованием полифторалкилакрилата не позволила получить частицы заданной морфологии. Образование сферических частиц ПАН со среднечисленным диаметром в диапазоне 0.2–0.5 мкм и узким распределением частиц по размерам (РЧР) с конверсией мономера около 70% обеспечивает только блок-сополимер. К сожалению, в работе не приводятся данные о кинетике процесса и ММР продукта реакции.
Развитием этих исследований стали работы X.-R. Teng [62, 63], посвященные осадительной полимеризации АН в сверхкритическом CO2. Авторы варьировали концентрацию мономера, инициатора, время, температуру и давление. Это позволило показать, что полимеризация АН в интервале концентраций мономера 6–14 мас. % протекает с низкой скоростью и приводит к образованию ПАН со средневязкостной ММ, равной (14–110) × 103, Mn = (5–15) × 103 и дисперсией Ð = = 3.0–3.2. Конверсия мономера не превышает 50%. ПАН-прекурсор, полученный формованием из раствора в ДМФА, имеет дефектную структуру и невысокие прочностные характеристики.
Более подробное влияние условий осадительной полимеризации АН на морфологию частиц исследовал М. Okubo с сотрудниками [64]. Авторам удалось найти условия, при которых достигается конверсия мономера, близкая к 100%, при этом образуется полимер с высокой степенью кристалличности и средневязкостной ММ около 50 × 103. Сами частицы однородные, сферической формы, микронного размера, чему способствовало интенсивное перемешивание реакционной среды.
Развитие идей осадительной и дисперсионной полимеризации АН в ск-CO2 предложено в работе [61]. Полимеризацию проводили в широком диапазоне давлений от 7.7 до 30 МПа и температуре 65°С в присутствии радикального инициатора ДАК. Для стабилизации образующихся частиц авторы использовали блок-сополимер на основе ПАН и полифторалкилакрилата, который обеспечивает образование сферических частиц субмикронного размера с узким РЧР:
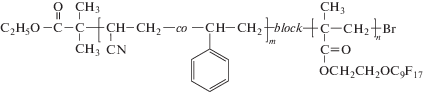
С ростом давления более высокие конверсии мономера, ММ полимера и узкое РЧР достигаются в дисперсионной, а не в осадительной полимеризации. При понижении давления, особенно вблизи Ркр (7.7–7.8 МПа), такое различие нивелируется, и в обоих случаях образуется высокомолекулярный кристаллический ПАН с конверсией мономера выше 90%, Mn = (130–190) × 103 и близким РЧР.
В радикальной полимеризации АН в сверхкритических средах следует ожидать образование атактического полимера. Это было подтверждено в работе [65], авторы которой провели сравнительное исследование микроструктуры цепи ПАН, образующегося в сверхкритическом СО2, в осадительной и суспензионной полимеризации. Анализ триадного и пентадного состава полимеров показал, что ПАН, синтезированный в сверхкритическом СО2, является атактическим и характеризуется бернуллиевским распределением триад и пентад. Содержание изотактических последовательностей в нем ниже, чем в образцах, полученных осадительной и суспензионной полимеризацией.
Сополимеризация АН в сверхкритическом СО2 описана в работах [66–68].
В работе [66] обсуждаются результаты осадительной сополимеризации АН с метилметакрилатом (ММА) и 2-хлорстиролом, инициированной ДАК при 70°С и 20 кПа в сверхкритическом СО2. Авторы уделили особое внимание составам мономерной смеси АН–сомономер, в которых содержание сомономера менее 10 мол. %. В условиях повышенного давления реакционная среда остается гомогенной в течение 3 ч полимеризации, после чего наблюдается выделение полимера в отдельную фазу. Конечный продукт полимеризации представляет собой порошок, мягкую твердую массу или гель в зависимости от природы сомономера и состава сополимера. При увеличении доли АН в смеси образуется порошкообразный продукт и повышается выход сополимера. Необычен тот факт, что при мольном содержании сомономера менее 10% среднечисленная ММ сополимера практически не зависит от состава мономерной смеси и составляет (50–60) × 103 и Ð = = 3–5, а при эквимолярном составе ММ резко возрастает, а ММР сужается (Ð = 1.5–2.0). Повышение доли сомономера приводит к уменьшению степени кристалличности сополимера и его набуханию в растворе СО2. Пластифицированный таким образом сополимер характеризуется пониженной температурой стеклования, что влияет и на термическое поведение сополимера.
Другим примером является осадительная гомо-, а также бинарная и тройная сополимеризация акрилонитрила с метилакрилатом и итаконовой кислотой или ее производными (монометил- и моноэтилитаконатом, моноамидом и моно-н-октиламидом итаконовой кислоты) в сверхкритическом СО2 при 65–80°С и 40 кПа [67]. В гомополимеризации повышение концентрации ДАК приводит к увеличению конверсии мономера и уменьшению ММ полимера, при этом ПАН характеризуется широким ММР (Ð = 3–5). Аналогичный результат получен для бинарных и тройных сополимеров. Преимуществом этого метода оказалось более эффективное использование реакционного объема (в 2–3 раза, с загрузками мономера до 50 об. %) по сравнению с растворной или традиционной гетерофазной полимеризацией. В условиях синтеза образуются мелкодисперсные частицы сферической формы, что делает очистку и последующую обработку полимера более простой. Однако в случае сополимеризации имеется существенный недостаток – образование сополимера с высокой композиционной неоднородностью. Это хорошо иллюстрируется результатами ДСК. Из приведенных авторами данных о термическом поведении бинарных и тройных сополимеров следует, что введение в цепь незначительного количества инертного мономера МА неожиданно резко понижает энергию активации циклизации по сравнению с ПАН, а добавление ускоряющего циклизацию сомономера (итаконовой кислоты или ее производных) не приводит к дополнительному уменьшению энергии активации циклизации.
Первый пример синтеза блок-сополимера ПАН с поливинилацетатом рассмотрен в работе [68] на примере ацетилацетоната кобальта (II) (Co(АcАc)2).
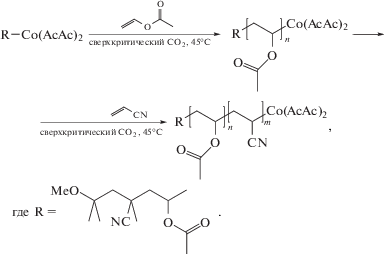
Вначале была проведена полимеризация винилацетата и доказано, что процесс протекает по “живому” механизму, т.е. ММ полимера линейно растет с конверсией, а дисперсия Ð ниже 1.1. С увеличением ММ поливинилацетата выше 103 нарушается его растворимость в реакционной среде. Полученный полимер способен выполнять функцию макроинициатора в полимеризации своего мономера и АН. В последнем случае для проведения контролируемого синтеза необходимо использовать олигомерный поливинилацетат. Это позволяет получить блок-сополимер с Mn ∼ ∼ 50 × 103 и Ð = 2.0 при конверсии АН ∼80%.
В целом (со)полимеризацию АН в среде сверхкритического CO2 можно рассматривать как перспективную технологию для получения (со)полимеров АН. Разработанные методы дают возможность в широком интервале регулировать ММ и состав сополимеров, а также получать статистические и блочные сополимеры. Легкость удаления растворителя за счет перевода диоксида углерода в газообразное состояние и высокая конверсия мономера, позволяющая избежать стадии очистки конечного продукта, а также возможность получения конечного продукта в виде тонкодисперсного порошка являются преимуществами этого метода синтеза.
Полимеризация в ионных жидкостях
Впервые применение ионных жидкостей в радикальной полимеризации описано в начале 2000-х гг. [69]. В настоящее время в ряде обзоров обобщено влияние ионных жидкостей на кинетику и механизм полимеризационных и поликонденсационных процессов [70–73].
Полимеризация АН в среде ионных жидкостей может протекать гомогенно или гетерофазно [54, 73]. При использовании в качестве ионных жидкостей солей 1,3-диалкилзамещенного имидазола
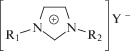 ,
,
где R1 и R2 = CnH2n+1, n = 1–4 (R1), n = 2–5 (R2); Y– = Cl–, Br–, BF$_{4}^{ - }$, PF$_{6}^{ - }$, (CF3SO2)2N–, полимеризация 20 и 50%-ных растворов АН протекает в гомогенных условиях, только в случае R1 = CH3 и R2 = C3H7, а Y– = Cl– или Br– [54]. При сравнении с полимеризацией в ДМФА скорость полимеризации и ММ полимера значительно выше, что, как полагают, связано с понижением значения энергии активации реакции роста и обрыва цепи, а также с уменьшением вероятности протекания побочных реакций. Температура стеклования ПАН, полученного в ионной жидкости и ДМФА, практически совпадает, а температура начала разложения существенно выше и зависит от ионной жидкости. Для R1 = R2 = C4H9, а Y– = PF$_{6}^{ - }$ она равна 340°С и R1 = CH3 и R2 = C3H7, а Y– = = (CF3SO2)2N– – 280°С, в то время как для ПАН, полученного в ДМФА, она составляет 220°С. Можно было бы предполагать, что повышение термостойкости полимеров связано с присутствием остаточной ионной жидкости. Однако эксперименты, проведенные в работе [73], не согласуются с этими соображениями: наличие даже небольшого количества ионной жидкости приводит к понижению температуры стеклования, т.е. она выполняет функцию пластификатора. Независимо от наличия или отсутствия ионной жидкости в продукте полимеризации термостойкость полимера, полученного в ДМФА, ниже. К сожалению, авторы не исследовали микроструктуру цепи и степень кристалличности полимера, и вероятной причиной различий могли бы быть изменения в указанных характеристиках.
Аналогичные результаты были получены для сополимеризации АН с ММА. При этом скорость сополимеризации и ММ сополимеров понижается при увеличении доли АН в мономерной смеси. Существенным моментом является различие составов сополимеров, образующихся на разных конверсиях, в ДМФА и в ионной жидкости, что указывает на разную активность мономеров при сополимеризации в данных средах.
Исследование гомополимеризации АН в растворе тетрафторбората 1-бутил-3-метилимидазолия R1 = CH3 и R2 = C4H9, а Y– = BF$_{4}^{ - }$ привело к аналогичным результатам [73]. ПАН, синтезированный в таких условиях, характеризуется Mn = = (30–50) × 103 и широким ММР (Ð > 3). Таким образом, более высокие скорость и ММ при полимеризации АН – общее явление, не зависящее от строения соли имидазолия.
Наиболее заметное развитие этого направления связано с контролируемой радикальной полимеризацией с переносом атома [74–78], что обусловлено использованием соединений переходных металлов и способностью ионной жидкости выполнять функцию лиганда и стабилизировать комплекс катализатора. Подробно этот вопрос будет обсуждаться ниже.
КОНТРОЛИРУЕМЫЙ СИНТЕЗ ПОЛИМЕРОВ АКРИЛОНИТРИЛА
Наряду с разработкой новых технологий синтеза (со)полимеров АН, основанных на известных механизмах полимеризации и методов ее проведения, в последние десятилетия усилия большого числа групп исследователей направлены на поиск способов управления молекулярными характеристиками ПАН. Сюда следует отнести работы в области анионной полимеризации и радикальной полимеризации с обратимой деактивацией цепи.
Анионная полимеризация
Анионная полимеризация интересна исследователям возможностью осуществления ее в режиме “живых” цепей, т.е. в условиях отсутствия реакций обрыва и передачи цепи. Этот вариант стал известен благодаря М. Szwarc в середине 1950-х гг. [79]. В настоящее время “живая” анионная полимеризация позволяет получать полимеры сложной архитектуры с заданной ММ, узким ММР, высокой стереорегулярностью и композиционной однородностью [80].
Первые сведения об анионной полимеризации АН появились в 1950–1960-е гг., когда было показано, что инициаторами для АН могут служить щелочные металлы, их амиды, алкоголяты, гидроокиси, металлоорганические соединения, четвертичные аммониевые основания и металлы в жидком аммиаке [81–89]. В нашей стране активные исследования гомо- и сополимеризации АН по анионному механизму развивались в 1960–1970-е гг. [90–98]. В этих работах было впервые обнаружено, что в ходе анионной полимеризации при температурах выше 0°С часто образуется окрашенный продукт, что возможно при образовании как сопряженных связей –C=N–, так и колец нафтилидинового типа:
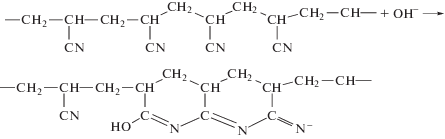
Были установлены основные кинетические параметры анионной полимеризации с участием инициаторов разного типа, найдены условия подавления побочных реакций обрыва и передачи цепи, описана твердофазная полимеризация АН, получен высокомолекулярный ПАН с М > 2 × 105, показано, что в анионной сополимеризации АН является более активным мономером, чем виниловые мономеры типа ММА и т.д.
Внедрение промышленных технологий синтеза ПАН и сополимеров АН, основанных на радикальных процессах, понизило интерес к анионной полимеризации АН, однако эти исследования продолжаются, хотя и с меньшей интенсивностью, чем изучение радикальных процессов.
Одной из задач, которую долгое время пытались решить в анионной полимеризации АН, – получение стереорегулярного ПАН. Такие попытки предпринимались неоднократно, но все они заканчивались синтезом атактического и зачастую не линейного, а разветвленного полимера [99–103]. Предполагалось, что причиной является кетениминная структура растущего аниона [104]. Удачным решением можно считать полимеризацию АН под действием диалкилов магния в неполярном растворителе (толуоле или ксилоле) при температуре выше 100°С [105]. На примере нескольких десятков инициаторов авторы провели систематическое исследование природы инициатора, температуры синтеза, природы и полярности растворителя и показали, что только диалкилы магния способны повысить стереорегулярность (изотактичность) ПАН; при этом понижение температуры синтеза приводило к увеличению доли гетеротриад, т.е. к потере регулярности структуры цепи. Развитием данной работы можно считать сообщение о синтезе высокоизотактического ПАН осадительной полимеризацией в ксилоле под действием дибутилмагния [106]. Недавно стереорегулярные сополимеры АН, акриламида и итаконовой кислоты были синтезированы в условиях твердофазной матричной полимеризации с участием безводных солей хлорида никеля или хлорида магния [107].
Продолжается поиск новых инициаторов. В указанных работах решаются разные задачи: синтез высокомолекулярного ПАН и/или поиск неметаллических инициаторов полимеризации.
Так, в работе [108] описан синтез ПАН с Mw ∼ ∼ 106 и Ð = 1.9–2.2 полимеризацией АН в ДМФА присутствии амидов лития, полученных взаимодействием бутиллития с аминами (диизопропиламином, диэтиламином, гексаметилдисилазаном, дициклогексиламином и 2,2,6,6-тетраметилпиперидином)
Авторы нашли условия, в которых побочные реакции изомеризации активного центра практически подавлены. Эти же авторы развили данную работу и осуществили анионную полимеризацию АН с такими же инициаторами в проточном микрореакторе, что позволило им получить полимер с более узким ММР (Ð ∼ 1.5), хотя и меньшей ММ (Mw = (400–800) × 103) [109].
Однокомпонентный биметаллический инициатор Ln2(OCH2CH2-NMe2)12(OH)2Na8 [Ln = Yb, Nd и Sm] был предложен в работе [110]. Он оказался эффективным в широком интервале температур от −78 до 50°C; полимеризация заканчивалась в течение ∼10 мин и приводила к образованию атактического ПАН со средневязкостной ММ, равной (20–40) × 103. Скорость полимеризации и выход полимера возрастали при увеличении полярности растворителя: ДМФА > толуол ∼ ∼ ТГФ > гексан.
Для инициирования анионной полимеризации кроме стандартных подходов использовали, например, электрохимическое генерирование анион-радикалов с помощью бензофенона и 2,2′-бипиридила [111]. Авторы доказали анионный механизм реакции и сосредоточили усилия на изучении реакции роста цепи, определили ее константу скорости и влияние на ее значение типа и концентрации поддерживающего электролита ((н-C4H9)4NClO4 или NaClO4) и инициатора.
Первые работы с использованием неметаллических инициаторов появились в начале 1990-х гг. [112]. Третичные амины, как оказалось, не способны инициировать полимеризацию АН, однако такая возможность у них появляется в комплексе с эпоксидом:
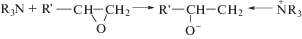
Это было продемонстрировано на примере 1,4-диазабицикло[2.2.2]октана и разных циклических окисей. Причем, если в качестве эпоксида использовать глицидилметакрилат или аллилглицидиловый эфир, то это позволяет получить макромономер, способный к полимеризации. Существенно, что полимеризация протекает в мягких условиях: в ДМСО при 40°С в течение 2–4 ч достигается конверсия мономера, близкая к 100%. К сожалению, авторы выбирали условия, в которых ММ образующегося полимера не превышает 104, а ММР оказывалось достаточно широким. Широкое ММР могло быть как следствием медленного инициирования, так и образованием разветвленного полимера.
Данное направление недавно получило развитие в работах российских исследователей из ИПФХ РАН [113, 114]. Авторам удалось существенно улучшить этот процесс. Было показано, что продукты взаимодействия этиленоксида и бициклических аминов, содержащих третичные атомы азота в вершине бициклической структуры
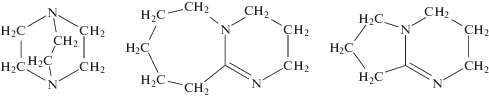
являются эффективными инициаторами анионной полимеризации акрилонитрила. Полимеризация акрилонитрила под действием системы этиленоксид–бициклический амин в полярном растворителе ДМСО при комнатной температуре протекает гомогенно в течение нескольких минут, а в слабо полярном растворителе ТГФ – гетерогенно с меньшей скоростью, в течение нескольких часов. Полимеризация успешно идет и в смеси этих растворителей в широком интервале температур (от комнатной до отрицательных значений). Разработанные инициирующие системы позволили существенно повысить ММ полимера (Mn = (25–480) × 103) при сохранении типичных значений дисперсии (Ð = 1.55–3.40), обусловленных реакциями внутри- и межмолекулярной передачи цепи. Синтез сополимеров АН с этиленоксидом, МА, этилакрилатом и диметилитаконатом был менее эффективным: выход и ММ оказались ниже, чем у гомополимера. Следует отметить, что изучение этих инициирующих систем находится еще на начальной стадии, поскольку отсутствуют данные о кинетических закономерностях полимеризации, топологии макромолекул и их стереорегулярности.
Другой пример неметаллического инициатора – тетраалкиламмониевые соли, например, тетрабутиламмоний карбазолид [115]. Однако они приводят к образованию окрашенного полимера, т.е. ПАН, содержащего в структуре сопряженные связи. В работе [116] описано применение дииодида самария для полимеризации АН, что позволило получить ПАН с Mw ∼ 35 × 103. Предполагаемый механизм полимеризации включает образование анион-радикала АН; он рекомбинирует и образует дианион, на котором дальше происходит рост цепи.
Позже в качестве инициаторов были предложены соединения трехвалентного фосфора, которые являются мягкими основаниями и способны присоединяться к С=С-связи АН без отщепления протона мономера. В отличие от третичных аминов фосфины способны инициировать анионную полимеризацию АН. Так, сообщается об использовании трифенилфосфина в полимеризации АН, однако ММ полимера была достаточно низкой [117]. Авторами был предложен цвиттер-ионный механизм полимеризации и образование активного центра на мономере, способного инициировать дальнейшую полимеризацию.
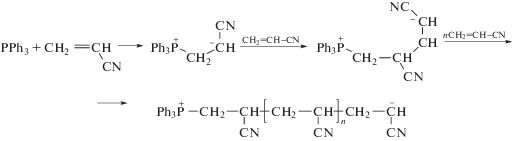
Эти идеи были развиты в работах [118–121]. Так, было показано, что высокомолекулярный ПАН образуется в присутствии триэтилфосфита при комнатной температуре в растворе ДМФА. Полученный прядильный раствор с концентрацией ПАН 30% пригоден для формования волокна. Тетраэтиламмоний бромид ускоряет полимеризацию, вероятно, за счет быстрого обмена катионами и замены фосфорсодержащего катиона на аммонийный.
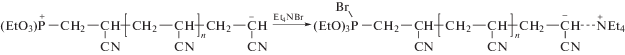
Также хорошо зарекомендовали себя в полимеризации инициаторы на основе трициклогексилфосфина.
Таким образом, неметаллические инициаторы анионной полимеризации представляют потенциальный интерес для практического применения. Достигнуты успехи в области синтеза относительно высокомолекулярного не только гомополимера АН, но и его сополимеров. Существенным ограничением этого направления является чувствительность к функциональным группам сомономеров, которую вряд ли удастся преодолеть, не прибегая к усложняющим приемам создания защиты требуемой группы.
Радикальная полимеризация с обратимой деактивацией цепи
Радикальная полимеризация может быть “живой”, т.е. протекать в безобрывном режиме, только в исключительных случаях [122]. Однако существуют возможности придания радикальной полимеризации черт “живой” анионной полимеризации. Основная идея такой радикальной полимеризации, которую принято называть радикальной полимеризацией с обратимой деактивацией цепи11, заключается в том, что в полимеризационную систему вводят специальные соединения, которые способны обратимо взаимодействовать с радикалами роста [123]. В результате макрорадикал становится неактивным (“спящим”), но в условиях реакции он активируется (“оживает”) и продолжает расти пока опять не прореагирует с введенной добавкой. Таким образом, макромолекула растет ступенчато, и чем чаще она “оживает” и “засыпает”, тем ближе закономерности радикальной полимеризации к “живой” анионной. В зависимости от способа активации макромолекул различают обратимое ингибирование, обратимый перенос атома и обратимую передачу цепи по механизму присоединения–фрагментации (ОПЦ) [124]. Основные черты этих процессов и их возможности обсуждаются в ряде обзоров [125–128]. Ниже мы рассмотрим их применение в контролируемом синтезе ПАН и сополимеров АН.
Полимеризация в присутствии стабильных и малоактивных радикалов. Обратимое ингибирование основано на обратимой реакции обрыва макрорадикалов ∼${\text{P}}_{n}^{ \bullet }$ с низкомолекулярными малоактивными или стабильными радикалами ${\text{X}}_{{}}^{ \bullet }$ – стабильными радикалами, инифертерами или кобальторганическими соединениями [129]:
Обратимые ингибиторы [130] и инифертеры [131] мало применяются в полимеризации АН по сравнению с другими вариантами контролируемой радикальной полимеризации, поскольку диссоциация связи Pn–X, необходимая для активации макрорадикалов, для большинства известных стабильных и малоактивных радикалов протекает с трудом. Исключением является реакция сополимеризации с более активным мономером, который с высокой вероятностью находится на конце радикала роста и образует лабильную связь с радикалом ${\text{X}}_{{}}^{ \bullet }$.
Тем не менее, за последние 10–20 лет есть отдельные достижения в этой области. Например, в работе [132] описано применение полиуретанового макроинифертера на основе тетрафенилэтана для полимеризации АН в ДМФА в интервале температур 60–80°С.
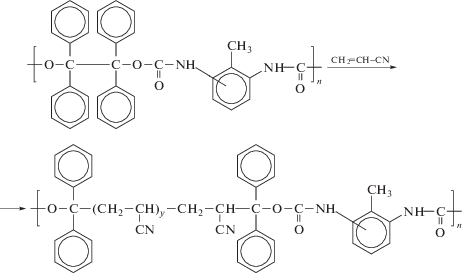
Скорость полимеризации характеризуется первым порядком по концентрации макроинифертера и полуторным по концентрации мономера. При учете того, что для инифертеров типично значение порядка скорости от 0 до 0.5 [133], это значение указывает на более сложный механизм процесса, чем предложенная авторами обратимая диссоциация связи АН–C(Ph)2. Процесс характеризуется чертами “живого”: авторы наблюдали линейный рост Mn полимера при увеличении конверсии мономера и относительно неширокое ММР конечного продукта полимеризации (Ð = = 1.6). При повторном использовании синтезированного ПАН в качестве макроинифертера его эффективность оказалась низкой: он медленно расходовался на протяжении полимеризации, в результате даже на глубоких конверсиях ММР полимера было бимодальным.
В отличие от инифертеров стабильные нитроксильные радикалы и нитроны более эффективны в контролируемом синтезе полимеров [134, 135]. Среди нитроксильных радикалов наиболее часто используют 2,2,6,6-тетраметил-1-пипeридиноксил (ТЕМПО) и его производные. Однако заметных успехов в гомополимеризации АН с участием нитроксильных радикалов достичь не удалось. Например, под действием ТЕМПО даже при высоких температурах (выше 120°С) образуются только короткие олигомеры, полимеризация затухает во времени, и предельная конверсия не превышает 15–20% [136]. Замена ТЕМПО на алкоксиамин, функционализированный сукцинимидной сложноэфирной группой, не позволяет повысить ММ (Mn < 14 × 103 и Ð = 1.14–1.26) и выход полимера [137].
Более интересные результаты получены в сополимеризации АН с виниловыми мономерами.
В работе [138] описана сополимеризация стирола и АН под действием ТЕМПО. Азеотропная сополимеризация (25 мас. % АН) протекает по “живому” механизму до глубоких конверсий в узком интервале температур 110–130°С и характеризуется линейным ростом Mn с увеличением конверсии мономера и не широким ММР (Ð ∼ ∼ 1.5–1.6). Такой результат объясняется высоким содержанием в смеси стирола и его более высокой реакционной способностью в сополимеризации (rАН = 0.08, rстирол = 0.43). Следствием этого на конце растущей цепи в широком интервале конверсий содержится стирол, который и обеспечивает реализацию механизма обратимого ингибирования. Результаты работы подтверждаются и независимыми квантово-химическими расчетами энергии диссоциации в алкоксиаминных аддуктах со стиролом и АН с учетом концевой и предконцевой модели, а также полярного, стерического и резонансного эффектов [139].
Развитие химии нитроксильных радикалов и появление новых радикалов и их аддуктов дало возможность повысить содержание АН в сополимере. Так, в работе [140] описано применение N-трет-бутил-α-изопропилнитрона в синтезе статистических и блок-сополимеров стирола и АН. В этом случае сополимер с содержанием ∼40 мол. % АН, Mn = = 35.5 × 103 и Ð = 1.3 удалось получить, воспользовавшись необычным приемом.
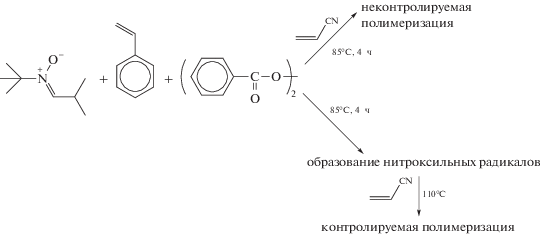
Авторы сначала при более низкой температуре генерировали образование нитроксильных радикалов в присутствии стирола и перекиси бензоила, после накопления нитроксилов в реакционной среде добавили АН и повысили температуру. Этот прием позволил осуществить контроль ММ и ММР полимера. Более того, полученный сополимер выполняет функцию макроинициатора и способен обеспечивать образование блок-сополимера со стиролом.
Более прочная связь в аддукте ПАН–нитроксил позволила найти интересное применение малым добавкам АН в полимеризации ММА под действием нитроксильных радикалов [141]. Основной проблемой в реализации “живого” механизма полимеризации метакрилатов с участием нитроксильных радикалов является реакция диспропорционирования между радикалом роста и нитроксилом. Однако введение в реакционную смесь, содержащую ММА, нитроксила SG-1 (N-трет-бутил-N-[1-диэтилфосфоно-(2,2-диметилпропил)]нитроксид) и его аддукта с метакриловой кислотой (BlocBuilder), даже небольшого количества АН позволила практически полностью подавить эту реакцию.
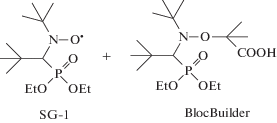
Причина этого заключается в уменьшении на несколько порядков константы диссоциации аддукта при переходе от ПММА–нитроксил к ПАН–нитроксил.
В дальнейшем возможности управления “живым” режимом полимеризации были развиты в работах М.Ю. Заремского на примере тройной сополимеризации АН, стирола и ММА и двух нитроксилов ТЕМПО и SG-1 [142, 143]. Было показано, что для сомономеров разной активности константа равновесия, показывающая возможность реализации “живого” механизма, определяется константой наиболее активного в полимеризации мономера. В тройке выбранных мономеров активность изменяется в ряду стирол > ММА > > АН. Так, в азеотропной терполимеризации (52 мол. % стирола, 18 мол. % ММА и 30 мол. % АН) механизм обратимого ингибирования осуществляется благодаря стиролу. При этом SG-1 обеспечивает лучший контроль ММР, чем ТЕМПО вследствие более высокого значения константы равновесия. В развитие этой концепции с участием тех же нитроксилов был осуществлен синтез градиентных сополимеров из мономерной смеси 30 мол. % стирола, 10 мол. % ММА и 60 мол. % АН. С помощью аддукта полистирола и SG-1 (Mn = 5.2 × 103) авторам удалось синтезировать градиентные сополимеры с Mn до 40 × 103 и Ð = 1.3.
В целом следует признать, что метод обратимого ингибирования мало пригоден для синтеза сополимеров АН для получения прекурсоров УВ. Он требует применения высоких температур, характеризуется значительной длительностью процесса и низким выходом полимера, а также не позволяет получить сополимеры с содержанием АН более 80 мол.% и высокой ММ.
Полимеризация с переносом атома. Данный процесс основан на обратимом взаимодействии металлоорганических соединений, главным образом галогенидов переходных элементов с алкилгалогенидами, в результате которого генерируется радикал, способный реагировать с мономером и обеспечивать рост цепи [144].
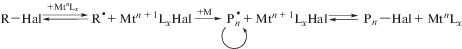 ,
,
где Мt – переходный металл валентности n, Hal – галоген, L – органический лиганд, R – алкил или арил, M – мономер, P$_{n}^{ \bullet }$ – макрорадикал.
Эволюция подходов к проведению полимеризации с переносом атома: прямая и обратная полимеризация с переносом атома, полимеризация с инициатором для постоянной регенерации активатора (ICAR ATRP) [145], полимеризация с активатором, генерируемым переносом электрона (AGET ATRP) [146], “живая” полимеризация с переносом одного электрона (SET-LRP) [147], полимеризация, активируемая электрохимически (e-ATRP) [148], фотоиндуцированная полимеризация без использования металлоорганических соединений (metal-free ATRP) [149] и фотоиндуцированная полимеризация с переносом электрона без использования металлоорганических соединений (PET-ATRP) [150], сопровождается не только поиском условий для контролируемого синтеза полимеров, но и расширением круга мономеров, катализаторов и реакционных сред.
Основные успехи в области синтеза гомо- и сополимеров АН полимеризацией с переносом атома обобщены в двух обзорах [151, 152]. К сожалению, структура и свойства (со)полимеров в обзорах не обсуждаются, что, вероятно, связано с отсутствием соответствующей информации в публикациях. Авторы обзоров уделили основное внимание влиянию природы катализатора, инициатора, лиганда и активатора, а также растворителя на скорость полимеризации и молекулярно-массовые характеристики полученных полимеров.
Следует отметить, что первые попытки использовать этот метод для получения ПАН предпринимались в конце 1990-х гг. [153, 154]. Вначале успехи ограничивались в основном синтезом полимеров с невысокой ММ (менее 104) и узким ММР, однако в начале 2000-х были найдены условия синтеза более высокомолекулярного ПАН [155–160]. К настоящему моменту времени предложен широкий набор катализаторов на основе главным образом солей меди и железа, и подходящие компоненты (инициаторы, лиганды, восстановители и т.д.) для синтеза ПАН и сополимеров АН широкого диапазона ММ с узким ММР, обобщенные в указанных выше обзорах [151, 152].
В настоящее время основные направления в синтезе ПАН полимеризацией с переносом атома сводятся к поиску условий для синтеза высокомолекулярного ПАН с узким ММР, макромолекулярному дизайну и созданию новых архитектур, катализаторов, не содержащих атомов металла и новых реакционных сред.
Замена соединений металлов фотоиндуцируемой полимеризацией с переносом атома (metal-free ATRP) – достаточно новое направление [161, 162]. Так, в полимеризации АН описано использование анилина и фенотиазинов, например, 10-фенилфенотиазина (1), 10-(4-метоксифенил)фенотиазина (2) или 10-(1-нафталенил)фенотиазина (3).
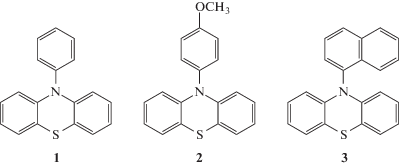
Основная идея этого механизма заключается в фотоактивации фенотиазина и при его последующем взаимодействии с алкилгалогенидом образуется алкильный радикал и фенотиазинильный катион-радикал.
Однако в случае АН данный метод пока приводит к образованию олигомеров, хотя и с узким ММР. Стоит отметить, что уже разработанные, стандартные подходы, такие как обратная полимеризация с переносом атома позволяют получать ПАН с более высокой ММ, например, с Mn ∼ ∼ (20–30) × 103 и Ð = 1.2 [163].
В области синтеза сверхвысокомолекулярного ПАН следует упомянуть работу [164], в которой с использованием ICAR ATRP удалось получить ПАН со средневязкостной массой выше 106.
Представляет практический интерес работа, в которой получают звездообразные полимеры на основе ПАН или сополимеров АН и МА с малым содержанием акрилата [165]. Для синтеза ПАН звездообразной архитектуры использовали прямой метод полимеризации с переносом атома и полифункциональные инициаторы. Авторы успешно синтезировали набор гомо- и сополимеров с разным числом лучей. Оказалось, что звездообразный ПАН (Mn = (10–70) × 103) не проявил необычных свойств в растворе ДМСО, характерных для полимеров нелинейной структуры: вязкость его раствора оказалась лишь немного меньше, чем вязкость раствора линейного ПАН. В отличие от него добавка небольшого количества метилакрилата существенно повысила гибкость полимера и понизила вязкость. Для звездообразного сополимера в отличие от линейного аналога более четко регистрировалась температура стеклования. Такие сополимеры могут быть интересны как добавки к линейным сополимерам, влияющие на реологию растворов ПАН.
Иллюстрацией новых возможностей метода в контролируемом синтезе ПАН является синтез блок-сополимерных частиц ПЭО–блок–ПАН дисперсионной полимеризацией, вызывающей самоорганизацию, в условиях ICAR ATRP [166]. Эта работа представляет исключительно академический интерес, однако она расширяет представления о способах реализации полимеризации с переносом атома.
Гораздо более перспективным направлением в полимеризации с переносом атома является применение ионных жидкостей [167, 168]. Его развитие связано с поиском новых “зеленых” растворителей и упрощением реакционной системы. Применение ионных жидкостей, обладающих высокой растворяющей способностью, в том числе неорганических соединений, в полимеризации с переносом атома позволило сократить число необходимых компонентов каталитической системы и облегчить стадию отделения соединений металла от продукта реакции. Другим достоинством указанных систем является возможность регенерации ионной жидкости и катализатора и их повторного использования.
Первые сведения об использовании ионных жидкостей в полимеризации АН с переносом атома появились в 2008 г. [169, 170].
Авторы применили как обратную, так и прямую реакцию. В первом случае использовали 1-бутил-3-метилимидазолия тетрафторборат, ДАК, FeCl3 и янтарную кислоту. Скорость полимеризации в ионной жидкости оказалась выше, чем в массе. Во втором случае применили ацетат, валерат и капроат 1-метилимидазолия, FeBr2 в качестве катализатора и 2-бромизобутират как инициатор. При этом скорость реакции в ионной жидкости и в растворе ДМФА совпадала. Однако, несмотря на хороший контроль ММ, в обоих случаях образовывался только олигомерный ПАН (Mn < 104, Ð < 1.2). Повысить его ММ удалось проведением пост-полимеризации, т.е. введением новой порции мономера по окончании полимеризации (Mn = (30–40) × 103, Ð = 1.2–1.3).
Один из вариантов использования ионных жидкостей как реакционной среды – микроэмульсия на основе ионной жидкости; для ее приготовления используют ионные ПАВ, например цетилтриметиламмоний бромид. Обратную полимеризацию с переносом атома для мономерной пары АН–стирол реализовали в микроэмульсии ионной жидкости 1-бутил-3-метилимидазолия гексафторфосфата с участием комплекса шестиводного гидрата FeCl3 и янтарной кислоты, а также перекиси бензоила в качестве инициатора [171]. Авторы доказали реализацию “живого” механизма в этих условиях и возможность получения сополимера с Mn ∼ 36 × 103, Ð = 1.3 постполимеризацией макроинициатора, образующегося в микроэмульсионной полимеризации. Наиболее любопытным результатом этой работы является изменение активности мономеров в сополимеризации при изменении состава мономерной смеси: с ростом содержания АН в смеси его активность понижается. Аналогичный результат изменения активности сомономеров наблюдали и в классической радикальной сополимеризации, проводимой в микроэмульсии ионной жидкости [172].
В другом варианте полимеризации с переносом атома (AGET ATRP) в дополнение к каталитическому комплексу на основе FeBr3, 2-бромизобутирата и аскорбиновой кислоты использовали ионные жидкости: ацетат, пропионат и бутират 1-метилимидазолия [173]. Авторам удалось осуществить “живую” полимеризацию АН в отсутствие и присутствии кислорода и получить ПАН с Mn > 20 × 103 и Ð < 1.2. Повторное применение ПАН в синтезе позволило получить продукт уже с Mn ∼ 70 × 103 и Ð ∼ 1.3.
При проведении SET-LRP брали Fe(0) и аналогичные инициатор и ионные жидкости: 2-бромизобутират и ацетат, пропионат и валерат 1-метилимидазолия [174] и получили результаты, близкие к описанным выше.
В целом сочетание преимуществ ионных жидкостей по сравнению с обычными органическими растворителями и водно-солевыми средами и достоинств полимеризации с переносом атома позволяет прогнозировать дальнейший рост интереса к синтезу сополимеров на основе АН данным методом для получения ПАН-прекурсоров.
Суммируя сказанное, можно сделать вывод о том, что полимеризация с переносом атома хотя и эффективнее процессов обратимого ингибирования с точки зрения практической реализации для синтеза (со)полимеров АН заданной ММ с узким ММР, но она также имеет ряд ограничений для синтеза ПАН-прекурсора. Во-первых, как и в анионной полимеризации, необходима высокая чистота реагентов, что связано с высокой чувствительностью каталитических систем к примесям и чувствительностью самих мономеров к катализаторам. Во-вторых, указанный метод не применим напрямую для синтеза сополимеров, содержащих кислотные и амидные группы. Кроме того, необходима очистка полимеров от металлсодержащих катализаторов, пагубно влияющих на дальнейшие стадии производства УВ. Однако применение ионных жидкостей и/или переход к неметаллическим катализаторам способны в будущем решить эти проблемы.
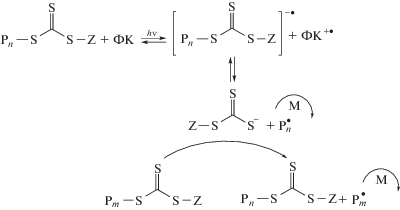
Полимеризация с обратимой передачей цепи по механизму присоединения–фрагментации. Данный процесс основан на использовании серосодержащих соединений общей формулы R–S–C(=S)–Z. Наряду с обычными для радикальной полимеризации элементарными реакциями инициирования, роста и обрыва цепи он включает реакции обратимой передачи цепи, в результате которых большинство образующихся макромолекул содержат в своей структуре концевые R и –S–C(=S)–Z группы от исходного ОПЦ-агента [175].
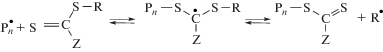
${{{\text{R}}}^{ \bullet }} + {\text{M}} \to {\text{P}}_{m}^{ \bullet }$
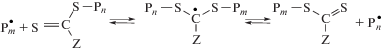
В отличие от обсуждавшихся выше процессов обратимого ингибирования и полимеризации с переносом атома ОПЦ-полимеризация толерантна к функциональным группам мономеров и растворителей и может осуществляться в широком интервале температур под действием вещественного инициирования, УФ- или γ-облучения [176, 177]. На сегодняшний день это единственный вариант контролируемой полимеризации, в котором удалось пройти всю цепочку – от синтеза (со)полимеров заданной ММ с узким ММР до получения прекурсора и УВ [178].
Впервые метод ОПЦ-полимеризации для синтеза ПАН был описан в 2003 г. [179]. Полимеризацию проводили в растворе этиленкарбоната при 60°С, используя 2-цианоэтилдитиобензоат и кумилдитиобензоат в качестве ОПЦ-агентов. Дитиобензоаты в обычных условиях вызывают замедление полимеризации многих виниловых мономеров [180], в результате авторам удалось получить лишь олигомерный ПАН с Mn ∼ 5 × 103 и Ð < 1.3.
Попытки синтезировать ПАН более высокой ММ методом ОПЦ предпринимались неоднократно [181–185], однако долгое время не удавалось превысить порог (20–30) × 103 при сохранении узкого ММР, кроме того, максимальное значение конверсии не превышало 50–60%. Впервые синтез ПАН с Mn > 105 и Ð < 1.5 был описан в работе [186]. Для этого потребовалось существенно понизить концентрацию ОПЦ-агента (фениловый эфир 4-[2-(карбазол-9-карбодитиоат)-2-метилпропионовой кислоты])
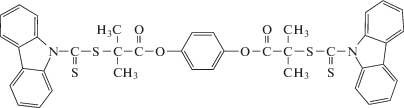
и, как следствие, инициатора ([АН] : [ОПЦ] : [ДАК] = = от 2500 : 2.5 : 0.5 до 30 000 : 2.5 : 0.5). Полимеризацию проводили в растворе ДМСО при АН : ДМСО = 1 : 2 (по объему). Удивительно, что при такой низкой концентрации инициатора ([АН] : [ДАК] = 30 000 : 0.5) за 48 ч конверсия АН достигла 60%, средневязкостная ММ составила 400 × 103, а дисперсия Ð = 1.3. Дополнительным введением кислоты Льюиса AlCl3 (0.01 мольный эквивалент по отношению к мономеру) удалось повысить долю изотактических триад ПАН с 25 до 34%. Электроформованием из раствора ПАН в ДМСО были получены нановолокна с диаметром в диапазоне 250–1100 нм в зависимости от молекулярной массы ПАН.
Альтернативный способ синтеза высокомолекулярного ПАН основан на фотоиндуцированной ОПЦ-полимеризации [187, 188]. Процесс базируется на способности серосодержащего соединения R–S–C(=S)–Z подвергаться гомолизу по связи C–S с образованием инициирующего радикала R• и менее активного радикала •S–C(=S)–Z, который обратимо взаимодействует с активными радикалами в системе. По сути, это механизм инифертерной полимеризации, предложенный Т. Otsu в 1981 г., и он аналогичен фотоактивированной полимеризации виниловых мономеров под действием бензилдитиокарбамата или тетраметилтиурамдисульфида [189, 190]. Очевидно, что в данном случае нет необходимости использовать радикальный инициатор, и полимеризацию можно вести в широком интервале температур, поскольку скорость распада аддукта определяется только мощностью облучения. Интерес к этому направлению, вероятно, связан с тем, что найдены подходящие соединения, способные к обратимому гомолизу в мягких условиях. Авторы с использованием обычного монофункционального тритиокарбоната – 2-циано-2-пропилдодецилтритиокарбоната под действием облучения с длиной волны 460–470 нм при комнатной температуре и [АН] : [ОПЦ] = 8000 синтезировали ПАН в этиленкарбонате с Mn = 220 × 103 и Ð = 1.2 [187]. Скорость процесса оказалась приемлемой: конверсия 60% достигнута за 10 ч полимеризации.
В другом варианте этого процесса [188] авторы использовали дитиобензоат 4-цианопентановой кислоты, который участвовал в окислительно-восстановительной реакции в присутствии TiO2 и образовывал радикалы, способные инициировать фотоиндуцированную ОПЦ-полимеризацию, условный механизм которой приведен на схеме. Данный процесс протекал в миниэмульсии и приводил к образованию олигомера с относительно узким ММР. К сожалению, авторы использовали существенно более низкое (в 40 раз) соотношение концентраций мономера и ОПЦ-агента, что не позволило до конца раскрыть потенциал этого процесса:
(ФК – фотокатализатор).
При наличии в системе обычного фотоинициатора вместо инифертерного реализуется обычный ОПЦ-механизм полимеризации. Такой вариант был использован в работе [189] на примере 1,2,3,5-тетракис-(карбазол-9-ил)-4,6-дицианобензола в качестве фотоинициатора и 2-цианопроп-2-ил-1-дитионафталата как ОПЦ-агента. Полимеризацией АН в ДМСО при [АН]:[ОПЦ]:[фотоинициатор] = 1500:1:0.075 и длине волны 458 нм за 4 ч были достигнуты конверсия ПАН 60% и Mn = 75 × 103.
Радиационная полимеризация с участием того же ОПЦ-агента в этиленкарбонате при комнатной температуре дает близкие результаты [190]. Однако в данном случае необходимо регулировать мощность дозы, чтобы предотвратить случайную деструкцию полимерной цепи. Авторы учитывали этот фактор, следствием чего явились более низкая скорость полимеризации и меньшая конверсия мономера, и как следствие, ММ была ниже.
Описанные выше результаты позволили развить новую стратегию синтеза полимеров – макроинициаторов с фоточувствительной концевой группой [191]. Авторы синтезировали ОПЦ-агент, S-бензил-O-(2-оксо-1,2-дифенилэтил)карбонодитиоат, который является эффективным ОПЦ-агентом и при 70°С в растворе ДМФА обеспечивает получение ПАН с узким ММР. Однако при облучении при 350 нм концевая группа в ПАН диссоциирует, и полимерный радикал в присутствии гексафторфосфата 1-этокси-2-метилпиридиния превращается в макрокатион, способный инициировать полимеризацию, например, бутил-винилового эфира:
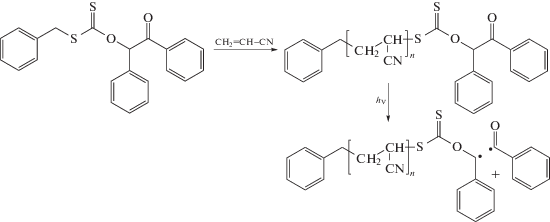
Такой подход позволяет получать функциональные полимеры, совмещая радикальную ОПЦ-полимеризацию с катионной.
Еще один интересный способ проведения ОПЦ-полимеризации описан в работе [192]. Авторы применили ОПЦ-механизм к полимеризации в водно-солевых средах (NaSCN и ZnCl2), используемых в промышленном синтезе ПАН. В присутствии дитиобензоата 4-цианопентановой кислоты, относительно стойкого в щелочных средах и ДАК, при [АН] : [ОПЦ] : [инициатор] = = 2000 : 1 : 0.4 авторам удалось получить ПАН с Mn = 60 × 103 и Ð = 1.4, однако в растворе хлорида цинка ОПЦ-механизм нарушался, вероятно, из-за комплексообразования кислоты Льюиса с растущим макрорадикалом [193].
Таким образом, были разработаны методы синтеза гомополимера АН с широким диапазоном ММ и функциональности концевых групп с достаточно узким ММР. Однако с точки зрения использования ПАН в качестве прекурсора углеродных волокон более важным является контролируемый синтез его сополимеров с невысоким содержанием сомономеров в цепи.
Первые успехи в синтезе высокомолекулярных сополимеров АН связаны с работами, посвященными получению сополимеров АН и бутадиена методом ОПЦ, в которых доля бутадиена была выше 50 мол. % [194–198]. В работе [194] бутадиен-нитрильный каучук получали растворной полимеризацией АН и 1,3-бутадиена в ДМАА при 100°С под действием моно- и бифункционального тритиокарбонатов и дитиоацетата:
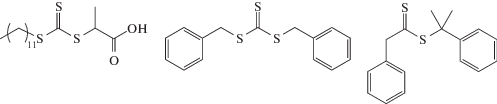
Это было первое достижение ОПЦ-процесса, поскольку ранее такие сополимеры получали только эмульсионной сополимеризацией. Авторы использовали эквимольное соотношение инициатора и ОПЦ-агента или двукратный избыток первого, и в таких условиях им удалось получить растворимый продукт с Mn до 60 × 103. Дисперсия сополимера увеличивалась в ходе полимеризации от 1.2 до 2.0. Более подробно данный процесс был исследован в работе [195] для азеотропной сополимеризации с участием монофункционального тритиокарбоната, изображенного выше, трех азоинициаторов и для широкого круга растворителей: диметилацетамида, хлорбензола, 1,4-диоксана, трет-бутанола, изобутиронитрила, толуола, триметилацетонитрила, диметилкарбоната, ацетонитрила, метилацетата, ацетона и трет-бутилметилового эфира с целью оптимизации условий проведения полимеризации при сохранении ее “живого” механизма. Природа растворителя, как оказалось, влияет только на скорость распада инициатора, что в свою очередь определяет общую скорость полимеризации. Однако при попытке повысить конверсию мономеров контроль ММР ухудшается и наоборот, синтез сополимера с высокой ММ и узким ММР происходит в условиях низкой скорости полимеризации. Решение данной проблемы было найдено [196], но сомнительно, чтобы его можно было реализовать в условиях производства. Синтез сополимеров проводили в присутствии монофункционального тритиокарбоната, содержащего алкинильную группу
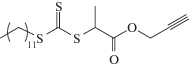
Сополимер с концевой алкинильной группой вступал в реакцию циклоприсоединения с 1,4-бис-(азидометилбензолом), что приводило к удвоению ММ сополимера. Таким образом, удалось получить линейные сополимеры с Mn ∼ 70 × 103 и Ð < 1.6. Тем же приемом, т.е. сочетанием ОПЦ-полимеризации и клик-химии, авторы воспользовались для синтеза разветвленного и сетчатого бутадиен-нитрильного каучука. Для этого они методом ОПЦ синтезировали терполимер АН (35 мол. %), 1,3-бутадиена (56 мол. %) и пропаргилметакрилата (9 мол. %) с Mn ∼ 3.9 × 103 и Ð = = 1.3 и подвергли его реакции циклоприсоединения с 1,4-бис-(азидометилбензолом). Данный прием был затем развит для получения блок-сополимеров линейного и звездообразного строения на основе бутадиен-нитрильного и стирол-нитрильного каучуков [197]. К сожалению, свойства указанных сополимеров в указанных работах не описаны. Однако этот пробел был заполнен в работе [198], в которой бутадиен-нитрильные сополимеры и их блок- и привитые сополимеры со стирол-нитрильным каучуком синтезировали азеотропной сополимеризацией с помощью монофункционального тритиокарбоната. Сополимеры бутадиена и АН имели две температуры стеклования: Тс = –24°С, характерную для статистического сополимера АН и бутадиена с содержанием АН 38 мол. %, и Тс = –66°С, близкую к значению для полибутадиена, полученного радикальной полимеризацией и содержащего звенья 1,4-цис, 1,4-транс- и 1,2-присоединения. При учете достаточно узкого ММР сополимеров такой результат указывает на их блок-градиентную структуру. По данным ДМА, блок- и привитые сополимеры (АН, бутадиена и стирола) характеризуются тремя релаксационными модами: в области –70…–80°С и –2…–6°С, соответствующей фазе мягкого блока – сополимера АН и бутадиена и в области 90–105°С, отвечающего фазе жесткого блока – сополимера АН и стирола.
В качестве сомономеров при синтезе сополимеров АН часто используют стирол, (мет)акрилаты, винилпиридин и другие сополимеры. Так, сополимеризацией стирола и АН в ДМФА по ОПЦ-механизму в присутствии кумилдитиобензоата получали статистические и диблок-сополимеры с контролируемой ММ и узким ММР (Ð = 1.3–1.5) [199]. В более поздней работе [200] была изучена активность стирола и АН при ОПЦ-сополимеризации в массе в присутствии 2-циано-2-пропилдитиобензоата и показано, что кажущаяся активность мономеров изменяется. Это связано с проявлением эффекта избирательной сольватации мономеров активным центром. Например, при мольной доле АН в мономерной смеси 0.9 активности мономеров равны rАН = 0.81 и rС = 0.09, а при его содержании 0.1 – rАН = 0.2 и rс = 0.35. Данное обстоятельство необходимо учитывать при синтезе сополимеров с заданным распределением звеньев в цепи. Был разработан метод синтеза композиционно-однородных бинарных сополимеров АН со стиролом, МА, н- и трет-бутилакрилатом их сополимеризацией в массе под действием бифункционального тритиокарбоната – дибензилтритиокарбоната, а также блок-статистических сополимеров на основе этих же мономеров [201]. Авторы показали, что сополимеризация и блок-полимеризация в массе происходит гомогенно при содержании сомономера 20 мол. % и выше образующиеся сополимеры характеризуются Mn = (20–40) × 103 и Ð = 1.2–1.3.
Направленный синтез ПС и его блок-сополимера с АН с бимодальным ММР осуществили в работе [202]. Авторы применили смесь моно- и бифункционального тритиокарбонатов. Следует заметить, что это весьма нетривиальный результат, поскольку функциональность ОПЦ-агента никак не должна влиять на ММ полимера.
Необычный вариант синтеза блок-сополимеров с АН предложен в работе [203]. Вначале был синтезирован поли(4-винилпиридин), содержащий концевую тритиокарбонатную группу, и затем он был использован как полимерный ОПЦ-агент в сополимеризации азеотропной смеси стирола и АН в растворе и эмульсии. В обоих случаях происходит образование блок-сополимера с узким ММР, причем в последнем случае образуется устойчивая дисперсия блок-сополимерных частиц.
Близким подходом воспользовались в работе [204]: блок-сополимер был получен дисперсионной полимеризацией в воде с использованием в качестве полимерного ОПЦ-агента полиэтиленоксида с дитиобензоатной концевой группой. В этом случае образовывалась дисперсия блок-сополимерных частиц диаметром 50–80 нм, в которых блок полиэтиленоксида выполнял функцию стабилизатора, а блок ПАН формировал ядро частиц.
Интерес исследователей привлекали и другие мономерные пары. Например, в работе [205] описана чередующаяся ОПЦ-сополимеризация β-пинена (rβ-пинен = 0) и АН (rАН = 0.66) с участием 2-цианпропил-2-ил дитиобензоата при 60°С в дихлорэтане. “Живой” механизм наблюдается только при содержании β-пинена не выше 25 мол. % и невысоких конверсиях, затем ОПЦ-механизм нарушается из-за деградационной передачи цепи на мономер. При добавлении к смеси кислоты Льюиса Et2AlCl тенденция к чередованию звеньев усиливается.
В работе [206] с использованием того же ОПЦ-агента в ДМФА синтезировали сополимеры АН с 2-(3-(6-метил-4-оксо-1,2,3,4-тетрагидропиримидин-2-ил)уреидоэтилметакрилатом (не более 10 мол. %), способные эффективно связывать ионы Hg2+ из водных растворов. Авторам удалось добиться удовлетворительного контроля ММ и получить набор сополимеров с Mn в диапазоне (20–40) × 103 и Ð = 1.2–1.6.
Линейные и гребнеобразные термочувствительные блок-сополимеры АН и N-изопропилакриламида были получены комбинацией ОПЦ-полимеризации и полимеризации с переносом атома [207]. Линейные блок-сополимеры были синтезированы ОПЦ-полимеризацией, а гребнеобразные – полимеризацией с переносом атома, причем на основе бромированного сополимера АН и 2-гидроксиэтилметакрилата, полученного ОПЦ-полимеризацией. В обоих случаях были синтезированы блок-сополимеры с узким ММР, обладающие термочувствительностью и поверхностной смачиваемостью.
Начиная с 2011 г., исследователи не только разрабатывали способы контролируемого синтеза ПАН и его сополимеров, но и начали изучать его свойства, в том числе поведение растворов (со)полимеров и процессы циклизации и термоокислительной стабилизации. Эти работы помогли понять, что привносит в свойства ПАН-прекурсора узкое ММР, композиционная однородность сополимеров и контроль за распределением звеньев в цепи.
Влияние ММ, ширины ММР, природы сомономера и состава сополимера на термическое поведение (со)полимеров, синтезированных в условиях ОПЦ-полимеризации, подробно рассмотрено в ряде работ [208–215].
Хорошо известно, что условия синтеза (например, природа растворителя, температура полимеризации, концентрация инициатора и т.д.) влияют на процессы термоокислительной стабилизации ПАН [216]. Причинами этого могут быть пластифицирующее действие остаточного растворителя, разная ММ полимера, примеси непрореагировавшего инициатора [217, 218]. Первые эксперименты показали, что ПАН с узким ММР, но с Mn < 20 × 103, подвергается циклизации при температуре на 40–50°С ниже, чем ПАН с широким ММР и более высокой ММ, полученный классической радикальной полимеризацией в том же растворителе [219]. Поведение олигомерного ПАН одной ММ зависит от природы концевых групп R исходного ОПЦ-агента: в случае бензильной концевой группы циклизация начинается при более высокой температуре, чем для сложноэфирной. Однако при увеличении ММ полимера данная зависимость исчезает. Для ПАН высокой ММ с узким ММР (ОПЦ), наоборот, циклизация начинается при более высокой температуре и протекает в более узком температурном интервале, чем для ПАН с широким ММР (“классика”) [213]. Систематическое изучение влияния ММ и ширины ММР выявило интересную закономерность: энергия активации реакции циклизации для ПАН с узким ММР на 40–90 кДж/моль ниже, чем с широким ММР, а величина предэкспоненты, позволяющая косвенно судить о молекулярности реакции, отличается на пять порядков и более, т.е. механизм инициирования реакции циклизации у этих полимеров, скорее всего, различается. Кроме того, экзо-эффект реакции циклизации у ПАН с узким ММР в ∼2.5 раза выше. Понижение температуры синтеза за счет использования окислительно-восстановительного или радиационного инициирования, по-видимому, дополнительно способствует уменьшению дефектов в полимерной цепи (таких как разветвления) и смещает экзо-эффект в сторону высоких температур, а его интенсивность возрастает [218]. Таким образом, можно высказать соображение о том, что контролируемая радикальная полимеризация уменьшает количество дефектов в структуре макромолекул, что приводит к более лавинообразному протеканию циклизации в гомополимерах.
При анализе термического поведения сополимеров задача осложняется тем, что к широкому ММР в классической радикальной сополимеризации добавляется конверсионная композиционная неоднородность. В связи с этим ОПЦ-полимеризация, способная устранить оба нежелательных фактора, является полезным инструментом для изучения циклизации сополимеров АН.
При сравнении термического поведения бинарных сополимеров АН с инертными мономерами МА, н-бутилакрилатом и ММА с узким ММР (Ð = 1.3–1.4) обнаружено, что, как и в “классических” сополимерах, при увеличении содержания сомономера от 2 до 10 мол. % циклизация начинается при более высоких температурах, а интенсивность экзо-эффекта уменьшается [210, 220].
Иная ситуация характерна для сополимеров с трет-бутилакрилатом, которые при повышенных температурах способны отщеплять изобутилен. В результате в макромолекуле появляются звенья акриловой кислоты, инициирующие циклизацию, и экзо-эффект смещается в область низких температур тем отчетливее, чем выше содержание в сополимере трет-бутилакрилата [220].
Сопоставление термического поведения сополимеров, содержащих звенья мономеров, ускоряющих циклизацию (акриламида или акриловой кислоты), полученных ОПЦ и обычной радикальной полимеризацией, показывает ту же тенденцию, что и для гомополимеров: температурный интервал циклизации сужается и смещается в область более высоких температур [212, 218, 221]. В таких сополимерах реализуются оба механизма циклизации – ионный (низкотемпературный) и радикальный (высокотемпературный) [16–25]. Более высокая композиционная однородность сополимеров, полученных методом ОПЦ, и их узкое ММР при определенных составах сополимеров приводят к изменению вклада этих механизмов и влияют на последовательность цепи сопряжения в ходе циклизации [212].
Кроме среднего состава сополимера на его свойства влияет характер распределения звеньев в цепи. Для сополимеров АН данный параметр определяет длину отрезка цепи сопряжения, т.е. дефектность и свойства термостабилизированного ПАН-прекурсора. Работы в этом направлении ведутся с использованием как классической радикальной полимеризации [222], так и ОПЦ-процесса [223–230].
В первом случае попытка оценить влияние длины последовательности звеньев АН в сополимере с МА на процессы циклизации была предпринята на примере синтеза сополимеров на ранних конверсиях классической радикальной сополимеризацией [222]. Это позволило получить набор сополимеров, различающихся средней длиной последовательности звеньев АН от ∼13 до ∼5. Увеличение содержания метилакрилата, т.е. уменьшение длины отрезка из звеньев АН, привело к уменьшению средней длины сопряженной структуры, повышению энергии активации окисления и уменьшению содержания окисленных структур в сополимере, подвергнутом стабилизации. Степень циклизации изменяется не монотонно и проходит через максимум при содержании метилакрилата в сополимере ∼8 мас. %. При увеличении степени циклизации повышаются термическая стабильность и коксовый остаток в полимере.
В ОПЦ-процессе таким исследованиям предшествовали работы китайских ученых, которые предложили теоретическую модель полунепрерывной ОПЦ-сополимеризации для обеспечения одинакового распределения мономерных звеньев по всей длине полимерной цепи [223], которую проверили при синтезе сополимеров стирола и бутилакрилата [224–226]. Применительно к сополимерам АН данный прием использовали в работах [221, 227].
При ОПЦ-сополимеризации АН (r1 = 0.39) и ускоряющего циклизацию N-изопропилакриламида (r2 = 0.72) в этиленкарбонате при 30°С в присутствии низкотемпературного азо-инициатора и 2-циано-2-пропилдодецилтритиокарбоната авторы вводили второй мономер дозировано на протяжении реакции с выбранной скоростью [227]. В продукте полимеризации доля второго мономера составляет 2.5–6.4 мол. %. Авторы показали, что медленное введение N-изопропилакриламида, с одной стороны, и его более высокая концентрация, с другой стороны, способствуют понижению тепловыделения при циклизации и расширению температурного интервала циклизации за счет смещения его начала в низкотемпературную область, по сравнению с единовременным введением мономеров в синтез. В целом более равномерное распределение в цепи сомономера, ускоряющего циклизацию, повышает скорость циклизации и увеличивает термостабильность полимера в инертной атмосфере при нагревании до 700°С.
Сополимеризацию АН (rАН = 0.49) с другим ускоряющим циклизацию мономером – акриловой кислотой (rАК = 2.5) проводили в ДМСО при 55°С с использованием в качестве инициатора персульфата калия и ОПЦ-агента 2-(додецилтиокарбонилтиоилтио)-2-метилпропионовой кислоты при разной скорости введения акриловой кислоты в полимеризацию [221]. Термическое поведение сополимеров близкого среднего состава и ММ, но с разным распределением звеньев в цепи различается. Так, увеличение времени введения акриловой кислоты в сополимеризацию приводит к повышению интенсивности высокотемпературного пика, отвечающего радикальному механизму циклизации, что связано с ростом длины последовательности звеньев АН. При ее более быстром добавлении в реакцию в сополимере усиливается вклад ионного механизма циклизации и суммарный экзо-эффект понижается. Микроструктура цепи повлияла также на значение энергий активации ионной и радикальной циклизации.
Таким образом, в настоящее время с помощью ОПЦ-полимеризации разработаны способы регулирования термического поведения сополимеров АН путем контроля их ММ, ММР, состава и распределения звеньев в цепи.
Перечисленные факторы влияют и на реологию растворов полимеров, на знании которой базируются условия для выбора режимов формования ПАН. Кривые течения ПАН и его бинарных (со стиролом или акриламидом) и тройных сополимеров (с метилакрилатом и итаконовой кислотой) в растворителях, ДМСО, ДМФА или ДМАА, которые часто используют в процессе формования из раствора, – типичные для полимеров [212, 228–230]. В широком интервале концентраций они являются ньютоновскими жидкостями; при высоких значениях напряжений сдвига, определяющихся ММ и ММР полимера, происходит переход в режим неустойчивого течения. Однако более узкое ММР сополимеров, синтезированных в условиях ОПЦ-полимеризации, служит причиной более низкой вязкости при той же концентрации полимера в растворе по сравнению с “классическим” полимером. Кроме того, на кривых течения область ньютоновского поведения (наибольшей ньютоновской вязкости) в них наблюдается в более широком интервале скорости сдвига. Анализ динамических свойств растворов ОПЦ-полимеров показывает, что гелеобразование в них происходит позже, чем в “классических” образцах. Таким образом, основным преимуществом метода ОПЦ с точки зрения формования является синтез полимеров с более узким ММР, что дает возможность использования более вязких растворов полимеров при формовании волокна.
Безусловно, наибольший интерес вызывают исследования, в которых пройдена вся цепочка – от синтеза полимера методом ОПЦ до получения из него углеродного волокна. В литературе есть всего два примера таких работ [210, 211, 214], однако они иллюстрируют широкие перспективы метода ОПЦ.
Сополимер АН и ММА с содержанием ММА 4 мол. % и Mn = 150 × 103 и Ð = 1.7 был синтезирован с использованием 2-циано-2-пропилдодецилтритиокарбоната в этиленкарбонате [210]. Формование вели “мокрым” способом из 24%-ного раствора сополимера в ДМФА. Авторы сформовали волокна ПАН-прекурсора плотной структуры, без пустот и круглой формы в поперечном сечении, которые имели прочность на разрыв 40–50 сН/текс, модуль упругости 900–1000 сН/текс, плотность 1.178 г/см3, степень кристалличности 80%, степень ориентации 87%. Авторы подобрали условия для термоокислительной стабилизации ПАН-прекурсора и карбонизации и получили углеродное волокно с прочностью на разрыв 2.5 ГПа.
В работе [214] предложен метод синтеза терполимеров АН, МА и итаконовой кислоты с Mn > > 300 × 103 и Ð < 1.2 методом ОПЦ в ДМСО под действием 2-циано-2-изопропилдитиобензоата, получения из него ПАН-прекурсора и углеродного волокна. Для сравнения авторы провели аналогичные операции для терполимера, синтезированного классической радикальной полимеризацией. Для синтеза ПАН авторы применили нестандартный подход. Полимеризацию проводили, задавая определенный профиль температуры реакции. Он включает короткий начальный период (менее 30 мин), в течение которого полимеризацию ведут при повышенной температуре (более 65°С) и длительный (15–20 ч), в ходе которого температура поддерживается ниже 60°С, что позволило достичь нетипичной для применения дитиобензоатов конверсии мономера ∼60%.
Формование волокон вели “мокрым” способом из 15%-ного раствора в ДМАА. Благодаря более узкому ММР вязкость раствора ОПЦ-полимера была существенно ниже, а волокно имело более плотную структуру. Механические характеристики ПАН-прекурсора, полученного методом ОПЦ, оказались заметно выше, чем “классического” прекурсора. Например, прочность на разрыв у первого была на 50% выше, а модуль упругости больше на 40%. Термоокислительную стабилизацию проводили в градиенте температур от 208 до 255°С, а карбонизацию в инертной атмосфере в интервале температур 400–1350°С. После карбонизации механические характеристики углеродного волокна, полученного из ОПЦ-полимера, остались выше, чем у образца сравнения.
Таким образом, контроль молекулярной структуры (со)полимеров АН в условиях ОПЦ-процесса можно рассматривать как перспективное направление для получения высокомодульного и высокопрочного углеродного волокна.
СОПОЛИМЕРЫ АКРИЛОНИТРИЛА, СПОСОБНЫЕ К ФОРМОВАНИЮ ИЗ РАСПЛАВА
Одна из причин, ограничивающих расширение области гражданского применения углеродных волокон, заключается в их относительно высокой стоимости. В связи с этим в последние десятилетия ведутся интенсивные разработки альтернативных, более экономичных способов получения ПАН-прекурсоров. Среди них, безусловно, лидирующим является формование волокна не из раствора, а из расплава. Данный метод не требует использования органических растворителей и позволяет существенно повысить концентрацию полимера при формовании. Кроме того, для него не пригодны (со)полимеры акрилонитрила, предназначенные для получения прекурсора формованием из раствора. Это связано с тем, что такие полимеры обладают достаточно высокой ММ и низким содержанием сомономеров, в результате чего процессы циклизации в них начинаются раньше перехода в вязкотекучее состояние.
Существует два основных способа понизить температуру плавления ПАН: получение (со)полимера с заданными молекулярными характеристиками и физическая модификация (со)полимера – пластификация [231, 232]. В первом случае прежде всего необходимо ввести в полимер определенные сомономеры в количестве, достаточном для нарушения кристаллической структуры ПАН. Кроме того, следует понизить ММ полимера до критических значений (обычно Mn < 50 × 103). Во втором – в качестве пластификаторов используют органические соединения, такие как N-ацетилморфолин, этиленкарбонат, спирты, а также воду. Ниже будут рассмотрены синтетические методы получения ПАН, способного к формованию из расплава.
Синтез таких сополимеров проводят классической радикальной полимеризацией в растворе, суспензии, эмульсии и осадительной полимеризацией в воде [233–236]. Последняя является более перспективной с точки зрения промышленного использования, поскольку приводит к образованию легко выделяемого продукта, не требующего дополнительной очистки. ММ полимера регулируют в основном с помощью агентов передачи цепи, а также температурой синтеза и соотношением мольных концентраций мономера и инициатора. Наиболее часто в качестве передатчиков используют меркаптаны, например додецилмеркаптан в количестве 0.05–0.70 мол. % [234], реже изопропанол [233].
Первый патент о получении (со)полимера АН и метакрилонитрила, переходящего в расплав без химического изменения структуры (циклизации), был опубликован в 1996 г. [237], спустя четыре десятилетия после первого упоминания об этих сополимерах как ПАН-прекурсорах [238]. Активные исследования в направлении синтеза и исследования свойств подобных сополимеров начались только в 2000-е гг. Все описанные в литературе сополимеры АН, способные к формованию из расплава, можно разделить на четыре группы: бинарные и тройные сополимеры АН с МА; терполимеры АН, МА и третьего сомономера, содержащего фоточувствительную функциональную группу; сополимеры АН с винилимидазолом; сополимеры АН с другими виниловыми мономерами.
Подробные исследования сополимеризации АН и МА для получения прекурсоров, формуемых из расплава, приведены в работах [233, 234, 239–241].
Было установлено, что при содержании МА ниже 10 мол. % даже при Mn ∼ 20 × 103 сополимер невозможно переработать из расплава. Время жизни расплава в данном случае достаточно мало, поскольку температура начала циклизации и образования лестничной структуры близка к температуре течения, и уже через несколько минут после плавления начинается нарастание вязкости вследствие циклизации. Выше этого порогового значения (10 мол. % МА) существенно изменяются свойства сополимера при повышенных температурах, такие как динамическая вязкость и модуль накопления [234]. Например, при 220°С значения динамической вязкости расплава и модуля упругости сополимера с 10 и 15 мол. % МА падают примерно на четыре порядка по сравнению с сополимерами, содержащими 2–7 мол. % МА. Увеличение содержания МА расширяет температурный интервал, в котором сополимер может формоваться из расплава, понижает энергию активации вязкого течения и температуру перехода в вязкотекучее состояние. Динамическая вязкость расплава гораздо чувствительнее к ММ, чем к составу сополимеров: при 220°С вязкость падает на три порядка при уменьшении ММ сополимера на порядок от 115 × 103 до 9 × 103. Аналогичного эффекта уменьшения вязкости можно добиться добавлением низкомолекулярного сополимера к высокомолекулярному. Стабильность расплава определяется не только температурой, но и составом и ММ сополимера; чем ниже последняя, тем позже по времени наблюдается прирост вязкости за счет реакции циклизации и образования пространственной структуры. Проведенные исследования позволили авторам предложить оптимальные с их точки зрения характеристики сополимера для получения ПАН-прекурсора формованием из расплава: доля МА в сополимере 15 мол. % и Mn ∼ 20 × 103.
Дополнительно понизить вязкость расплавов сополимера АН и МА можно с использованием СО2 в качестве пластификатора, предварительно насыщая им сополимер в достаточно жестких условиях в течение длительного времени [242] или путем его адсорбции непосредственно во время экструзии [243].
Другим способом получения сополимеров АН с МА, способного к переработке из расплава, является микроинкапсулирование в него материала с обратимыми фазами22 [244]. Для этого авторы использовали осадительную сополимеризацию АН и МА (15 мол. %) в воде при 30°С с участием меркаптана и через 2 ч полимеризации добавили в реакционную систему новую порцию инициатора и от 5 до 25 мас. % н-октадекана (материала с обратимыми фазами). В данном случае был получен сополимер с Mn ∼ 50 × 103 и широким ММР (Ð > 2.5). Увеличение концентрации микроинкапсулированного октадекана понижает индекс расплава и одновременно повышает температуру стеклования сополимера от 88 (в отсутствие октадекана) до 92°С (25 мас. % октадекана). Сформованное из расплава при 200°С волокно, содержащее октадекан, имеет микропористую структуру. Результатом этого является понижение прочности на разрыв (от 3.2 до 1.0 сН/дтекс) и разрывного удлинения (от 26 до 10%) с ростом содержания октадекана. В последующей работе авторы повысили содержание микроинкапсулированного октадекана до 40% и показали, что сополимер с М ∼ ∼ 30 × 103 и содержанием МА 15 мол. %, способен плавится при 206°С [245].
В описанных выше исследованиях использовали разные варианты получения сополимеров. При сравнении вязкости расплавов сополимеров близкой ММ и состава, но синтезированных разными способами, оказалось, что она различается. Такой результат обусловлен тем, что активность мономеров в радикальной сополимеризации существенно меняется при переходе от гомогенного к гетерофазному методу синтеза [129]. В итоге степень разупорядоченности макромолекул сополимеров, влияющая на свойства расплава полимера, меняется в зависимости от того, насколько равномерно внедряется МА в полимерную цепь. Влияние условий синтеза на поведение расплавов сополимеров АН и МА изучено в работе [233]. К сожалению, одновременно изменялись разные параметры – ММ сополимера и ММР, и это не позволило проследить за влиянием микроструктуры цепи на свойства расплава. Данный вопрос до сих пор остается открытым. Анализ результатов работы [233] позволяет сделать вывод о том, что основной фактор, кроме состава сополимера, влияющий на вязкость расплава, – ММР сополимера. Увеличение доли высокомолекулярной фракции приводит к заметному росту вязкости и сужению температурного интервала стабильности расплава.
Описанные выше разработки позволили получить полые волокна формованием сополимеров АН и МА (15 мол. % МА) из расплава с удовлетворительными физико-механическими характеристиками: Тс = 96°С, предел прочности на разрыв 16 сН/дтекс, деформация 18.7%, модуль упругости 3 ГПа [246].
Очевидно, что при термостабилизации волокон из сополимеров АН и МА вначале будет происходить их плавление, а затем циклизация. Следовательно, чтобы использовать их в качестве прекурсоров углеродного волокна, необходимо было разработать специальный подход.
В качестве первого приема использовали введение сомономеров, ускоряющих циклизацию. Предполагалось, что будут найдены условия, при которых сополимер можно будет переработать из расплава, а затем при более высокой температуре быстро подвергнуть термоокислительной стабилизации. Так, было предложено ввести в сополимер АН и МА малое количество третьего сомономера – акриловой кислоты, метакриловой кислоты или акриламида в мольном отношении АН : МА:сомономер от 85 : 11 : 4 до 85 : 14 : 1 [241]. Более подробно влияние содержания третьего мономера изучили на примере итаконовой кислоты. Все сополимеры получали сополимеризацией в растворе ДМФА с использованием додецилмеркаптана. Очевидно, что добавление ускорителя циклизации должно было понизить температурный интервал стабильности расплава по сравнению с бинарным сополимером АН–МА с тем же содержанием АН. Динамическая вязкость расплава в интервале температур 200–220°С определяется количеством сомономера и его природой. При 200°С расплавы остаются стабильными в течение более 30 мин, а при 220°С вязкость начинает возрастать быстрее вследствие реакции циклизации и образования сопряженных и пространственных структур. Скорость ее нарастания изменяется в ряду итаконовая кислота > акриламид > метакриловая кислота > акриловая кислота. Анализ кинетики изменения вязкости расплава позволил оценить энергию активации циклизации и константу скорости реакции и определить оптимальные составы терполимеров. Например, введение 3 мол. % итаконовой кислоты оказалось приемлемым для получения терполимера, сохраняющего способность к переработке через расплав при 200°С и подвергающегося циклизации с высокой скоростью при 220°С.
В недавней работе для этой же цели использовали диметилитаконат [247]. Авторы сравнили сополимеры АН и МА и АН и диметилитаконата с одинаковым содержанием сомономера 5, 10 и 15 мол. %. Выбранные условия синтеза – нетипичные для поставленных задач, поскольку сополимеры АН и МА характеризовались Mn ∼ 105 и Ð ∼ 2.0, а сополимеры АН и диметилитаконата Mn ∼ (40–60) × 103 и Ð ∼ 1.1–1.7. Согласно выводам авторов, сополимер с содержанием диметилитаконата 15 мол. % предпочтительнее, чем сополимер с 15 мол. % МА более высокой ММ, так как он плавится без циклизации при более низкой температуре (190°С). Однако время изотермической циклизации при любой температуре у него выше, чем у сополимера с МА.
Вероятно, более удобным способом для проведения стабилизации ПАН-прекурсора, полученного формованием из расплава, является его электронно-лучевое облучение. В работе [248] описано получение волокна формованием расплава сополимера АН и МА (15 мол. % МА) при 190°С, его электронно-лучевое облучение дозой 1500 кГр, последующая ступенчатая стабилизация до температуры 250°С и карбонизация при 1200°С. Полученное УВ характеризовалось пределом прочности 1.4 ГПа, модулем упругости 110 ГПа и разрывным удлинением 1.3%.
Циклизация, индуцированная разными вариантами облучения и приводящая к образованию ПАН лестничной структуры, была описана и в других работах [249–252]. Например, такого эффекта можно добиться путем УФ-облучения сополимера АН и МА [249]. Как и в описанном выше случае, это приводит к возникновению радикалов и частичной сшивке полимера. Для сравнения авторы использовали коммерческие сополимеры АН и МА, полученные формованием из раствора (6 мол. % МА) и из расплава (12 мол. % МА). Оба образца подвергали УФ-облучению, затем термоокислительной стабилизации и карбонизации и на каждом этапе исследовали физико-механические характеристики волокон. Карбонизованное волокно, полученное из расплавного ПАН, характеризовалось пределом прочности 0.3 ГПа, модулем упругости 60 ГПа и разрывным удлинением 0.6%. Эти характеристики оказались ниже, чем для углеродного волокна из сополимера, полученного формованием из раствора.
Более эффективным может быть другой прием, основанный на введении в сополимер АН и МА фоточувствительного мономера, который под действием света определенной длины волны способен переходить в возбужденное состояние и генерировать радикалы на полимерной цепи. К таким мономерам относятся 4-бензофенилвинилкарбонат
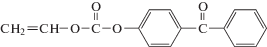 ,
,
4-бензофенилвинилкарбамат
 ,
,
2-винилоксикарбонилоксиметилантрахинон
и винилоксикарбонилбензоиновый эфир
 [253].
[253].
В работе [235] описано получение терполимера АН, МА и акрилоилбензофенона эмульсионной полимеризацией с использованием меркаптана с содержанием 14 мол. % МА и 1 мол. % акрилоилбензофенона
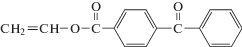
Авторы сравнили свойства бинарного сополимера АН и МА (Mn = 26 × 103) с тем же содержанием АН и терполимеров разной ММ (Mn = 16 × 103 и 22 × 103). Важным результатом работы стало доказательство того, что включенный в цепь акрилоилбензофенон является достаточно термостойким и при нагревании до 200–260°С не претерпевает никаких изменений. Изучение хемореологии сополимера и терполимеров позволило показать, что наличие фоточувствительного мономера способствует понижению скорости нарастания вязкости за счет образования полимера сетчатой структуры и не влияет на стабильность расплава. Таким образом, можно ожидать, что подобные терполимеры могут представлять интерес как прекурсоры для получения углеродных волокон.
В работе [254] сформованные из расплава ПАН-прекурсоры, полученные с использованием аналогичных со- и терполимеров, были подвергнуты УФ-облучению и изучено изменение их химической структуры. Бинарные сополимеры сохраняют свои свойства после облучения. В отличие от них терполимеры при облучении меняют химическую структуру, что проявляется, в частности, в изменении окраски, и тем интенсивнее, чем выше мощность облучения. Механизм этой реакции можно представить следующим образом:
Авторы провели систематическое исследование данного процесса методами УФ- и ИК-спектроскопии, однако за этим не последовало изучения стабилизации и карбонизации и механических свойств волокон.
Развитием этой тематики стало получение УВ из терполимера АН, МА и акрилоилбензофенона с мольным соотношением 85 : 14 : 1, синтезированного эмульсионной полимеризацией и сформованного из расплава, подвергнутого УФ-облучению, термостабилизации и карбонизации [255]. Углеродное волокно имело предел прочности 0.6 ГПа, модуль упругости 130 ГПа и разрывное удлинение 0.4%. Однако синтез аналогичного терполимера суспензионной полимеризацией и те же манипуляции привели к получению углеродного волокна с пределом прочности 0.6 ГПа, модулем упругости 7.3 ГПа и разрывным удлинением 0.4% [256]. Следует отметить, что в последнем случае авторы не использовали меркаптан для регулирования ММ и, возможно, это стало причиной более низких прочностных характеристик УВ. Очередная попытка получения матов из углеродных волокон была предпринята в работе [257]. Прекурсор терполимера (АН : МА : акрилоилбензофенон = 85 : 14 : 1, синтезирован эмульсионной полимеризацией) был использован для получения нетканых матов, которые подвергли УФ-обработке, термостабилизации и карбонизации при 1500°С. Предел прочности составил 1 сН/текс, модуль упругости 110 сН/текс, разрывное удлинение 1.3%.
Приведенные результаты позволяют рассматривать данный подход – синтез терполимера с фоточувствительным мономером как перспективный вариант получения ПАН-прекурсора и углеродного волокна из него. Однако он требует оптимизации как в способе синтеза и составе терполимера, так и в выборе условий для получения углеродного волокна.
Другой потенциальный вариант прекурсора углеродного волокна – это сополимеры АН и 1-винилимидазола (ВИМ) [258]. При их сополимеризации в растворе образующиеся сополимеры имеют чередующееся или близкое к нему строение (rАН = 0.12, rВИМ = 0.24) [259]. Такие сополимеры имеют относительно невысокую ММ, что достигается использованием меркаптана в качестве агента передачи цепи, способны переходить в вязкотекучее состояние и затем циклизоваться на воздухе с образованием лестничной структуры. Оптимальный состав сополимера был определен при систематическом исследовании влияния доли винилимидазола на температуру стеклования, коксовый остаток, вязкоупругие свойства расплава при разной температуре, стабильность расплава и т.д. [258]. Наилучшие результаты показал сополимер, содержащий 18 мол. % винил-имидазола, расплав которого устойчив при 192°С, а выше 210°С подвергается циклизации с высокой скоростью.
Дальнейшие исследования были направлены на поиск оптимальных условий для формования указанных сополимеров из расплава: например, понижение температуры формования или времени, необходимого для формования. Для этой цели использовали обычные пластификаторы (этиленкарбонат, пропиленкарбонат или этиленгликоль), полиэдральный олигомерный силсесквиоксан (октафенил- и октаизобутилсилсесквиоксан) и новый олигомер на основе акрилонитрила и метил-2-(1Н-имидазол-1-ил)акрилата (20 мол. %) с Mn = (2–5) × 103 [260]
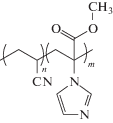
Последний оказал наиболее значимое влияние на свойства расплава. Так, добавка 8 мас. % олигомера приводит к понижению температуры стеклования сополимера АН и винилимидазола (Mn ∼ 50 × 103, синтезирован аналогично работе [258], Тс = 115°С) на 40°С и двукратному уменьшению напряжения сдвига. Температура, при которой начинается циклизация, изменяется всего на 5–10°С по сравнению с исходным значением (280°С). Это позволяет существенно расширить температурный интервал переработки сополимера из расплава. Формование волокна проводили при 180–192°С, а термоокислительную стабилизацию при нагревании от 100 до 300°С со скоростью 1 град/мин. Оказалось, что олигомер встраивается в лестничную структуру циклизующегося полимера. Карбонизация при 1000°С привела к получению углеродных волокон с пределом прочности 1.9 ГПа и модулем упругости 190 ГПа.
Результатом проведенных исследований явился патент [261], в котором описан способ получения углеродного волокна на основе ПАН-прекурсора – сополимера АН и 1-винил-, 4-винил, 2-винил- и 1-метил-2-винилимидазола (70–80 мол. % АН) с добавками 5–10 мас. % олигомера акрилонитрила-со-N-имидазолакрилата c Mn = (1–2) × 103 с прочностными характеристиками, аналогичными работе [258].
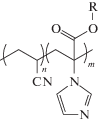 ,
,
где R=H, CH3, CnH2n+ 1.
ПАН-прекурсор, полученный формованием сополимера АН и винилимидазола из раствора, нашел необычное применение. Известно, что окись азота NO способна обратимо взаимодействовать с акцепторами и образовывать диазениумдиолат. Скорость этой реакции в случае ПАН невелика, однако введение звеньев винилимидазола в сополимер повышает способность АН реагировать с NO [262]:
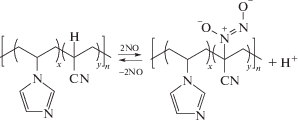
Такие волокна, аккумулирующие и затем выделяющие NO, представляют интерес в хирургии для заживления ран. В данном случае высвобождаемый NO препятствует их инфицированию. Оказалось, что волокно, полученное формованием сополимера АН и винилимидазола из расплава, запасает больше NO, чем волокно, полученное электроформованием того же сополимера или гомополимера АН. При этом дополнительная модификация волокна поликапролактоном позволила осуществить контролируемое высвобождение NO в течение нескольких суток.
Среди других мономеров, которые использовали для получения сополимеров АН, способных переходить в вязкотекучее состояние, следует упомянуть N,N′-ди(2-пропил)акрилоамидины (N,N′-ди(2-пропил)акрилоамидин и N-2-пропил-N′-трет-бутилакрилоамидины) [263]:
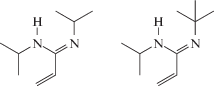
Акрилоамидины не оказывают заметного влияния на температуру стеклования, но изменяют термическое поведение сополимеров с АН. В инертной атмосфере температура начала циклизации понижается от 198°С для ПАН, до 178°С при добавлении в полимер 1 мол. % N,N′-ди(2-пропил)акрилоамидина и до 138°С при его содержании 19 мол. %. На воздухе реакция более экзотермичная и начинается для сополимеров с 20 мол. % акрилоамидинов и Mn ∼ 25 × 103 при 140–170°С в зависимости от заместителей при азоте. В отсутствие добавок сополимеры переходят в вязкотекучее состояние выше 150°С, а при добавлении 10 мас. % ДМФА температура, при которой возможно формование из расплава, понижается до 130°С. Отталкиваясь от этих результатов, авторы получили прекурсор, провели его термоокислительную стабилизацию и карбонизовали в инертной атмосфере нагреванием волокон от комнатной температуры до 1400 или 1800°С со скоростью 10 град./мин. Полученное углеродное волокно характеризовалось пределом прочности 0.9 ГПа, модулем упругости 85–100 ГПа и разрывным удлинением 1.2%.
Другим примером является использование метакрилонитрила [237]. В радикальной сополимеризации он более активен, чем АН [264], и расходуется в сополимеризации с более высокой скоростью. Данный фактор оказывает решающее значение на свойства ПАН-прекурсора, поэтому один из способов регулирования свойств прекурсора ‒ поддержание постоянства состава мономерной смеси в ходе синтеза сополимера, что обеспечивается разной скоростью введения мономеров в сополимеризацию. ММ задается добавками меркаптанов или спиртов. Сополимеры АН и МАН с содержанием последнего 25–50 мас. % и относительно невысокой ММ, полученные эмульсионной полимеризацией, обладают способностью переходить в вязкотекучее состояние до начала циклизации и могут быть использованы для переработки из расплава.
В целом анализ литературы свидетельствует о том, что технологии получения ПАН-прекурсора из расплава еще несовершенны. Основные требования к таким сополимерам заключаются в невысокой ММ и наличии не менее 10 мол. % сомономера. Однако оптимальная химическая природа такого сомономера, требования к молекулярной структуре цепи (характер распределения звеньев в цепи и длина отрезков последовательности АН) до сих пор не установлены, и эта область, по-видимому, будет активно развиваться в ближайшие годы.
АНАЛИЗ ПАТЕНТНОЙ ЛИТЕРАТУРЫ
Обзор российских и зарубежных патентов, посвященных получению гомо- и сополимеров акрилонитрила, используемых для изготовления ПАН прекурсоров углеродного волокна, за последние тридцать лет показывает, что исследователи концентрировали усилия на модификации условий проведения уже известных процессов (новые инициаторы, растворители/среды, мономеры), а также на разработке новых механизмов полимеризации. Работы в области синтеза сополимеров АН, предназначенных для использования в других областях (например, в качестве термопластов), основаны в принципе на тех же синтетических подходах [265–295 ], но в данном обзоре рассматриваться не будут.
Радикальная полимеризация
Российскими и зарубежными учеными были разработаны новые способы осуществления (со)полимеризации акрилонитрила в массе [296], растворе [297–299], эмульсии [300], осадительной полимеризацией в воде [301–305] и в сверхкритическом CO2 [306–308].
Так, в патенте [309] описано применение новых геминальных бисгидропероксидов [310, 311] общей формулы
 ,
,
где R1 – низший алкил, R2 – Н или СН3, R3 – Н или низший алкил, R4 – Н или низший алкил, либо R1 и R4 – разветвленный алкил. В этом случае несшитый ПАН, растворимый в ДМФА и ДМАА, получают полимеризацией в массе в присутствии геминальных бисгидропероксидов при умеренной температуре (50–60°С). Применение таких инициаторов экономически выгодно, поскольку их получают из промышленно производимого сырья, такого как кетоны и водные растворы пероксида водорода. Кроме того, проведение полимеризации в массе мономера, а не в растворе, удешевляет и упрощает технологию получения и выделения полиакрилонитрила, снижает экологическую нагрузку на окружающую среду.
Полимеризацию в растворе использовали [297] для получения сополимеров акрилонитрила с акрилатом натрия или смесью акрилата натрия с метилакрилатом, или смесью акрилата натрия с метилакрилатом и итаконовой кислотой в 45–52%-ном водном растворе роданида натрия при 40–90°С. В этом случае авторы заменяли более дорогой мономер винилацетат, дешевым – акрилатом натрия с целью получения хорошо окрашиваемого ПАН-волокна.
В патенте [298] описан способ синтеза сополимеров акрилонитрила с содержанием сомономера не более 10% и Mw = (80–120) × 103 радикальной полимеризацией в растворе ДМСО или ДМФА под действием ДАК или азобисизовалериановой кислоты, аналогично данным патента [299], или осадительной полимеризацией в воде в присутствии тиоцианата натрия, хлорида цинка или перхлората натрия. Особенностью этого метода является способ синтеза мономера – акрилонитрила из глицерола. Формование волокна проводят мокрым способом, используя подходящий органический растворитель. По достижении оптимального диаметра волокна его подвергают термоокислительной стабилизации и затем карбонизации.
Эмульсионная полимеризация крайне редко используется для синтеза сополимеров АН. Тем не менее, описано получение волокнообразующих сополимеров акрилонитрила с эфирами акриловой и метакриловой кислоты эмульсионной полимеризацией в присутствии катионного или анионного ПАВ под действием радиационного инициирования [300]. Это позволяет осуществлять полимеризацию в интервале 5–60°С и получать сополимеры с ММ до 1.5 × 106 с конверсией 80–95%. Одно из преимуществ данного метода – высокая скорость полимеризации и возможность регулировать ММ полимера.
Более распространенным способом гетерофазной полимеризации является осадительная полимеризация, которую чаще проводят в воде [301–304]. Так, описана осадительная полимеризация акрилонитрила с метилакрилатом и сомономером, содержащим кислотную группу (акриловая, метакриловая и итаконовая кислоты), с использованием традиционной окислительно-восстановительной системы на основе персульфата калия и метабисульфита натрия в интервале температур от 40 до 60°С и pH 2–3 [301]. Особое внимание исследователи уделяли чистоте мономеров и воды, полагая, что указанные факторы способствуют повышению ММ и химической чистоты сополимеров АН, которые можно использовать для получения прекурсоров углеродного волокна.
В другом варианте осадительной полимеризации АН применяют окислительно-восстановительную систему персульфат калия и бисульфит натрия в сочетании с сульфатом железа(II) [302]. Это позволяет добиться высокой скорости процесса при умеренных температурах: продолжительность реакции составляет 5–60 мин при 45–60°C. Такой способ обеспечивает получение ПАН с Mn в интервале (110–125) × 103 и дисперсией Ð = 2–3.
Для осадительной сополимеризации АН с винилацетатом и виниловой карбоновой кислотой (или ее производными) в качестве инициаторов применяют также смесь персульфата аммония, бисульфита натрия и сульфата железа(II) [303]. Полимеризацию проводят при 50–70°C и pH от 2 до 5. Содержание акрилонитрила в системе составляет 20–30 мас. % в расчете на массу воды. Преимущество этого метода – достаточно низкая стоимость исходного сырья. Похожий вариант описан в патенте [304], в котором осуществлена осадительная сополимеризация АН с акриловой или метакриловой кислотой (их эфирами или амидами) и итаконовой кислотой под действием персульфата аммония и бисульфита либо сульфита натрия в интервале температур 30–80°C в течение 1–10 ч. Концентрации компонентов были подобраны таким образом, чтобы получить сополимеры с Mn = (17.5–26) × 103 и дисперсией Ð = = 2.7–2.9.
Осадительную сополимеризацию АН с метакриловой кислотой и акриламидом инициировали также смесью персульфата аммония, гидросульфита аммония и сульфата железа(II) при 50°С и рН 3 [305]. Особенностью этого способа является многостадийное выделение полимера, что может быть оправдано только высокими прочностными характеристиками получаемого углеродного волокна.
Необычный вариант осадительной полимеризации предложен в патенте [312], в котором сополимеризацию полярных мономеров, в частности, акрилонитрила с метакриловой кислотой проводят в смеси подходящих растворителей. Температуру полимеризации выбирают выше нижней критической температуры растворения полимера. Таким образом удается направленно регулировать относительную активность мономеров в сополимеризации, получать сополимеры разной микроструктуры с М = 103–105 и узким ММР.
Осадительная сополимеризация АН в сверхкритическом диоксиде углерода описана в патентах [306–308]. По мнению авторов, преимуществами данного способа получения являются экологичность, экономичность и энергоэффективность. Солимеризация акрилонитрила с итаконовой кислотой или ее производными инициируется традиционными инициаторами, например ДАК, при 65–80°C. Отличием перечисленных систем от осадительной полимеризации в воде является чрезвычайно широкое ММР. Для решения этой проблемы было предложено использовать тиолы (этантиол), которые понижают дисперсию сополимера на 40–45% [307].
Радикальная полимеризация с обратимой деактивацией цепи
Установление общих закономерностей радикальной полимеризации с обратимой деактивацией цепи позволило разработать и запатентовать ряд интересных методов синтеза (со)полимеров акрилонитрила с контролируемой ММ и относительно узким ММР.
Радикальную полимеризацию с применением нитроксильных радикалов использовали авторы патента [265], в котором упоминается широкий круг сомономеров, включая акрилонитрил. Стоит отметить, что это изобретение охватывает сополимеры различного строения: гомо-, диблок-, триблок-сополимеры, мультиблоки, градиентные сополимеры. Основным мономером, обеспечивающим контролируемый синтез сополимеров различного строения на основе акрилонитрила, является стирол и его производные; полимеризацию проводят при температурах выше 100°С.
Использование радикальной полимеризации с переносом атома для синтеза сополимеров АН предложено в патентах [313–315]. Особенностью метода является использование соединений переходных металлов, что не позволяет применять их напрямую для полимеризации виниловых кислот и амидов. Стоит отметить, что в указанных патентах нет информации об очистке полимеров от катализатора и свойствах ПАН-прекурсора или углеродного волокна на его основе. Так, описана сополимеризация акрилонитрила, метилакрилата и диметилитаконата в растворе ДМСО при 60–70°С с использованием инициатора – четыреххлористого углерода и сложной каталитической системы [313, 314]. Каталитическая система состояла из катализатора – бромида меди(I) и двух лигандов – трис-(2-пиридилметил)амина и трис-[2-(диметиламино)этил]амина, а также активирующего агента глюкозы. Такой способ позволяет получить сополимер с Mn > 60 × 103 и узким ММР (Ð < 1.5).
Разработан метод сополимеризации акрилонитрила со стиролом (или его производными) с переносом атома при 60–90°С в среде этиленкарбоната, пропиленкарбоната, ДМСО, ДМФА или их смеси [315]. Катализатором реакции полимеризации является соль меди с органическим лигандом (2,2-бипиридином или его производным). На первой стадии синтезируют макроинициатор на основе сополимера акрилонитрила с виниловым сомономером в течение 0.1–20 ч. Содержание акрилонитрила в макроинициаторе должно составлять от 10 до 80 мол. %, а Mw = (0.5–50) × 103. Если принять массовое содержание акрилонитрила и его сомономеров за 100 мас. ч., то содержание макроинициатора в системе должно составлять 0.5–10 мас. ч., а растворителя 500–2000 мас. ч. По заявлению авторов, этот способ позволяет получать сополимеры акрилонитрила и стирола, растворы которых обладают низкой вязкостью из-за подавления структуризации (гелеобразования).
Оказался востребованным в получении ПАН-прекурсора и метод ОПЦ-полимеризации [307, 316, 317]. Так, описана полимеризация АН в среде ДМСО, ДМФА или этиленкарбоната в присутствии тритиокарбонатов в условиях радиационного излучения при комнатной температуре [316]. Из приведенных растворителей этиленкарбонат наиболее безопасен для окружающей среды и одновременно обеспечивает хороший контроль полимеризации. Процесс состоит из двух стадий – на первом этапе длительностью от 1 до 10 ч получают олигомерный ОПЦ-агент с Mw = (1.3–3.9) × 103. На втором этапе, продолжительность которого также составляет от 1 до 10 ч, проводят полимеризацию АН под действием этого агента. Условия синтеза выбирают таким образом, чтобы конечный ПАН характеризовался Mw < 55 × 103 и Ð < 1.5.
Более широкий спектр растворителей использован в патенте [317]. ОПЦ-сополимеризацию акрилонитрила с виниловыми кислотами, виниловыми сложными эфирами, виниловыми амидами и имидазолами в присутствии тритиокарбонатов авторы осуществляли в среде не только органического растворителя, но и водных растворов хлорида цинка или тиоцианата натрия. Полимеризацию инициируют органической перекисью или азосоединением при 40–85°C. Предпочтительное содержание растворителя в системе составляет 72–95 мас. %; доля АН в мономерной смеси – более 90 мас. %, содержание инициатора и ОПЦ-агента – менее 1 мас. % в расчете на мономер. Получаемые по этому способу сополимеры имеют Mw = (60–500) × 103 и Ð < 2.
В патенте [318] описан способ получения (со)полимеров АН с участием дитиобензоатов и тритиокарбонатов, в котором обозначены концентрационные условия для получения полимеров заданной ММ. Так, мольное соотношение концентраций сомономеров и ОПЦ-агента должно быть не менее 1 : 1, АН и ОПЦ-агента – не менее 400 : 1 и ОПЦ-агента и инициатора от 5 : 1 до 10 : 1. Такие условия обеспечивают высокую ММ (не менее 200 × 103) и узкое ММР сополимеров (Ð < 1.3). Авторы полагают, что полученные таким способ ПАН-прекурсоры позволяют существенно повысить физико-механические свойства (модуль упругости и прочность на разрыв) углеродных волокон, полученных на их основе, по сравнению с углеродными волокнами, полученными на основе ПАН-прекурсоров с меньшей молекулярной массой и/или более широким ММР.
Разработан способ синтеза сополимера АН в сверхкритическом CO2 в присутствии дибензилтритиокарбоната [307]. Гетерофазный характер полимеризации не оказывает какого-либо влияния на дисперсию продукта при полимеризации в среде диоксида углерода, т.е. не позволяет контролировать ММР.
Анионная полимеризация и полимеризация с переносом группы
Способы получения сополимеров акрилонитрила, макромолекулы которых образованы по анионному механизму приведены в патентах [319–321].
Разработана новая инициирующая система анионного типа, которая, по утверждениям авторов, не вносит в полимеры трудноудаляемые примеси [319, 320]. Авторы проводили сополимеризацию АН с кислородсодержащими мономерами в ДМСО, ТГФ или их смесях при температуре от –20 до +80°C. Инициирующая система не включает в себя атомы металлов и содержит только атомы С, Н, N и O, например окись этилена – 2,2-диазабициклооктан или смесь окиси пропилена и 2,2-диазабициклооктана. Предложенный способ позволяет получать олигомеры и разветвленные полимеры в широком диапазоне ММ: Mn = = (6–400) × 103 и Ð = 1.2–3.5.
Традиционные металлсодержащие анионные инициаторы применяли в работе [321] для синтеза сверхвысокомолекулярного волокнообразующего ПАН. Полимеризацию АН проводили в ДМФА в присутствии низкой концентрации 1,2-бис-диэтиламино-2-оксоэтанолата лития. Исследователи продемонстрировали возможность осуществления полимеризации при высокой концентрации мономера (до 2.8 моль/л) в условиях умеренно низких температур (до –20°С). При этом удалось максимально ограничить протекание побочных реакций и избежать процессов циклизации и гелеобразования. Средневязкостная молекулярная масса полученных гомополимеров акрилонитрила составляла (570–840) × 103.
Наконец, в работе [322] использовали полимеризацию с переносом группы для синтеза ПАН. Полимеризацию проводили в среде ДМФА, ДМАА или ДМСО при температуре – 20…–60°C под действием катализатора – фторида тетрабутиламмония и инициатора – триметилцианосилана (или 1-метокси-1-(триметилсилокси)-2-метил-1-пропена). Данный метод позволяет получать стереорегулярные полимеры акрилонитрила с Mw = (50–300) × 103 и Ð = 1.2–1.5.
ЗАКЛЮЧЕНИЕ
Рассмотренные в обзоре современные подходы к синтезу (со)полимеров АН, предназначенных для использования в качестве прекурсоров углеродного волокна, позволяют высказать некоторые соображения о перспективах их промышленного использования.
Первый вопрос, который возникает: какие требования должны предъявляться к сополимерам АН? Анализ литературы показывает, что свойства ПАН-прекурсора определяются его химическим составом, ММ и ММР и средней длиной последовательности звеньев АН. С точки зрения синтеза сополимеров АН с заданной молекулярной структурой наиболее перспективным вариантом является ОПЦ-полимеризация. Она толерантна к функциональным группам мономеров, ее можно использовать в гомогенной и гетерофазной полимеризации, она решает проблему контроля ММР и композиционной неоднородности сополимеров. Широкий спектр ММ образующихся сополимеров от десятков до сотен тысяч позволяет предложить ОПЦ-процесс для получения ПАН-прекурсоров, формуемых как из раствора, так и из расплава. Первое подтверждается данными анализа патентной литературы и описанным получением углеродных волокон на основе ОПЦ-сополимеров. Второе направление пока не реализовано на практике. Применение других вариантов контролируемой радикальной или анионной полимеризации на сегодняшний день не ушло дальше синтеза сополимеров и исследования их термического поведения или реологии растворов.
Применение “зеленых” сред может, на наш взгляд, представлять интерес для создания ПАН-прекурсора, формуемого из расплава.
Например, при сополимеризации АН в сверхкритическом СО2 сополимер может адсорбировать некоторое количество СО2, который способен пластифицировать полимеры. Соответственно будет понижаться температура стеклования и облегчаться переход сополимера в вязкотекучее состояние. Кроме того, после завершения реакции СО2 легко удаляется и конечный полимерный продукт не требует очистки. Традиционно в сверхкритическом СО2 получают сополимеры высокой ММ с широким ММР, но применение меркаптанов в качестве регуляторов ММ в этом случае могло бы решить данную проблему.
Перспективным может быть сочетание полимеризации с переносом атома с использованием ионных жидкостей. Тогда выигрышным является традиционное для таких систем образование сополимеров с относительно невысокой ММ и возможность более легкого отделения катализатора от полимера за счет высокой растворимости первого в ионной жидкости.
Актуальны работы по поиску новых недорогих сомономеров, которые позволяют регулировать реологию растворов/расплавов сополимеров и их циклизацию и термоокислительную стабилизацию.
Наконец, представляют интерес исследования по контролю распределения сомономеров в цепи ПАН, которое наряду с другими факторами определяет дефектность стабилизированных и карбонизованных волокон. Решением этой задачи может быть сополимеризация с постоянной или регулируемой скоростью введения мономеров в сополимеризацию в сочетании с механизмом обратимой деактивации цепи.
В целом известные из открытых источников способы получения углеродных волокон на основе ПАН-прекурсора относятся к сегменту среднепрочных и среднемодульных волокон. Можно надеяться на то, что новые синтетические подходы, развитые в последние десятилетия, позволяющие осуществлять контроль молекулярной структуры полимеров и минимизировать дефекты в их строении, откроют новые возможности для создания углеродных волокон с повышенными прочностными характеристиками.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-17004-мк).
Список литературы
Roberts T. // Materials Technology Publications. Watford, UK, 2006. P. 10, 93.
http://cs2.toray.co.jp/news/toray/en/newsrrs02.nsf/ 0/B126D9D8433675EE49257D11001770C5.
Houtz R.C. Pat. 2404727A USA. 1944.
Knudsen J.P., Ucci P.A. Pat. 3088793A USA. 1956.
Mackenzie H.D., Reeder F. Pat. 944,217 Britain. 1963.
Schmidt W.G. Pat. 2923694A USA. 1955.
Shindo A. Pat. 28287 Japan. 1959.
Carbon Fibers and Their Composites / Ed. by P. Morgan. New York: Taylor and Francis, 2005.
Chand S. // J. Mater. Sci. 2000. V. 5. P. 1303.
Carbon Fiber Composites / Ed. by D.L. Chung. Newton: Butterworth-Heinemann, 1994.
Liu J., He L., Ma S., Liang J., Zhao Y., Fong H. // Polymer. 2015. V. 61. P. 20.
Dalton S., Heatley F., Budd P.M. // Polymer. 1999. V. 40. P. 5531.
Gupta A., Harrison I.R. // Carbon. 1997. V. 35. P. 809.
Dalton S., Heatley F., Budd P.M. // Polymer. 1999. V. 40. P. 5531.
Ju A., Guang S., Xu H. // Carbon. 2013. V. 54. P. 323.
Huang X. // Materials. 2009. V. 2. P. 2369.
Edie D.D. // Carbon. 1998. V. 36. P. 345.
Bajaj P., Paliwal D.K., Gupta A.K. // J. Appl. Polym. Sci. 1993. V. 49. P. 823.
Bansal R.C., Donnet J.B. // Compr. Polym. Sci. 1990. V. 6. P. 501.
Ouyang Q., Cheng L., Wang H., Li K. // Polym. Degrad. Stab. 2008. V. 93. P. 1415.
Beltz L.A., Gustafson R.R. // Carbon. 1996. V. 34. P. 561.
Bhanu V., Rangarajan P., Wiles K., Bortner M., Sankarpandian M., Godshall D., Wilkes G. // Polymer. 2002. V. 43. P. 4841.
Tsai J.S., Lin C.H. // J. Appl. Polym. Sci. 1991. V. 43. P. 679.
Bajaj P., Roopanwal A. K. // J. Macromol. Sci., Polym. Revs. 1997. V. 37. P. 97.
Rwei S.P., Way T.F., Hsu Y.S. // Polym. Degrad. Stab. 2013. V. 98. P. 2072.
Siti N.A.M.J., Rusli D., Ishak A. // Int. J. Chem. Eng. Appl. 2012. V. 3. № 6.
Siti N.A.M.J., Rusli D., Ishak A. // Materials. 2014. V. 7. P. 6207.
Min B.G., Son T.W., Kim B.C., Jo W.H. // Polym. J. 1992. V. 24. P. 841.
Krishnan G.S., Burkanudeen A., Murali N., Phadnis H. // eXPRESS Polym. Lett. 2012. V. 6. № 9. P. 729.
Ju A.-Q., Li M.-J., Luo M., Ge M.-Q. // Chinese Chem. Lett. 2014. V. 25. № 9. P. 1275.
Henrici-Olive G., Olive S. // Adv. Polym. Sci. 1980. V. 32. P. 123.
Tsai J.S., Lin C.H. // J. Mater. Sci. 1991. V. 26. P. 3996.
Fitzer E., Frohs W., Heine M. // Carbon. 1986. V. 24. P. 387.
Wangxi Z., Jie L., Gang W. // Carbon. 2003. V. 41. P. 2805.
Bansal R.C., Donnet J.B. // Compr. Polym. Sci. 1990. V. 6. P. 501.
Saha B., Schatz G.C. // J. Phys. Chem. B. 2012. V. 116. P. 4684.
Tsai J.S., Lin C.H. // J. Mater. Sci. 1991. V. 26. P. 3996.
Thomas W.M. // Adv. Polym. Sci. 1961. V. 2. P. 401.
Garcia-Rubio L.H., Hamielec A.E. // J. Appl. Polym. Sci. 1979. V. 23. № 5. P. 1397.
Борт Д.Н., Зверева Г.Ф., Кучанов С.И. // Высокомолек. соед. А. 1978. Т. 20. № 8. С. 1827.
Srinivasan N.T., Santappa M. // Makromol. Chem. 1958. B. 26. № 1. S. 80.
Izumi Z. // J. Polym. Sci., Polym. Chem. 1967. V. 5. № 3. P. 469.
Edwin Moore D., Gustav Parts A. // Die Makromol. Chem. 1960. V. 37. № 1. P. 108.
Гольдфейн М.Д. // Высокомолек. соед. А. 1990. Т. 32. № 11. С. 2243.
Mathakiya I., Vangani V., Rakshit A.K. // J. Appl. Polym. Sci. 1998. V. 69. № 2. P. 217.
Wiles K.B., Bhanu V.A., Pasquale A.J., Long T.E., McGrath J.E. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 12. P. 2994.
Hou C., Ying L., Wang C. // J. Mater. Sci. 2005. V. 40. № 3. P. 609.
Hou C., Sun C., Ying L., Wang C. // J. Appl. Polym. Sci. 2005. V. 96. P. 483.
Bisht H.S., Chatterjee A.K. // J. Macromol. Sci. C. 2001. V. 4. № 3. P. 139.
Shavit N., Konigsbuch M., Oplatka A. Pat. 3345350A USA. 1963.
Shaffer K.A. // Trends Polym. Sci. 1995. V. 3. P. 146.
Canelas D.A // Adv. Polym. Sci. 1997. V. 133. P. 103.
Kendall J.L. // Chem. Rev. 1999. V. 99. P. 543.
Vygodskii Ya.S., Mel’nik O.A., Lozinskaya E.I., Shaplov A.S. // Polymer Science A. 2005. V. 47. № 3–4. P. 122.
Span R., Wagner W. // J. Phys. Chem. 1996. V. 25. P. 1509.
Supercritical Fluid Extraction /Ed. by M. McHugh, Newton: Butterworth-Heinemann, 1994.
Knox D.E. // Pure Appl. Chem. 2005. V. 77. P. 513.
Zetterlund P.B., Aldabbagh F., Okubo M. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 3711.
Shiho H. // Macromolecules. 2000. V. 33. P. 1565.
Yang Y., Dong Q., Wang Z. // J. Appl. Polym. Sci. 2006. V. 102. P. 5640.
Wang Z., Yang Y.J., Dong Q. // Polymer. 2006. V. 47. P. 7670.
Teng X.-R. // J. Appl. Polym. Sci. 2003. V. 87. P. 1393.
Teng X.-R., Shao H.-L., Hu X.-C. // J. Appl. Polym. Sci. 2002. V. 86. P. 2338.
Okubo M., Fujii S., Maenaka H, Minami H. //Colloid Polym. Sci. 2003. V. 281. P. 964.
Teng X.-R., Hu X.-C., Shao H.-L. // Polym. J. 2002. V. 34. № 7. P. 534.
Yeo S., Kiran E. // Macromolecules. 2004. V. 37. P. 8239.
Shlyahtin A.V., Nifant’ev I.E., Bagrov V.V., Lemenovskii D.A., Tavtorkin A.N., Timashev P.S. // Green Chem. 2014. V. 16. P. 1344.
Kermagoret A., Chau N.D.Q., Grignard B., Cordella D., Debuigne A., Jérôme C., Detrembleur C. // Macromol. Rapid Commun. 2016. V. 37. P. 539.
Harrison M., MacKenzie S., Haddleton D.M. // Chem. Commun. 2002. P. 2850.
Kubisa P. // Progr. Polym. Sci. 2009. V. 34. № 12. P. 1333.
Ionic Liquids: Applications and Perspectives / Ed. by A. Kokorin. InTech, 2011.
Шаплов А.С. Дис. … канд. хим. наук. М.: ИНЭОС РАН, 2005.
Cheng L., Zhang Y., Zhao T. // Macromol. Symp. 2004. V. 216. P. 9.
Hou C., Qu R., Ji C., Sun C., Wang C. // J. Polym. Sci., Polym. Chem. 2008. V. 46. № 8. P. 2701.
Hou C., Qu R., Ji C., Sun C., Wang C. // Polymer. 2008. V. 49. P. 3424.
Chen H., Meng Y., Liang Y., Lu Z., Lu P. // J. Mater. Res. 2009. V. 24. P. 1880.
Chen H., Liang Y., Liu D., Tan Z. // Mater. Sci. Eng. C. 2010. V. 30. № 4. P. 605.
Wang G.-X., Lu M., Hou Z.-H., Wu H. // J. Polym. Res. 2013. V. 20. P. 80.
Szwarc M. // Nature. 1956. V. 178. № 4543. P. 1168.
Hirao A., Goseki R., Ishizone T. // Macromolecules. 2014. V. 47. № 6. P. 1883.
Higginson W.C., Wooding N.S. // J. Chem. Soc. 1952. P. 760.
Foster F.C. // J. Am. Chem. Soc. 1952. V. 74. P. 2299.
Imoto M., Kinoshita M. // J. Chem. Soc. Japan, Industr. Chem. Sect. 1958. V. 61. P. 452.
Frankel M., Ottolenghi A., Albeck M., Zilkha A. // J. Chem. Soc. 1959. P. 3858.
Zilkha A., Feit B.A., Frankel M. // J. Chem. Soc. 1959. P. 928.
Zilkha A., Feit B.A., Frankel M. // J. Polym. Sci. 1961. V. 49. P. 231.
Zahn A., Schäfer P. // Makromol. Chem. 1959. B. 30. S. 225.
Overberger C.G., Juki H., Urakawa N. // J. Polym. Sci. 1960. V. 45. P. 127.
Cundall R.B., Eley D.D., Worrall J. // J. Polym. Sci. 1962. V. 58. P. 869.
Аскаров М.А., Трубицына С.Н. // Высокомолек. cоед. 1963. Т. 5. № 8. С. 1235.
Ерусалимский Б.Л., Кулевская И.В. // Высокомолек. cоед. 1965. Т. 7. № 1. С. 184.
Берлин А.А., Кочетов Е.В., Ениколопян Н.С. // Высокомолек. cоед. 1965. Т. 7. № 1. С. 187.
Панайотов И.М., Обрешков А.Т. // Высокомолек. cоед. 1965. Т. 7. № 2. С. 366.
Кочетов Е.В., Берлин А.А., Ениколопян Н.С. // Высокомолек. cоед. 1966. Т. 8. № 6. С. 1018.
Красулина В.Н., Новоселова А.В., Орлова Г.А. // Высокомолек. cоед. А. 1970. Т. 12. № 5. С. 1029.
Кочетов Е.В., Берлин А.А., Масальская Е.М., Ениколопян Н.С. // Высокомолек. cоед. А. 1970. Т. 12. № 5. С. 1118.
Кубасова Н.А., Динь Д.С., Гейдерих М.А., Шишкина М.В. // Высокомолек. cоед. А. 1971. Т. 13. № 1. С. 162.
Коротков А.А., Красулина В.Н., Новоселова А.В. // Высокомолек. cоед. А. 1971. Т. 13. № 12. С. 2661.
Matsuzaki K., Uryu T., Okada M., Shiroki H. // J. Polym. Sci. A-1. 1968. V. 6. P. 1475.
Inoue Y., Nishioka A. // Polym. J. 1972. V. 3. P. 149.
Brandy C., Gervaris D., Vairon J.P. // Eur. Polym. J. 1984. V. 20. P. 619.
Raynal S. // Eur. Polym. J. 1986. V. 22. P. 559.
Hirofumi O., Kunio H., Kenji K. // Polym. J. 1993. V. 25. № 3. P. 245.
Bach H.C., Knorr R.S. // Encyclopedia of Polymer Science and Engineering. New York: Wiley, 1985. V. 1. P 339.
Kamide K., Ono H., Hisatani K. // Polym. J. 1992. V. 24. № 9. P. 917.
Zhang F., Xu Z.-X., Jin R.-G. // Acta Polymerica Sinica. 2008. V. 1. № 11. P. 1102.
Krishnan G.S., Burkanudeen A., Murali N., Phadnis H. // eXPRESS Polym. Lett. 2012. V. 6. № 9. P. 729.
Shi X., Jiang J. // Chinese Chem. Lett. 2019. V. 30. № 2. P. 473.
Shi X., Jiang J. // Polymer Science B. 2019. V. 61. № 6. P. 511.
Cao Y., Li J.M., Wang Y.R., Yao Y.M., Shen Q. // Chinese Sci. Bull. 2011. V. 56. № 26. P. 2780.
Miertuš S., Kysel’ O., Májek P. // Chem. zvesti. 1979. V. 33. № 2. P. 167.
Ikeda I., Mitsumoto S., Yamada Y., Suzuki K. // Polym. Int. 1991. V. 26. P. 115.
Estrin Y.I., Tarasov A.E., Grishchuk A.A., Chernyak A.V., Badamshina E.R. // RSC Adv. 2016. V. 6. P. 106064.
Estrin Y.I., Grishchuk A.A., Tarasov A.E., Perepelitsina E.O., Badamshina E.R. // Polymer Science B. 2016. V. 58. № 1. P. 19.
Reetz M.T., Knauf T. // Pat. 5 494 983 USA. 1996.
Zhang T., Jiang L., Sun W., Shen Z., Ma S. // Eur. Polym. J. 2002. V. 38. P. 2329.
Jaaks V., Eisenbach C.D., Kern W. // Makromol. Chem. 1972. B. 161. S. 139.
Ogawa T., Quintana P. // J. Polym. Sci., Polym. Chem. 1975. V. 13. P. 2517.
Ogawa T., Romero J. // Eur. Polym. J. 1975. V. 13. P. 419.
Schaper F., Foley S.R., Jordan R.F. // J. Am. Chem. Soc. 2004. V. 126. P. 2114.
Terán N., Estrada M.R., Ogawa T. // J. Macromol. Sci., Pure Appl. Chem. 2005. V. 42. P. 1679.
Каплан А.М., Стояченко И.Л., Голубев В.Б., Гольданский В.И. // Высокомолек. соед. Б. 1975. Т. 17. № 2. С. 259.
Qiu J., Charleux B., Matyjaszewski K. // Polimery. 2001. V. 46. P. 453.
Fundamentals of Controlled/Living Radical Polymerization (Polymer Chemistry Series) / Ed. by N.V. Tsarevsky. Oxford: Royal Society of Chemistry, 2013.
Bisht H.S., Chatterjee A.K. // J. Macromol. Sci., Polym. Revs. 2001. V. 41. № 3. P. 139.
Braunecker W.A., Matyjaszewski K. // Progr. Polym. Sci. 2007. V. 32. № 1. P. 93.
Matyjaszewski K., Xia J. // Chem. Revs. 2001. V. 101. I. 9. P. 2921.
Moad G., Rizzardo E., Thang S.H. // Austral. J. Chem. 2009. V. 62. № 11. P. 1402.
Handbook of Radical Polymerization / Ed by K. Matyjaszewski, T.P. Davis. Hoboken: Wiley-Interscience, 2002.
Fukuda T., Terauchi T., Goto A., Ohno K. // Macromolecules. 1996. V. 29. P. 6393.
Tharanikkarasu K., Radharishnan G.J. // Polym. Sci., Polym. Chem. Ed. 1996. V. 34. P. 1723.
Tharanikkarasu K., Sundar S., Thankam C.V., Radhakrishnan G. // J. Polym. Mater. 1998. V. 15. I. 3. P. 243.
Otsu T. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 12. P. 2121.
Fukuda T., Terauchi T., Goto A., Ohno K., Tsujii Y., Miyamoto T. // Macromolecules. 1996. V. 29. I. 20. P. 6393.
Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science / Ed. by D. Gigmes. Oxford: Royal Society of Chemistry, 2015.
Grishin D.F. // Polymer Science A. 2008. V. 50. № 3. P. 221.
Benoit D., Chaplinski V., Braslau R., Hawker C.J. // J. Am. Chem. Soc. 1999. V. 121. P. 3904.
Brinkmann-Rengel S., Niessner N. // ACS Symp. Ser. 2000. V. 768. P. 394.
Megiel E., Kaim A. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 1165.
Detrembleur C., Sciannamea V., Koulic C., Claes M., Hoebeke M., Jérôme R. // Macromolecules. 2002. V. 35. P. 7214.
Nicolas J., Brusseau S., Charleux B. // J. Polym. Sci., Polym. Chem. 2010. V. 48. P. 34.
Zaremski M.Yu., Plutalova A.V., Eremeev I. // Macromol. Theory Simul. 2016 V. 25. P. 413.
Zaremski M., Eremeev I., Garina E., Borisova O., Korolev B. // J. Polym. Res. 2017. V. 24. P. 151.
Wang J.-S., Matyjaszewski, K. // J. Am. Chem. Soc. 1995. V. 117. P. 5614.
Jakubowski W., Matyjaszewski K. // Macromolecules. 2005. V. 38. № 10. P. 4139.
Jakubowski W., Min K., Matyjaszewski K. // Macromolecules. 2006. V. 39. P. 39.
Percec V., Guliashvili T., Ladislaw J.S., Wistrand A., Stjerndahl A., Sienkowska M.J., Sahoo S. // J. Am. Chem. Soc. 2006. V. 128. № 43. P. 14156.
Tang W., Matyjaszewski K. // Macromolecules. 2006. V. 39. P. 4953.
Treat N.J., Sprafke H., Kramer J.W., Clark P.G., Barton B.E., Read de Alaniz J., Hawker C.J. // J. Am. Chem. Soc. 2014. V. 136. № 45. P. 16096.
Konkolewicz D., Schroder K., Buback J., Bernhard S., Matyjaszewski K. // ACS Macro Lett. 2012. V. 1. P. 1219.
Гришин Д.Ф., Гришин И.Д. // Успехи химии. 2015. Т. 84. № 7. С. 712.
Grishin D.F., Grishin I.D. // Fibre Chem. 2019. V. 50. № 6. P. 514.
Matyjaszewski K., Jo S.M., Paik H., Gaynor S.G. // Macromolecules. 1997. V. 30. № 20. P. 6398.
Matyjaszewski K., Jo S.M., Paik H., Shipp D.A. // Macromolecules. 1999. V. 32. № 20. P. 6431.
Lazzari M., Chiantore O., Mendichi R., López-Quintela M.A. // Macromol. Chem. Phys. 2005. V. 206. № 14. P. 1382.
Dong H., Tang W., Hou C., Liu J.S., Wang C. // Polym. Int. 2006. V. 55. № 2. P. 171.
Ho C., Qu R., Ying L., Wang C.W. // J. Appl. Polym. Sci. 2006. V. 99. P. 32.
Hou C., Qu R.J., Ji C.N., Wang C.W. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 219.
Matyjaszewski K. // Macromolecules. 2007. V. 40. P. 2974.
Jiang J., Lu X., Lu Y.J. // J. Appl. Polym. Sci. 2010. V. 116. P. 2610.
Pan X., Lamson M., Yan J., Matyjaszewski K. // ACS Macro Lett. 2015. V. 4. P. 192.
Liang E.-X., Zhong M., Hou Z.-H., Huang Y., He B.-H., Wang G.-X., Liu L.-C., Wu H. // J. Macromol. Sci., Pure Appl. Chem. 2016. V. 53. № 4. P. 210.
Li S., Wang Y., Ma L., Zhang X., Dong S., Liu L., Zhou X., Wang C., Shi Z. // Designed Monomers Polymers. 2019. V. 22. № 1. P. 180.
Huang Z., Chen J., Zhang L., Cheng Z., Zhu X. // Polymers. 2016. V. 8. P. 59.
Pitto V., Voit B.I., Loontjens T.J.A., van Benthem R.A.T.M. // Macromol. Chem. Phys. 2004. V. 205. P. 2346.
Wang G., Wang Z., Lee B., Yuan R., Lu Z., Yan J., Pan X., Song Y., Bockstaller M.R., Matyjaszewski K. // Polymer. 2017. V. 129. P. 57.
Tsarevsky N.V., Matyjaszewski K. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 5098.
Tsarevsky N.V., Matyjaszewski K. // Chem. Rev. 2007. V. 107. P. 2270.
Hou C., Qu R., Ji C., Sun C., Wang C. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 2701.
Hou C., Qu R., Sun C., Ji C., Wang C., Ying L., Jiang N., Xiu F., Chen L. // Polymer. 2008. V. 49. P. 3424.
Wang G.-X., Lu M., Hou Z.-H., Wu H. // J. Polym. Res. 2013. V. 20. P. 80.
Lee K.C., Gan L.M., Chew C.H., Ng S.C. // Polymer. 1995. V. 36. P. 3719.
Chen H., Liang Y., Liu D., Tan Z., Zhang S., Zheng M., Qu R. // Mater. Sci. Eng. C. 2016. V. 30. P. 605.
Ma J., Chen H., Zhang M., Yu M. // J. Polym. Sci., Polym. Chem. 2012. V. 50. P. 609.
Chiefari J., Chong Y.K., Ercole F., Krstina J., Jeffery J., Le T.P.T., Mayadunne R.T.A., Meijs G.F., Moad C.L., Moad G., Rizzardo E., Thang S.H. // Macromolecules. 1998. V. 31. I. 16. P. 5559.
You Y.Z., Hong C.Y., Bai R.K., Pan C.Y., Wang J. // Macromol. Chem. Phys. 2002. V. 203. P. 477.
Millard P.-E., Barner L., Stenzel M.H., Davis T.P., Barner-Kowollik C., Müller A.H.E. // Macromol. Rapid Commun. 2006. V. 27. I. 11. P. 821.
Cai J.Y., McDonnell J., Brackley C., O’Brien L., Church J.S., Millington K., Phair-Sorensen N. // Materials Today Communications. 2016. V. 9. P. 22.
Tang C., Kowalewski T., Matyjaszewski K. // Macromolecules. 2003. V. 36. № 23. P. 8587.
Moad G., Rizzardo E., Thang S.H. // Aust. J. Chem. 2012. V. 65. P. 985.
An Q., Qian J., Yu L., Luo Y., Liu X. // J. Polym. Sci., Polym. Chem. 2005. V. 43. P. 1973.
Liu X.-H., Zhang G.-B., Lu X.-F., Liu J.-Y., Pan D., Li Y.-S. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 490.
Liu X.-H., Li Y.-G., Lin Y., Li Y.-S. // J. Polym. Sci., Polym. Chem. 2007. V. 45. P. 1272.
Liu X.-H., Zhang G.-B., Li B.-X., Bai Y.-G. // Eur. Polym. J. 2008. V. 44. P. 1200.
Aqil A., Detrembleur C., Gilbert B., Jerome R. // Chem. Mater. 2007. V. 19. P. 2150.
Niu S., Zhang L., Zhu J., Zhang W., Cheng Z., Zhu X. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 1197.
Li J., Ding C., Zhang Z., Zhu J., Zhu X. // React. Funct. Polym. 2017. V. 113. P. 1.
He J.-Y., Lu M. // J. Macromol. Sci., Pure Appl. Chem. 2019. V. 56. № 5. P. 443
Huang Z., Zhang L., Cheng Z., Zhu X. // Polymers. 2017. V. 9. P. 4.
Zhang S., Yin L., Wang J., Zhang W., Zhang L., Zhu X. // Polymers. 2017. V. 9. P. 26.
Uyar Z., Turgut F., Arslan U., Durgun M., Degirmenci M. // Eur. Polym. J. 2019. V. 119. P. 102.
Kopeć M., Krys P., Yuan R., Matyjaszewski K. // Macromolecules. 2016. V. 49. № 16. P. 5877.
Кабанов В.А., Зубов В.П., Семчиков Ю.Д. Комплексно-радикальная полимеризация. М.: Букинист, 1987.
Kaiser A., Brandau S., Klimpel M., Barner-Kowollik C. // Macromol. Rapid Commun. 2010. V. 31. P. 1616.
Dürr C.J., Emmerling S.G.J., Kaiser A., Brandau S., Habicht A.K.T., Klimpel M., Barner-Kowollik C. // J. Polym. Sci., Polym. Chem. 2012. V. 50. P. 174.
Dürr C.J., Emmerling S.G.J., Lederhose P., Kaiser A., Brandau S., Klimpel M., Barner-Kowollik C. // Polym. Chem. 2012. V. 3. P. 1048.
Dürr C.J., Hlalele L., Kaiser A., Brandau S., Barner-Kowollik C. // Macromolecules. 2013. V. 46. P. 49.
Enríquez-Medrano F.J., Soriano-Corral F., Acuña-Vázquez P., Cabrera-Álvarez E.N., Saade-Caballero H., Castañeda-Facio A., López L.V., de León-Gómez R.D. // J. Mex. Chem. Soc. 2014. V. 58. № 2. P. 194.
Fan D., He J., Xu J., Tang W., Liu Y., Yang Y. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 2260.
Alam M.M., Peng H., Jack K.S., Hill D.J.T., Whittaker A.K. // J. Polym. Sci., Polym. Chem. 2017. V. 55. P. 919.
Chernikova E.V., Poteryaeva Z.A., Plutalova A.V. // Polymer Science B. 2014. V. 56. № 2. P. 109.
Jiang H., Zhang L., Qin J., Zhang W., Cheng Z., Zhu X. // J. Polym. Sci., Polym. Chem. 2012. V. 50. № 19. P. 4103.
Božović-Vukić J., Mañon H.T., Meuldijk J., Koning C., Klumperman B. // Macromolecules. 2007. V. 40. № 20. P. 7132.
Kim J.S., Jeon H.J., Lee K.M., Im J.N., Youk J.H. // Fibers Polymers. 2010. V. 11. № 2. P. 153.
Li A.-L., Wang Y., Liang H. Lu J. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 2376.
Wang W., Bai L., Chen H., Xu H., Tao Q. // Polym. Bull. 2018. V. 75. P. 4327.
Wan L.-S., Lei H., Ding Y., Fu L., Li J. Xu Z.-K. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 92.
Chernikova E.V., Poteryaeva Z.A., Belyaev S.S., Nifant’ev I.E., Shlyakhtin A.V., Kostina Yu.V., Cherevan’ A.S., Efimov M.N., Bondarenko G.N., Sivtsov E.V. // Polymer Science B. 2011. V. 53. № 7–8. P. 391.
Chernikova E.V., Poteryaeva Z.A., Belyaev S.S., Sivtsov E.V. // Russ. J. Appl. Chem. 2011. V. 84. P. 1031.
Spörl J.M., Ota A., Beyer R., Lehr T., Müller A., Hermanutz F., Buchmeiser M.R. // J. Polym. Sci., Polym. Chem. 2014. V. 52. P. 1322.
Spörl J., Hermanutz F., Buchmeiser M. // Int. Fiber J. 2014. V. 28. № 6. P. 24.
Chernikova E.V., Kishilov S.M., Plutalova A.V., Kostina Yu.V., Bondarenko G.N., Baskakkov A.A., Il’yin S.O., Nikolaev A.Yu. // Polymer Science B. 2014. V. 56. № 5. P. 553.
Moskowitz J.D., Abel B.A., McCormick C.L., Wiggins J.S. // J. Polym. Sci., Polym. Chem. 2016. V. 54. P. 553.
Cai J., McDonnell J.Y., Brackley C., O’Brien L. // Materials Today Commun. 2016. V. 9. P. 22.
Chernikova E.V., Toms R.V., Prokopov N.I., Duflot V.R., Plutalova A.V., Legkov S.A., Gomzyak V.I. // Polymer Science B. 2017. V. 59. № 1. P. 28.
Khayyam H., Jazar R.N., Nunna S., Golkarnarenji G., Badii K., Mousa Fakhrhoseini S., Naebe M. // Progr. Mater. Sci. 2020. V. 107. P. 100575.
Chernikova E.V., Kostina Yu.V., Efimov M.N., Prokopov N.I., Gerval’d A.Yu., Toms R.V., Nikolaev A.Yu., Shkirev M.D. // Polymer Science B. 2015. V. 59. № 2. P. 116.
Томс Р.В., Прокопов Н.И., Гервальд А.Ю., Черникова Е.В. // Пласт. массы. 2016. № 5–6. С. 21.
Черникова Е.В., Потеряева З.А., Беляев С.С., Сивцов Е.В. // Журн. прикл. химии. 2011. Т. 84. № 6. С. 1010.
Черникова Е.В., Потеряева З.А., Плуталова А.В., Баскаков А.А. // Пласт. массы. 2014. № 7–8. С. 33.
Toms R.V., Balashov M.S., Shaova A.A., Gerval’d A.Yu., Prokopov N.I., Plutalova A.V., Grebenkina N.A., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 2. P.
Hao J., Liu Y., Lu C. // Polym. Degrad. Stab. 2018. V. 147. P. 89.
Wang R., Luo Y., Li B., Sun X., Zhu S. // Macromol. Theory Simul. 2006. V. 15. P. 356.
Sun X., Luo Y., Wang R., Li B.-G., Liu B., Zhu S. // Macromolecules. 2007. V. 40. P. 849.
Sun X., Luo Y., Wang R., Li B.-G., Zhu S. // AIChE J. 2008. V. 54. P. 1073.
Li X., Liang S., Wang W.-J., Li B.-G., Luo Y., Zhu S. // Macromol. React. Eng. 2015. V. 9. P. 409.
Moskowitz J.D., Wiggins J.S. // Polymer. 2016. V. 84. P. 311.
Kaur J., Millington K., Cai J.Y. // J. Appl. Polym. Sci. 2016. V. 133. № 48. P. 44273.
Ilyin S.O., Baskakov A.A., Chernikova E.V., Kulichikhin V.G. // Russ. Chem. Bull., Int. Ed. 2017. V. 66. № 4. P. 711.
Skvortsov I.Yu., Toms R.V., Prokopov N.I., Chernikova E.V., Kulichikhin V.G. // Polymer Science A. 2018. V. 60. № 6. P. 793.
Gupta A.K., Paliwal D.K., Bajaj P. // J. Appl. Polym. Sci. 1998. V. 70. № 13. P. 2703.
Jamil S., Daik R., Ahmad I. // Materials. 2014. V. 7. № 9. P. 6207.
Bhanu V.A., Rangarajan P., Wiles K., Bortner M., Sankarpandian M., Godshall D., Glass T.E., Banthia A.K., Yang J., Wilkes G., Baird D., McGrath J.E. // Polymer. 2002. V. 43. P. 4841.
Rangarajan P., Yang J., Bhanu V., Godshall D., McGrath J., Wilkes G., Baird D. // J. Appl. Polym. Sci. 2002. V. 85. P. 69.
Bortner M.J., Bhanu V., McGrath J.E., Baird D.G. // J. Appl. Polym. Sci. 2004. V. 93. P. 2856.
Gao X.-Y., Han N., Zhang X.-X., Yu W.-Y. // J. Mater. Sci. 2009. V. 44. P. 5877
Curatolo B.S., Li G.S. Eur. pat. 0492909B1. 1990.
Weinstock H.H., Jr, Nesty G.A. Pat. 2692875A USA. 1949.
Yang J., Banthia A.K., Godshell D., Rangarajan P., Glass T.E., Wilkes G.L., Baird D.G., McGrath J.E. // Polym. Prepr. 2000. V. 41. P. 59.
Bhanu V.A., Wiles K.B., Banthia A.K., Mansuri A., Sankarpandian M., Rangarajan P., Glass T.E., Baird D.G., Wilkes G.L., McGrath J.E. // Polym. Prepr. 2001. V. 42. P. 663.
Rangarajan P., Bhanu V., Godshall D., Wilkes G., McGrath J., Baird D. // Polymer. 2002. V. 43. P. 2699.
Bortner M.J., Baird D.G. // Polymer. 2004. V. 45. P. 3399.
Wilding M.D., Baird D.G. // Polym. Eng. Sci. 2009. V. 49. P. 1990.
Gao X.-Y., Han N., Zhang X.-X., Yu W.-Y. // J. Mater. Sci. 2009. V. 44. P. 5877.
Han N., Zhang X.-X., Wang X.-C. // J. Appl. Polym. Sci. 2007. V. 103. P. 2776.
Peng W., Han N., Tang X., Liu H., Zhang X. //Adv. Mater. Res. 2011. V. 332–334. P. 339.
Rwei S.-P., Way T.-F., Chiang W.-Y., Tseng J.-C. // Textile Res. J. 2018. V. 88. № 13. P. 1479.
Lee J.H., Jin J.-U., Park S., Choi D., You N.-H., Chung Y., Ku B.-C., Yeo H. // J. Industr. Eng. Chem. 2018. V. 71. P. 112.
Paiva M.C., Kotasthane P., Edie D.D., Ogale A.A. // Carbon. 2003. V. 41. P. 1399.
Bullock R.E. // Fiber Sci. Technol. 1974. V. 7. № 2. P. 157.
Dietrich J., Hirt P., Herlinger H. // Eur. Polym. J. 1996. V. 32. № 5. P. 617.
Badawy S.M., Dessouki A.M. // J. Phys. Chem. B 2003. P. 107. V. 41. P. 11273
Czech Z. // Int. J. Adhesion Adhesives. 2007. V. 27. P. 195.
Naskar A.K., Walker R.A., Proulx S., Edie D.D., Ogale A.A. // Carbon. 2005. V. 43. P. 1065.
Mukundan T., Bhanu V.A., Wiles K.B., Johnson H., Bortner M., Baird D.G., Naskar A.K., Ogale A.A., Edie D.D., McGrath J.E. // Polymer. 2006. V. 47. P. 4163.
Wang Y.-Z., Wang S.-G., Liu J.-L. // Key Eng. Mat. 2014. V. 575–576. P. 151.
Walker R.A., Naskar A.K., Ogale A.A. // Plastics, Rubber and Composites. 2006. V. 35. № 6–7. P. 242.
Deng W., Lobovsky A., Iacono S.T., Wu T., Tomar N., Budy S.M., Long T., Hoffman W.P., Smith D.W. // Polymer. 2011. V. 52. P. 622.
Pekel N., Rzaev Z.M.O., Güven O. // Macromol. Chem. Phys. 2004. V. 205. P. 1088.
Batchelor B.L., Mahmood S.F., Jung M., Shin H., Kulikov O.V., Voit W., Novak B.M., Yang D.J. // Carbon. 2016. V. 98. P. 681.
Yang D.J., Batchelor B., Samsuddin Faisal Mahmood, Smith D.W., Wenjin Deng, Hyunkyu Shin, Minhye Jung. Pat. 10435821 USA. 2015.
Lowe A., Deng W., Smith D.W., Balkus K.J. // Macromolecules. 2012. V. 45. P. 5894.
König S., Clauss M.M., Giebel E., Buchmeiser M.R. // Polym. Chem. 2019. V. 10. P. 4469.
Kamide K., Hisatani K. // Polym. J. 1992. V. 24. № 12. P. 1377.
Engelbrecht L.A., Noordam A. Pat. 7858694 B2 USA. 2005.
Parker D.K., Feher F.J., Mahadevan V. Pat. 7592409 B2 USA. 2003.
Tsuji R., Hiiro T. Pat. 7094833 B2 USA. 2002.
Preti D., Rossi A.G., Nocci R., Vecchini N. Pat. 5807928 USA. 1996.
Pober K.W., Starks C.M. Pat. 5405916 USA. 1991.
Tazaki T., Kuramoto M. Pat. 5391671 USA. 1990.
Tagami T., Imai Y., Kakimoto M., Kiyohara O., Narushima H. Pat. 5342895 USA. 1990.
Bertsch R.J. Pat. 5312956 USA. 1991.
Arrighetti S., Brancaccio A., Cesca S., Giuliani G. Pat. 4145378 USA. 1976.
Fanta G.F., Stout E.I., Doane W.M. Pat. 4134863 USA. 1976.
Hradil J., Stamberg J., Coupek J. Pat. 4067825 USA. 1975.
Alberts H., Bartl H., Prinz R. Pat. 4056671 USA. 1975.
Howe K.L. Pat. 3963807A USA. 1972.
Pat. 101608005A China. 2008.
Пат. 2325733C1 Россия. 2007.
Eur. pat. EP1935915A1 2006.
Pat. 1911974B China. 2006.
Wenz E., Buchholz V., Eichenauer H., Wolf U, Born R.-J., Jansen U., Dietz W. Pat. 20030130433A1 USA. 2002.
Пат. 1562852A1 СССР 1987.
Kaaruhansu Z. Pat. 55050100 Japan. 1979.
Wenzel F., Bruemmer H.-D., Krieg M. Pat. 4241203A USA. 1978.
Jo W.H., Kim T., Hwang I.C., Kwon N.H. Pat. 20110201756A1 USA. 2010.
Parikh D.R., Rikhoff R.S., Harris L.D., Vanis V.V. Pat. 2011085202A1 France. 2011.
Pat. 1986635B China. 2006.
Пат. 2391356C1 Россия. 2008.
Пат. 2008111361A Россия. 2006.
Пат. 2009108986A Россия. 2009.
Пат. 98101121A Россия. 1995.
Пат. 2113444C1 Россия. 1990.
Пат. 2091403C1 Россия. 1994.
Пат. 2032695C1 Россия. 1992.
Пат. 2393173C2 Россия. 2008.
Пат. 2017865C1 Россия. 1992.
Plee D. Pat. 20100047153A1 USA. 2008.
Cerf M., Colombie D., N’Zudie T. Pat. 20040068069A1 USA. 2000.
Пат. 2422467C2 Россия. 2009.
Пат. 2084463C1 Россия. 1993.
Pat. 106589191B China. 2015.
Pat. 104231158B China. 2013.
Pat. 101161694A China. 2007.
Hirota N., Sinmen Y., Matsuyama N., Nii T., Shibatani H. Pat. 2010013014A Mexico. 2009.
Пат. 2560173C2 Россия. 2013.
Пат. 2528395C2 Россия. 2012.
Пат. 2550873C2 Россия. 2013.
Пат. 2393173C2 Россия. 2008.
Terent’ev A.O., Kutkin A.V., Platonov M.M., Ogibin Y.N., Nikishin G.I. // Tetrahedron Lett. 2003. V. 44. P. 7359.
Terent’ev A.O., Platonov M.M., Ogibin Yu.N., Nikishin G.I. // Synth. Commun. 2007. V. 37. № 8. P. 1281.
Caneba G., Dar Y. Pat. 20050250919A1 USA. 2002.
Пат. 2627264C1 Россия. 2016.
Пат. 2697882C1 Россия. 2018.
Aoki J., Okuda H., Ise M. Pat. 2014141608A Japan. 2002.
106749807B China. 2016.
Пат. 2016129965A Россия. 2014.
Yun J. Pat. 9777081B2 USA. 2014.
Пат. 2014108588A Россия. 2014.
Пат. 2565767C2 Россия. 2014.
Пат. 2376319C2 Россия. 2007.
106589193B China. 2016.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)