

Высокомолекулярные соединения (серия С), 2020, T. 62, № 1, стр. 66-80
Пэгилированные системы в фармацевтике
Н. Н. Порфирьева a, Р. И. Мустафин a, *, В. В. Хуторянский a, b, **
a Институт фармации, Казанский государственный медицинский университет
420126 Казань, ул. Фатыха Амирхана, 16, Россия
b Reading School of Pharmacy, University of Reading
PO box 224 Whiteknights, Reading RG66AD, United Kingdom
* E-mail: rouslan.moustafine@gmail.com
** E-mail: v.khutoryanskiy@reading.ac.uk
Поступила в редакцию 24.05.2019
После доработки 10.06.2019
Принята к публикации 24.06.2019
Аннотация
Обзор посвящен использованию полиэтиленгликоля и конъюгации с полиэтиленгликолем (ПЭГилирование) для создания новых лекарственных форм и модификации биомолекул. Рассмотрены особенности ПЭГилированных наночастиц, липосом, белков, ферментов, малых молекул лекарств и полиэлектролитов, а также их влияние на системную доставку лекарств, включая преодоление различных биологических барьеров и адгезию к слизистым тканям (мукоадгезию).
ВВЕДЕНИЕ
Одним из путей повышения эффективности фармакотерапии является усовершенствование способов доставки лекарственных веществ. Перспективным в этом направлении развития считается процесс модификации молекул различными полимерными соединениями. Так, в мировой практике накоплен опыт получения конъюгатов лекарств путем присоединения к ним инертной макромолекулы полиэтиленгликоля. Данные конъюгаты ПЭГ с лекарственными веществами обеспечивают не только лучшую переносимость, но также обладают улучшенным фармакокинетическим профилем, способствуют более глубокому проникновению молекул и обеспечивают дополнительную защиту от действия протеолитических ферментов. Безопасность использования ПЭГ при создании систем доставки лекарственных веществ подтверждена управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (US Food and Drug Administration, FDA) как субстанции, разрешенной к использованию в медицине (производство лекарственных препаратов), продуктах питания и косметологии.
В настоящем обзоре рассмотрены физико-химические свойства полиэтиленгликоля, способы его присоединения к различным материалам фармацевтического назначения (ПЭГилирование), особенности поведения ПЭГилированных материалов в биологических системах и их применение в фармацевтике.
СИНТЕЗ И СВОЙСТВА ПОЛИЭТИЛЕНГЛИКОЛЯ
Впервые полимеризация этиленоксида и получение полиэтиленгликоля в присутствии гидроксидов щелочных металлов или хлорида цинка были осуществлены Вюрцем в 1863 году [1]. Позже, в 1929 г. Штаудингер и Швейцер синтезировали ПЭГ с разной молекулярной массой для вискозиметрических исследований, а уже в 1930 году было налажено производство ПЭГ путем присоединения этиленоксида к этиленгликолю в присутствии оснований:
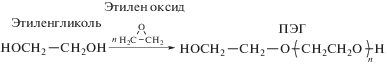
В настоящее время в промышленности ПЭГ синтезируют путем анионной полимеризации с раскрытием кольца этиленоксида, инициированной нуклеофильной атакой гидроксид-иона на эпоксидное кольцо [2]. Использование гликолей в качестве инициаторов является более предпочтительным, так как при таком способе возможно получение ПЭГ с низкой полидисперсностью (см. схему синтеза ПЭГ).
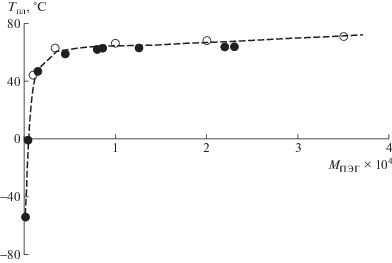
Полимеры окиси этилена представляют собой водорастворимые материалы, молекулярная масса которых может достигать значений 106. Полимеры с молекулярной массой до 4 × 104 принято называть полиэтиленгликолями, а более высокомолекулярные – полиэтиленоксидами [3, 4]. В зависимости от ММ полимеры окиси этилена могут представлять собой бесцветные жидкости, воскообразные либо твердые материалы. На рис. 1 представлена зависимость температуры плавления Тпл полиэтиленгликоля от его молекулярной массы. Зависимость построена по данным, взятым из работ [5, 6].
Полимеры окиси этилена растворимы во многих органических растворителях (бензоле, четыреххлористом углероде, хлороформе, диметилформамиде, ацетонитриле) и хорошо растворимы в воде, причем способность к растворению закономерно снижается с увеличением молекулярной массы [3, 7].
В водных растворах ПЭГ является типичным неионным полимером, для которого характерна способность к образованию различного рода комплексов с некоторыми веществами полимерной и неполимерной природы. Так, в слабокислых средах ПЭГ может образовывать интерполимерные комплексы с поликарбоновыми кислотами, например полиакриловой (ПАК) или полиметакриловой кислотой (ПМАК) посредством водородных связей [8–12]. Способность к формированию интерполимерных комплексов проявляется лишь выше критической молекулярной массы, составляющей ∼103 [9]. Известны также комплексы ПЭГ и ПЭО с малыми молекулами, например фенолами, ионами щелочных металлов, мочевиной и йодом [13]. При определенных соотношениях взаимодействующих компонентов возможно образование тройных комплексов, с участием двух полимеров и низкомолекулярных веществ, как это показано в работе [14] на примере комплексов ПМАК–ПЭО–аминазин.
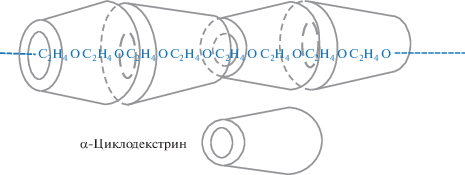
Интересной особенностью ПЭГ является его способность к образованию комплексов включения с циклодекстринами. Отсутствие боковых заместителей в цепи, а также сравнительно гидрофобная природа групп –CH2–СН2– позволяет его макромолекулам нанизывать многочисленные циклические молекулы циклодекстрина, приводя к формированию агрегатов и гелей. Ниже приведена структура комплексов, образующихся в смесях ПЭГ и α-циклодекстрина (перепечатана из работы [15] с разрешения “American Chemical Society”):
В литературе также имеются сведения об образовании гелей и многослойных пленок с участием ПЭГилированных коллоидных частиц на основе органосиликатов при вовлечении их в процессы комплексообразования с ПАК [16] и α-циклодекстрина [17].
Наличие в ПЭГ двух концевых гидроксильных групп позволяет создавать макромолекулы как разветвленной, так и линейной структуры. Возможно получение сшитых форм ПЭГ, образующих в воде гидрогели [18], а также синтез ПЭГ с различными функциональными группами (малеимиды, метакрилаты, акрилаты и другие) за счет модификации концевых гидроксильных групп [19]. Таким образом, благодаря структуре ПЭГ возможно получение и гетерофункциональных производных. Например, конъюгат ПЭГ, содержащий альдегидные и тиольные концевые группы, может быть создан путем полимеризации с 3,3-диэтокси-1-пропанолом, который формирует пропиональдегид после кислотного гидролиза; полимеризацию возможно остановить при помощи метансульфонилхлорида с превращением в этилдитиокарбонат и свободный тиол [2, 20, 21].
Высокое содержание атомов кислорода в макромолекулах ПЭГ позволяет им связываться с несколькими молекулами воды. Подобный эффект влечет за собой формирование “водного облака” вокруг модифицированных молекул, что значительно повышает их гидродинамический радиус [22].
ПЭГ является биосовместимым полимером с очень низкой иммуногенностью (способностью вызывать иммунный ответ), антигенностью (способностью связываться с антителами) и отсутствием токсичности. Он быстро и без структурных изменений выводится in vivo. Так, при М ∼ ∼ 2 × 104 ПЭГ преимущественно удаляется с мочой, а при большей молекулярной массе выводится вместе с мочой и калом [18]. Однако низкомолекулярные олигомеры распадаются in vivo под действием алкогольдегидрогеназы до токсичных метаболитов, ввиду чего в пищевых продуктах, косметике и фармацевтике используют ПЭГ с М > 103, безопасность которых подтверждена многочисленными исследованиями [23].
Первое упоминание об использовании ПЭГилированных систем доставки лекарственных веществ датируется 1977 годом, когда Davis и Abuchowski описали процесс присоединения ПЭГ к бычьему сывороточному альбумину [24]. Впоследствии, в 1990 году FDA утвердило первый модифицированный белковый продукт Adagen®, представляющий собой ПЭГилированный фермент аденозиндеаминазы, направленный на лечение тяжелых форм вируса иммунодефицита [25]. С тех пор полиэтиленгликоль все больше привлекает внимание исследователей в качестве потенциального агента для создания систем с улучшенной доставкой лекарств.
ПЭГИЛИРОВАННЫЕ ЛИПОСОМЫ
Долгие годы использование наночастиц в медицине и физиологии было ограничено тем, что иммунная система распознавала их, как чужеродные объекты и подвергала атаке. Однако в начале 80–90 гг. ХХ века были успешно предприняты попытки покрытия наночастиц ПЭГ [26, 27], благодаря чему увеличилось время их циркуляции в кровотоке. Уже в 1995 г. FDA одобрило первый препарат на основе ПЭГилированных липосом – Doxil®. Липосомы “Doxil Stealth®” увеличивают биодоступность доксорубицина в 90 раз выше в течение одной недели после инъекции, при этом увеличивается время циркуляции в крови и период полувыведения (трое-четверо суток) [28]. Положительные свойства таких липосом связаны с особенностью их строения. Так, приведено строение обычных липосом (А) и липосом, содержащих ПЭГ на поверхности (Б):
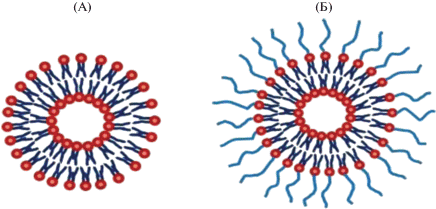
Видно, что липосомы состоят из фосфолипидов, представляющих собой дифильные молекулы, имеющие гидрофильную полярную головку и гидрофобные неполярные углеводородные цепи. Фосфолипидные молекулы способны образовывать в воде бислойные липидные мембраны [29]. Соответственно, водорастворимые лекарственные вещества могут попадать во внутреннее водное пространство липосом, а гидрофобные – в липидный бислой. ПЭГ, включенный в липидный бислой, образует гидратированную оболочку, которая надежно защищает липосомы от агрегации и фагоцитоза, тем самым тормозя преждевременное высвобождение лекарственного вещества в ходе циркуляции [30, 31]. Более того, оболочка из ПЭГ способствует снижению адсорбции опсонинов, таких как комплемент C3a и C3b, фибронектин и иммуноглобулин (преимущественно класса G) на поверхности липосом [32, 33].
ПЭГилирование липосом может быть достигнуто различными способами: физическим адсорбированием полимера на поверхности пузырьков, ковалентным присоединением реакционноспособных ПЭГ-производных к поверхности предварительно полученных липосом или включением ПЭГ-липидного конъюгата вместе с другими фосфолипидами во время получения липосом [32, 34]. В клинической практике широко используется метод закрепления полимера в липосомальной мембране; одним из таких примеров выступает конъюгат ПЭГ с дистеароилфосфатидилэтаноламином (ДСФЭА) [35, 36].
Важными параметрами при получении ПЭГилированных систем являются молекулярная масса и структура фрагмента ПЭГ, которые должны быть связаны с липосомой. При повышении молекулярной массы ПЭГ до 750 и выше в ПЭГилированных липосомах можно наблюдать их более длительную циркуляцию в крови. Значит, изменение длины цепи ПЭГ дает увеличение времени циркуляции при системном введении. Одним из таких примеров можно назвать использование ПЭГилированных липосом с М = (1.9 и 5.0) × 103, состоящих из сфингомиелина/яичного фосфатидилхолина/холестерола/ДСФЭА–ПЭГ, которые продемонстрировали увеличение времени пребывания в крови по сравнению с короткими молекулами ПЭГ (М = 750 и 120) [35]. Отмечено, что повышенное содержание ПЭГ в системе предотвращает агрегацию липосом в крови, тогда как липосомы, полученные с более низким содержанием ПЭГ, со временем агломерируются [37]. В работах [38, 39] методами компьютерного моделирования был исследован молекулярный механизм защитного действия ПЭГ. Показано, что сравнительно малое число макромолекул ПЭГ способно образовывать защитный слой на поверхности липосом, предотвращающий опсонизацию белков. Предложены подходы для рассчета оптимальной концентрации ПЭГ на поверхности липосом для достижения их защитного эффекта при различной молекулярной массе.
В связи с тем что ПЭГилированные липосомы способны направленно доставлять лекарственное вещество, снижать общетоксическое действие и увеличивать время циркуляции в крови, их активно применяют для создания систем доставки противоопухолевых препаратов [28, 34, 40]. Установлено, что при достаточно длительном периоде полувыведения таких липосом, небольшая их часть будет пассивно накапливаться в опухоли, избегая при этом здоровые ткани [41–43].
В настоящее время на стадии клинических испытаний находятся ПЭГилированный липосомальный цисплатин (SPI-077TM от “Alza Corporation” и LipoplatinTM от “Regulon Inc.”) и липосомальный полусинтетический аналог камптотецина (S-CKD602, “Alza Corporation”) [34, 44–46]. Активно ведутся работы по разработке ПЭГилированных липосом с такими функциональными группами, как малеимиды, которые дают возможность использовать их в качестве различных лигандов для векторной доставки лекарственных веществ [47, 48], либо обеспечивать адгезию к слизистой ткани (мукоадгезия), что применимо для трансмукозальной доставки лекарств [49]. Также осуществляются исследования ПЭГилированного липосомального рекомбинантного FVIII для лечения гемофилии A [50].
ПЭГИЛИРОВАННЫЕ БЕЛКИ И ФЕРМЕНТЫ
Обработка белков ПЭГ является клинически доказанным подходом к увеличению периода полувыведения и уменьшению иммуногенности протеиновой терапии [51–54]. Данный подход способствует повышению молекулярной массы белков, маскирует их поверхность, улучшает фармакодинамику (механизм действия лекарства на организм) и фармакокинетику (химические превращения лекарства в организме) [55].
Получение ПЭГилированных белков обычно достигается посредством химической реакции между надлежащим образом активированным фрагментом ПЭГ и реакционноспособными группами белка, которые включают боковые цепи аминокислот (лизина, цистеина, гистидина, аргинина, аспарагиновой кислоты, глутаминовой кислоты, серина, треонина и тирозина) или N-концевую аминогруппу и С-концевую карбоновую кислоту [56]. Наиболее широко при этом используется монометиловый эфир ПЭГ (м-ПЭГ), один конец которого содержит метильную группу, а другой – гидроксильную, непосредственно участвующую в связывании белка [22]. Применяется и разветвленная форма ПЭГ (м-ПЭГ-2) в ПЭГилированных протеинах, у которой имеются две полимерные цепи, связанные одним активным фрагментом:
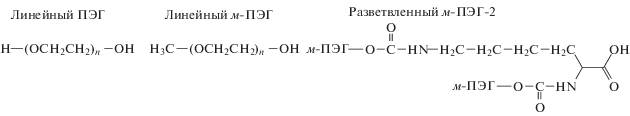
Конъюгаты ПЭГ с разветвленной формой, в результате бóльшего экранирования поверхности, обладают длительным периодом полувыведения, в отличие от линейной [57–59].
Широкое применение ПЭГилированных конъюгатов и результаты поздних стадий клинических испытаний подтвердили важность химического состава и строения ПЭГ. Например, для ПЭГилированных белков первого поколения было характерно использование линейных макромолекул ПЭГ с низкой молекулярной массой, что приводило к возникновению побочных реакций, слабым связям с полипептидами и недостаточной селективностью при модификации [2, 52].
Первоначально для получения активных макромолекул ПЭГ применяли хлорид циануровой кислоты (2-,4-,6-трихлортриазин), который реагировал с каталазой и бычьим сывороточным альбумином, в результате чего увеличивалось время циркуляции и снижалась иммуногенность, по сравнению с исходными белками [24, 60]. Использовали также ПЭГ-дихлортриазин, способный вступать в реакцию с нуклеофильными функциональными группами (тирозин, лизин, цистеин, гистидин и другие), с замещением одного из атомов хлора и получением продукта с сохранением заряда на вторичном амине [61]. Возможность реакции с оставшимся атомом хлора в итоге приводила к сшиванию белковых молекул, имеющих в своем составе нуклеофильные остатки, что отрицательно сказывалось на свойствах ПЭГилированных белков [2].
Используемые методы получения производных ПЭГ в первых работах (за исключением некоторых [62]) не позволяли получать чистые монофункциональные конъюгаты с высокой молекулярной массой, поэтому в дальнейшем получили развитие новые подходы для модификации белков с образованием специфических мест связи.
Реагенты, содержащие ПЭГ, второго поколения исследований являются N-концевыми или тиольными, которые демонстрируют избирательную селективность по отношению к лизину. Лизин можно назвать наиболее распространенной аминокислотой. Он может составлять до 10% от общей аминокислотной последовательности в белках.
Среди первых представителей второго поколения выделяется м-ПЭГ-пропиональдегид [63], полученный путем модификации N-концевой аминогруппы [64]. Таким образом, полной селективности не наблюдалось, однако обширная гетерогенность, проявляющаяся в отношении модификации лизина, была значительно снижена [2]. Neulasta®, ПЭГилированный гранулоцитарный колониестимулирующий фактор (ГКСФ), одобренный в 2002 г., был получен с помощью ковалентной конъюгации ПЭГ с N-концевым метионильным остатком [51]. Модификация N-концевого амина в основном происходит путем восстановительного аминирования [2, 64, 65] и по праву считается способом образования специфических мест связи с белком [51, 64, 66–69].
Другим способом образования специфических связей с белком является присоединение молекул цистеина к белку. Благодаря присутствию тиольных групп в данной аминокислоте возможно образование ковалентной связи в присутствии других нуклеофилов, что минимизирует потерю биологической активности [51, 52, 70]. Более того, цистеин может быть добавлен к полипептидам именно в том месте, где они необходимы с помощью генной инженерии [70, 71]. Примером образования специфических мест связей с белком посредством цистеина можно назвать ПЭГилированный продукт “CIMZIA®” (“UCB Pharma S.A.”, Бельгия), получивший одобрение в 2008 году (таблица 1) [34, 51, 72].
Таблица 1.
| Наименование ПЭГилированного продукта (производитель) | ПЭГилированный конъюгат | Место связи ПЭГ с молекулой | МПЭГ × 10–3 | Год утверждения | Обоснование |
|---|---|---|---|---|---|
| Adagen® (“Enzon Inc”) | ПЭГ-адемаза | Лизин | 5 | 1990 | увеличение периода полувыведения |
| Oncaspar® (“Enzon Inc”) | ПЭГ-аспаргаза (L-аспарагиназа) | Лизин | 5 | 1994 | увеличение периода полувыведения, снижение иммуногенности |
| PegIntron® (“Schering-Plough Co.”) | ПЭГ-интерферон (интерферон альфа-2b) | Гистидин, цистеин, лизин, серин, тирозин, гистидин | 12 | 2000–2001 | увеличение периода полувыведения, снижение иммуногенности |
| Neulasta® (“Amgen”) | ПЭГ-филграстим (ГМКСФ) | N-концевая молекула белка | 20 | 2002–2003 | увеличение периода полувыведения |
| Somavert® (“Pfizer Inc”) | ПЭГ-висомант (генно-инженерный аналог человеческого гормона роста) | Лизин, N-концевая молекула белка | 5 | 2003 | антагонист рецептора гормона роста, увеличение периода полувыведения |
| Mircera® (“Hoffman-La Roche Ltd.”) | CERA (эпоэтин-β) | Лизин | 30 | 2007 | увеличение периода полувыведения |
| Cimzia® (“UCB Pharma S.A.”) | Цертолизумаб ПЭГ (anti-TNF-aFab’) | C-концевая молекула белка | 40 | 2008 | увеличение периода полувыведения, снижение иммуногенности |
| Krystexxa® (“Savient Pharmaceuticals”) | ПЭГ-лотиказа (рекомбинантная уриказа) | Лизин | 10 | 2010 | увеличение периода полувыведения |
Еще одним примером метода образования ПЭГилированных белков можно назвать окисление карбогидратных остатков или N-концевого серина или треонина [73]. Ниже приведены основные подходы, используемые при получении таких белков:
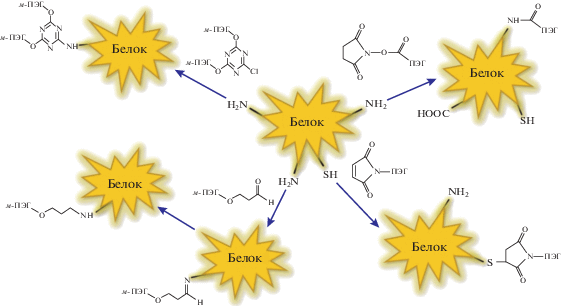
Сравнительные исследования белков с их ПЭГилированными аналогами в целом демонстрируют неизменность структуры белка при его обработке с помощью ПЭГ – данные спектроскопии ЯМР, кругового дихроизма и УФ-спектрофотометрии [53, 74–80]. Ярким тому примером являются исследования конъюгатов, содержащих и не содержащих ПЭГ с М = 5 × 103, методом ЯМР [77], а также сравнение четырех различных моноинсулиновых конъюгатов с помощью метода кругового дихроизма [74, 78].
Интересный подход к использованию свойств ПЭГилированных белков был предложен в работе Y. Hayashi с соавторами [81], где лекарственные формы были получены в виде мукоадгезивных гелей на основе интерполимерных комплексов двух ПЭГилированных продуктов (папаина и соевого ингибитора трипсина) с ПАК. Образование этих гелей наблюдалось при рН 3.0 из-за формирования интерполимерных комплексов между звеньями ПЭГ и макромолекулами ПАК, стабилизированных водородными связями. При повышении рН среды до 7.4 имела место диссоциация интерполимерных комплексов, что приводило к высвобождению связанных с ПЭГ белков из гелей. Авторами рассмотрены перспективы использования таких систем для доставки ПЭГилированных белков в носовую полость, глаза и влагалище.
Вместе с тем использование ПЭГ для модификации белков имеет и отрицательные стороны; из-за большой молекулярной массы в высоких дозах возможно его накопление в печени [82]. Недавние исследования на животных также показали образование антител против ПЭГ при введении ПЭГилированных белков [51, 83–85].
ПЭГИЛИРОВАННЫЕ ЧАСТИЦЫ ДЛЯ СИСТЕМНОГО ДЕЙСТВИЯ
В настоящее время накоплен опыт использования наночастиц, покрытых ПЭГ, для системного действия с целью преодоления биологических барьеров и лечения различного рода заболеваний. Детально рассмотрено влияние таких физико-химических факторов, как молекулярная масса ПЭГ, поверхностная плотность, свойства ядра наночастиц, время повторного введения на эффективную доставку лекарств и генов [28, 86–88]. Например, чтобы избежать взаимодействия с белками сыворотки крови и клетками мононуклеарной фагоцитарной системы (МФС), представляющей собой физиологическую защиту клеток, обладающих способностью поглощать и переваривать чужеродный материал, оболочка с ПЭГ должна обеспечивать достаточно толстый слой для экранирования поверхности наночастиц. Отмечено, что увеличение молекулярной массы ПЭГ, встроенного в мицеллы предотвращает агрегацию и адсорбцию компонентов крови, что приводит к увеличению времени их циркуляции in vivo [89]. Ключевую роль в конечном биораспределении играет размер частиц. Крупные частицы, которые не могут быть усвоены почками, имеющие гидродинамический радиус более 200 нм, обладают более высокой скоростью клиренса (процесс очищения биологических жидкостей или тканей организма), в отличие от частиц с меньшим радиусом, независимо от того, являются ли они ПЭГилированными или нет [90, 91]. ПЭГилированные наночастицы с гидродинамическим радиусом менее 150 нм вызывают повышенное поглощение в костном мозге кроликов, в то время как частицы диаметром 250 нм в основном локализируются в селезенке и печени с незначительной долей поглощения костным мозгом [91, 92]. Наиболее широко используемым методом определения поглощения частиц in vivo можно назвать применение радиоактивного 14С или гистологические исследования инкапсулированного индия [91–96]. Изучение конечного уровня биораспределения и клиренса частиц in vitro наиболее часто проводится с помощью метода проточной цитометрии и измерения флуоресценции, связанной с клетками [91, 97, 98]. Показано, что на эффективность переноса системно вводимых векторов генов in vivo на основе ПЭГилированных полимеров влияет также форма наночастиц [99]. Стержнеобразная и червеобразная формы обладают более высокой экспрессией трансгена по сравнению со сферической, предполагая уменьшение поглощения удлиненных наночастиц клетками МФС. ПЭГилированные наночастицы используют для увеличения времени циркуляции частиц и накопления в опухоли, а также для снижения иммуногенности аденовирусов и увеличения времени циркуляции вирусных генных векторов, что дает предпосылки для использования содержащих ПЭГ покрытий в новых направлениях фармакотерапии [100, 101].
ПЭГИЛИРОВАННЫЕ ЧАСТИЦЫ В КАЧЕСТВЕ НОСИТЕЛЕЙ С УСИЛЕННОЙ ПРОНИЦАЕМОСТЬЮ
Доставка лекарств непосредственно к месту локализации очага заболевания, избегая при этом системного действия, способна также значительно улучшить фармакотерапию. Однако при местном применении лекарственных препаратов существует ряд препятствий, среди которых так называемые “биологические барьеры”, ограничивающие их полноценное действие. Таким барьером являются слизистые оболочки, которые покрывают поверхность органов дыхания, мочевой, половой и пищеварительной систем, а также поверхности глаз. Они надежно защищают эпителий, обладая высокой адгезией к патогенам [102–105], частицам окружающей среды и системам доставки лекарств [102, 106, 107], способствуя их быстрому удалению. Таким образом, защитные свойства слизистых оболочек ограничивают возможности доставки лекарственных веществ и генов через их поверхность. Проникновение частиц глубоко через слизистый барьер без разрушения защитных функций может способствовать улучшению профилактического и терапевтического лечения [102], чего можно достичь посредством конъюгации лекарственных веществ с ПЭГ. Для получения ПЭГилированных коллоидных частиц могут быть использованы те же принципы и химические реакции, что и для получения ПЭГилированных белков, ферментов и липосом.
Одни из первых исследований в области проникновения ПЭГилированных систем через слизистые оболочки были проведены на основе изучения диффузии наночастиц в образцах свежей цервиковагинальной слизи человека. Показано, что наночастицы размером до 500 нм могут быстро диффундировать, если они плотно покрыты низкомолекулярным ПЭГ [106, 108]. Позднее, на примере наночастиц на основе сополимеров молочной и гликолевой кислот (ПМГК) было также подтверждено, что их проникновение через слизь обеспечивает улучшенную доставку в цервикальный канал, при этом использование ПЭГилированных покрытий увеличивает скорость диффузии [109]. Наночастицы с покрытием на основе плюроника F-127 (триблок сополимера ПЭГ–полипропиленгликоль–ПЭГ) обеспечили значительное улучшение и более равномерное покрытие поверхности шейки матки и влагалища у мышей [28], что дает дополнительные преимущества при доставке лекарств. Сами по себе плюроники способны образовывать мицеллярные структуры с более гидрофобным ядром, состоящим из полипропиленгликоля, и гидрофильной ПЭГ-содержащей опушкой. Многие лекарственные препараты, обладающие низкой растворимостью в воде, могут быть солюбилизированы в растворах плюроников, и наличие ПЭГ-содержащей поверхности в таких системах оказывает существенное влияние на проницаемость через биологические мембраны [110].
Положительные свойства ПЭГилированных наночастиц также были обнаружены при доставке в слизистую оболочку носа [111]. Отмечено, что слизь с высокой степенью вязкости накапливается в пазухах носа у больных с хроническим риносинуситом. В этом случае терапия наночастицами должна обеспечить проникновение в слизистую, во избежание элюирования во время промывания, и достичь лежащих ниже эпителиальных клеток. Таким образом, была измерена скорость диффузии наночастиц полистирола и их ПЭГилированных конъюгатов в свежей слизи пациентов с хроническим риносинуситом в обычной и полипозной форме. Установлено, что ПЭГилированные частицы размером до 200 нм способны легко проникать во все образцы слизи с обычной формой хронического риносинусита и почти в половину образцов от пациентов с полипом носа.
Измерение скорости диффузии частиц применяли также для оценки проникающей способности через высоковязкую мокроту, которая препятствует эффективной доставке лекарственного вещества и гена в виде наночастиц в легкие у пациентов с муковисцидозом – наследственным заболеванием, характеризующимся поражением желез внешней секреции и тяжелыми нарушениями функций органов дыхания [112]. Частицы диаметром до 200 нм, которые были покрыты низкомолекулярным ПЭГ, перемещались через неразбавленную мокроту до 90 раз быстрее, чем частицы аналогичного размера без покрытия. Напротив, транспорт частиц размером 500 нм с ПЭГ и без него был сильно затруднен. Используя результаты определения скорости переноса частиц различных размеров в сочетании с 3D-масштабированием препятствии, показано, что размер пор составляет примерно 140 ± 50 нм. Значит, наночастицы диаметром до 200 нм способны проходить данный барьер, при этом ПЭГилированные покрытия увеличивают скорость доставки лекарственных веществ.
Преодоление естественных защитных функций глаза также является актуальной проблемой для местной доставки лекарств. Исследование барьерных свойств роговицы глаза было проведено с помощью флуоресцентно-меченных частиц, помещенных на поверхность коровьей роговицы [113]. Результаты эксперимента продемонстрировали, что основным барьером для проникновения частиц является эпителий роговицы. Предварительная ее обработка β-циклодекстрином обеспечивает более высокую проницаемость низкомолекулярных соединений, таких как флуоресцеин натрия, но не улучшает проникновение наночастиц и более крупных молекул. Воздействие ПЭГилированных органосиликатных наночастиц с молекулярной массой ПЭГ 750 на поверхность глаза показало их способность к мукоадгезии. Однако использование ПЭГ с более высокой молекулярной массой (М = 5 × 103) позволяет наночастицам проникать через роговицу после удаления эпителиального слоя [113].
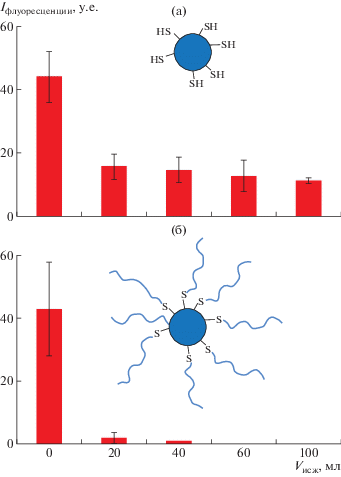
Ранее авторами работы [114] было проведено сравнительное исследование мукоадгезивных свойств и удерживания тиолированных органосиликатных наночастиц и их ПЭГилированного аналога на слизистой поверхности роговицы глаз коров in vitro. Установлено, что тиолированные частицы обладают высокой мукоадгезивной способностью из-за образования ковалентных связей (–S–S-мостиков) c остатками цистеина, присутствующими в муцине. ПЭГилированные наночастицы показали отсутствие адгезии к поверхности слизистой оболочки глаз (рис. 2; перепечатан из работы [114] c разрешения “American Chemical Society”).
Рис. 2.
Удерживание тиолированных органосиликатных наночастиц (а) и их ПЭГилированного аналога (б) на поверхности роговицы глаз коровы в потоке искусственной слезной жидкости (исж) в экспериментах in vitro.
В работе [115] исследовано интравитреальное (внутриглазное) введение наночастиц полистирола размером до 510 нм, которое продемонстрировало быстрое проникновение в стекловидное тело при покрытии их ПЭГ, в то время как движение наночастиц диаметром 1190 нм было сильно ограничено вне зависимости от химического состава поверхности.
Потенциальные преимущества ПЭГилированных частиц также были показаны на здоровой и воспаленной слизистой оболочке кишечника. Нано- и микрочастицы ПМГК, и эти же частицы, покрытые хитозаном, демонстрировали низкое проникновение в образцах здоровых тканей. ПЭГилированные наночастицы ПМГК обладают высокой способностью проникновения через здоровую (2.3%) и воспаленную слизистую оболочку (5.3%). Примечательно, что ПЭГилированные микрочастицы показали повышенную проницаемость в воспаленной слизистой кишечника (3.3%) по сравнению со здоровой (4.1%) [116].
ПЭГИЛИРОВАННЫЕ ПОЛИЭЛЕКТРОЛИТЫ И ИХ КОМПЛЕКСЫ ФАРМАЦЕВТИЧЕСКОГО НАЗНАЧЕНИЯ
Некоторые полимеры, применяемые в различных фармацевтических технологиях, при их конъюгации с ПЭГ приобретают ряд ценных свойств, не характерных для исходных материалов. Подтверждением тому служат многочисленные сведения о ПЭГилированных производных хитозана, катионного полисахарида, имеющего ряд уникальных физико-химических и биофармацевтических свойств. Конъюгацию ПЭГ с хитозаном можно осуществить посредством его реакции с ПЭГ-содержащими альдегидными, карбоксильными, карбонатными, иодидными, эпоксидными, акрилатными, N-гидроксисукцинимидными и сульфонатными группами [117].
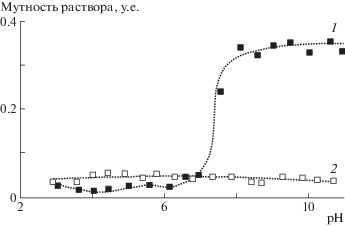
Присоединение ПЭГ к хитозану в этом случае происходит, как правило, с участием его аминогрупп. В литературе также имеются сведения о присоединении ПЭГ через гидроксильные группы хитозана [117]. Как правило, это приводит к существенным изменениям характера растворимости хитозана в воде. Так, исходный хитозан обычно теряет свою растворимость в воде при рН > 6.5 из-за депротонирования аминогрупп и потери положительного заряда на его макромолекулах [118]. ПЭГилированный хитозан при достаточной степени замещения и молекулярной массе ПЭГ не проявляет рН-зависимую растворимость и остается в растворе даже при высоких значениях рН (рис. 3). Перепечатано из [119] с разрешения автора.
Рис. 3.
Мутность раствора хитозана (1) и его ПЭГилированного производного (2) в зависимости от рН. Мутность раствора измерена при 400 нм.
Несмотря на то, что хитозан является биосовместимым материалом, конъюгация его с ПЭГ улучшает его токсикологические характеристики и снижает цитотоксичность по отношению к различным клеткам [117].
Сведения о фармацевтических применениях ПЭГилированного хитозана включают использование данных производных для получения наночастиц в качестве носителей лекарственных веществ. Наличие в таком ПЭГилированном хитозане многочисленных остаточных аминогрупп сохраняет его полиэлектролитные свойства и позволяет получать различные комплексы с анионными фармакологически активными полисахаридами, пептидами, белками и олигонуклеотидами. Например, в работе [120] сообщалось о получении наноносителей на основе интерполиэлектролитных комплексов ПЭГилированного хитозана и анионного полисахарида гепарина, которые вызывали процесс программируемой клеточной гибели (апоптоз) раковых клеток B-16–F-100 in vitro. Частицы интерполиэлектролитных комплексов имели сферическую форму и размеры порядка 200 нм. Они сохраняли отличную коллоидную стабильность в плазме крови благодаря стерической стабилизации, обеспечиваемой короной ПЭГ. В другой работе [121], X. Yang с соавторами были получены наноносители противоракового лекарственного вещества (метотрексат), путем его комплексообразования с ПЭГилированным хитозаном. Эти носители представляли собой наночастицы до 300 нм, в которых содержание метотрексата составляло от 21 до 95% в зависимости от степени ПЭГ-замещения в хитозане и соотношения полиэлектролит : лекарственное вещество.
Известно также о ПЭГилированных катионных производных хитозана, поли(L-лизина) и полиэтиленимина в качестве носителей плазмидных ДНК, коротких интерферирующих РНК (siRNA) и олигонуклеотидов [122–124]. Введение ПЭГ при этом может способствовать улучшению способности полимеров к трансфекции (процесс введения нуклеиновой кислоты в клетки), увеличению времени циркуляции и стабильности к хранению поликомплексов с носителями генетической информации.
В литературе также описаны ПЭГилированные анионные полимеры фармацевтического назначения. Так, например, M. Alibolandi с соавторами [125] были получены ПЭГилированные производные ацетилированной карбоксиметилцеллюлозы, к которым был химически присоединен противораковый препарат 7-этил-10-гидроксикамптотецин. Данные конъюгаты образовывали наночастицы, дополнительно модифицированные присоединением аптамеров РНК, которые позволяли осуществлять узнавание специфических рецепторов CD-133 (проминин-1), присутствующих на поверхности раковых клеток. Данные носители продемонстрировали повышенное поглощение и цитотоксичность по отношению к раковым клеткам HT-29 по сравнению с клетками CHO, у которых нет рецепторов CD-133.
В своей работе J.M. Shin с соавторами [126] получили ПЭГилированные производные гиалуроновой кислоты, которые были использованы в качестве носителей овальбумина как модельного антигена. В качестве связующего звена между макромолекулами гиалуроновой кислоты и ПЭГ были использованы группы, подвергающиеся ферментативному расщеплению под действием матричной металлопротеазы 9 (MMP-9). Это придавало полученному конъюгату свойства, чувствительные к MMP-9, что приводило к отщеплению короны ПЭГ при попадании в орган-мишень и эффективному поглощению этой системы раковыми клетками TC-1.
Таким образом, конъюгация некоторых полиэлектролитов с ПЭГ позволяет получать полимеры с уникальными свойствами, существенно отличающимися от исходных материалов. ПЭГ-содержащие полиэлектролиты, как правило, сохраняют свои полиэлектролитные свойства и способны к образованию комплексов с противоположно заряженными полимерами, биополимерами и малыми молекулами. Эти системы могут быть использованы при создании наноносителей лекарственных препаратов.
ПЭГИЛИРОВАННЫЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ
Альтернативным способом использования ПЭГ является его конъюгация с лекарственным веществом, приводящая к снижению иммуногенности, токсичности и низкому усвоению макрофагами и, как следствие, длительному кровообращению [127–129]. В частности, данный процесс был использован для солюбилизации и доставки противоопухолевого лекарственного препарата – паклитаксела (таксола). Так, получены водорастворимые сложные эфиры 2-таксола ПЭГ и показано, что они действуют в качестве пролекарств в экспериментах in vitro. Установлено, что присоединение ПЭГ к лекарствам или пролекарствам требует использования полимера с М ≥ 30 × 103 для предотвращения быстрого выведения ПЭГ-содержащих соединений [128]. Другим примером является получение пролекарства с использованием противопухолевого препарата камптотецина (пегамотекана) [130]. Камптотецин конъюгировали с ПЭГ (М = 40 × 103) посредством связи аланинового эфира в присутствии диизопропилкарбодиимида. Активной частью молекулы камптотецина, ответственной за конформационные изменения между лактоном и карбоксилатными формами, выступает гидроксильная группа в двадцатом положении. В результате синтеза данная группа блокируется аланиновой связью, которая стабилизирует молекулу в активной конформации лактона [131]. Пегамотекан прошел первую фазу клинических исследований, однако был исключен из испытаний во время изучения во второй фазе из-за недостаточной эффективности [132, 133].
Возможным ограничением использования ПЭГ в качестве носителя лекарственного вещества является наличие всего двух реакционноспособных групп на полимерную цепь, что ограничивает возможность высокой загрузки лекарства. В качестве решения данной проблемы была предложена дендритная структура концевой цепи ПЭГ. Компания “Enzon Pharmaceuticals, Inc.” разработала конъюгат SN-38, представляющий собой активный метаболит камптотецина с использованием ПЭГ, имеющий в своем составе четыре концевых группы [131, 134].
Иным способом получения ПЭГилированных пролекарств можно назвать использование ангидридных связей между молекулами лекарства и ПЭГ [127]. Однако положительный результат при этом можно ожидать у лекарственных веществ, имеющих в своем составе по меньшей мере одну реакционноспособную карбоксильную группу. Данный способ был разработан для доставки ПЭГилированных лекарств в качестве мукоадгезивных систем, поскольку муцин на слизистых оболочках обладает выраженным свойством текучести и наносимый препарат подвержен быстрой утилизации с поверхности слизистой оболочки, независимо от мукоадгезионной силы [127, 135]. Так, модельным лекарственным веществом служил индометацин, имеющий в своем составе карбоксильную группу и используемый для доставки лекарств в глаза, как нестероидный противовоспалительный препарат. Приведена структура комплексов, образованных конъюгатом м-ПЭГ–ангидрид–индометацин и ПАК, и их диссоциация в слезной жидкости (перепечатано из работы [127] с разрешения издательства “Elsevier”):
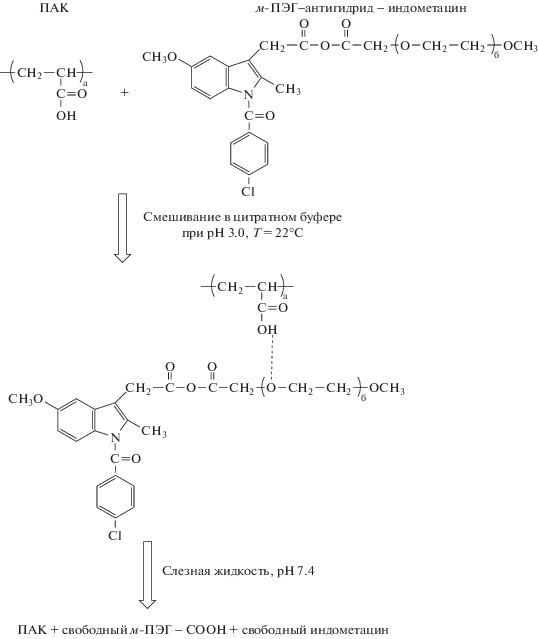
Впоследствии, на основе ПЭГилированного индометацина были получены комплексы с ПАК, предназначенные для диссоциации во время набухания состава, при контакте с поверхностью слизистых оболочек (pH 7.4). В результате, полученные комплексы при достижении поставленных условий высвобождают ПЭГ-индометацин, который далее гидролизуется до свободного лекарственного вещества и ПЭГ [127].
В этой связи, накоплен опыт образования систем “неразлагаемый полимер–лекарственное вещество”. В частности, был синтезирован и охарактеризован in vitro ПЭГ-содержащий галоперидол для селективной доставки в центральную или периферическую нервную систему, моделируя дофаминергическую систему, представляющую собой совокупность взаимосвязанных нейронов, секретирующих в качестве трансмиттера дофамин. Полученный конъюгат продемонстрировал превосходную стабильность при испытании in vitro. Проведены исследования связывания радиолиганда, которые выявили, что способность соединяться с дофаминовым рецептором D-2 у ПЭГилированного галоперидола выражена в меньшей степени по сравнению со свободным лигандом. Теоретическое моделирование продемонстрировало, что конъюгат может резко ограничить прохождение галоперидола через гематоэнцефалический барьер (полупроницаемый барьер между кровью и нервной тканью). Совокупность представленных результатов позволяет сделать вывод о необходимости проведения исследований ПЭГилированного галоперидола in vivo в контексте разработки системы, способной селективно (периферически или центрально) моделировать дофаминергическую систему [136].
Активные исследования в области ПЭГилированных лекарств нашли отражение в получении первого перорального опиоидного антагониста – ПЭГ-налоксегола (“Movantik™”), используемого для лечения опиоид-индуцированной дисфункции кишечника, сохраняя при этом центральный анальгетический эффект опиоидов. Таким образом, данный конъюгат прошел все клинические испытания и был одобрен управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) [134, 136, 137].
Список литературы
Herzberger J., Niederer K., Pohlit H., Seiwert J., Worm M., Wurm F.R., Frey H. // Chem. Rev. 2016. V. 116. № 4. P. 2170.
Roberts M.J., Bentley M.D., Harris J.M. // Adv. Drug Deliv. Rev. 2002. V. 54. № 4. P. 459.
Кабанов В.А. Энциклопедия полимеров. М.: Советская энциклопедия, 1974. Т. 2. С. 427.
Кнунянц И.Л. Химия: Большой энциклопедический словарь. М.: Большая российская энциклопедия, 1988.
Pielichowski K., Flejtuch K. // Polym. Adv. Technol. 2002. V. 13. P. 690.
Majumdar R., Alexander K.S., Riga A.T. // Pharmazie. 2010. V. 65. № 5. P. 342.
Бектуров Е.А., Бакауова З.Х. Синтетические водорастворимые полимеры в растворах. Алма-Ата: Наука, 1981.
Кабанов В.А., Паписов И.М. // Высокомолек. соед. А. 1979. Т. 21. № 2. С. 243.
Bekturov E.A., Bimendina L.A. // Adv. Polym. Sci. 1981. V. 41. P. 99.
Tsuchida E., Abe K. // Adv. Polym. Sci. 1982. V. 45. P. 1.
Кеменова В.А., Мустафин Р.И., Алексеев К.В., Скородинская А.М., Зезин А.Б., Тенцова А.И., Кабанов В.А. // Фармация. 1991. Т. 60. № 3. С. 67.
Hydrogen-Bonded Interpolymer Complexes: Formation, Structure and Applications / Ed. by V.V. Khutoryanskiy, G. Staikos. Hackensack: World Scientific Publ. Co. Pte. Ltd., 2009.
Бектуров Е.А., Хамзамулина Р.Э., Бакауова З.Х., Кудайбергенов С.Е., Джумадилов Т.К., Чердабаев А.Ш., Акаубеков М.А. Молекулярные комплексы полимеров. Алма-Ата: Наука, 1988. С. 176.
Аксенова Н.И., Кеменова В.А., Харенко А.В., Зезин А.Б., Бравова Г.Б., Кабанов В.А. // Высокомолек. cоед. A. 1998. Т. 40. № 3. С. 403.
Harada A., Li J., Kamachi M. // Macromolecules. 1993. V. 26. № 21. P. 5698.
Irmukhametova G.S., Fraser B.J., Keddie J.L., Mun G.A., Khutoryanskiy V.V. // Langmuir. 2012. V. 28. № 1. P. 299.
Serres-Gómez M., González-Gaitano G., Kaldybekov D.B., Mansfield E.D.H., Khutoryanskiy V.V., Isasi J.R., Dreiss C.A. // Langmuir. 2018. V. 34. № 36. P. 10591.
Geckil H., Xu F., Zhang X., Moon S., Demirci U. // Nanomedicine. 2010. V. 5. № 3. P. 469.
Wilems T.S., Lu X., Kurosu Y.E., Khan Z., Lim H.J., Smith Callahan L.A. // J. Biomed. Mater. Res. A. 2017. V. 105. № 11. P. 3059.
Akiyama Y., Osuka H., Nagasaki Y., Kato M., Kataoka K. // Bioconj. Chem. 2000. V. 11. P. 947.
Harris J.M., Kozlowski A. Pat. 5672662A.USA. 1997.
Никитин И.Г., Байкова И.Е., Гогова Л.М. // Лечебное дело. 2005. № 4. С. 18.
Working P.K., Newman M.S., Johnson J., Cornacoff J.B. // ACS Symp. Ser. 1997. V. 680. P. 45.
Abuchowski A., McCoy J.R., Palczuk N.C., van Es T., Davis F.F. // J. Biol. Chem. 1977. V. 252. № 11. P. 3582.
Weissig V., Pettinger T.K., Murdock N. // Int. J. Nanomedicine. 2014. V. 9. P. 4357.
Arturson P., Laakso T., Edman P. // J. Pharm. Sci. 1983. V. 72. № 12. P. 1415.
Tan J.S., Butterfield D.E., Voycheck C.L., Caldwell K.D., Li J.T. // Biomaterials. 1993. V. 14. № 11. P. 823.
Suk J.S., Xu Q., Kim N., Hanes J., Ensign L.M. // Adv. Drug Deliv. Rev. 2016. V. 99. P. 28.
Raikov A.O., Hashem A., Baryshnikova M.A. // Russ. J. Biother. 2016. V. 15. № 2. P. 90.
Blume G., Cevc G. // Biochim. Biophys. Acta. 1993. V. 1146. № 2. P. 157.
Vert M., Domurado D. // J. Biomater. Sci. Polym. Ed. 2000. V. 11. № 12. P. 1307.
Immordino M.L., Dosio F., Cattel L. // Int. J. Nanomed. 2006. V. 1. № 3. P. 297.
Cattel L., Ceruti M., Dosio F. // J. Chemother. 2004. V. 16. Suppl. 4. P. 94.
Milla P., Dosio F., Cattel L. // Curr. Drug Metab. 2012. V. 13. № 1. P. 105.
Allen T.M., Hansen C., Martin F., Redemann C.,Yau-Young A. // BBA – Biomembr. 1991. V. 1066. № 1. P. 29.
Allen C., Dos Santos N., Gallagher R., Chiu G.N.C., Shu Y., Li W.M., Johnstone S.A., Janoff A.S., Mayer L.D., Webb M.S., Bally M.B. // Biosci. Rep. 2002. V. 22. № 2. P. 225.
Braeckmans K., Buyens K., Bouquet W., Vervaet C., Joye P., De Vos F., Plawinski L., Doeuvre L., Angles-Cano E., Sanders N.N., Demeester J., De Smedt S.C. // Nano Lett. 2010. V. 10. № 11. P. 4435.
Torchilin V.P., Omelyanenko V.G., Papisov M.I., Bogdanov A.A., Trubetskoy V.S., Herron J.N., Gentry C.A. // Biochim. Biophys. Acta. 1994. V. 1195. № 1. P. 11.
Torchilin V.P., Papisov M.I. // J. Liposome Res. 1994. V. 4. № 1. P. 725.
Deshpande P.P., Biswas S., Torchilin V.P. // Nanomedicine. 2013. V. 8. № 9. P. 1509.
Allen T.M., Hansen C., Rutledge J. // Biochim. Biophys. Acta. 1989. V. 981. № 1. P. 27.
Allen T.M., Chonn A. // FEBS Lett. 1987. V. 223. № 1. P. 42.
Klibanov A.L., Maruyama K., Torchilin V.P., Huang L. // FEBS Lett. 1990. V. 268. № 1. P. 235.
Seetharamu N., Kim E., Hochster H., Martin F., Muggia F. // Anticancer Res. 2010. V. 30. № 2. P. 541.
Boulikas T. // Exp. Opin. Inv. Drugs. 2009. V. 18. № 8. P. 1197.
Zamboni W.C., Ramalingam S., Friedland D.M., Edwards R.P., Stoller R.G., Strychor S., Maruca L., Zamboni B.A., Belani C.P., Ramanathan R.K. // Clin. Cancer Res. 2009. V. 15. № 4. P. 1466.
Li T., Takeoka S. // Int. J. Nanomed. 2013. V. 8. P. 3855.
Golkar N., Tamaddon A.M., Samani S.M. // J. Liposome Res. 2015. V. 26. № 2. P. 113.
Kaldybekov D.B., Tonglairoum P., Opanasopit P., Khutoryanskiy V.V. // Eur. J. Pharm. Sci. 2018. V. 111. P. 83.
Di Minno G., Cerbone A.M., Coppola A., Cimino E., Di Capua M., Pamparana F., Tufano A., Di Minno M.N. // Haemophilia. 2010. V. 16. № 1. P. 2.
Ginn C., Khalili H., Lever R., Brocchini S. // Future Med. Chem. 2014. V. 6. № 16. P. 1829.
Kolate A., Baradia D., Patil S., Vhora I., Kore G., Misra A. // J. Control. Release. 2014. V. 192. P. 67.
Pasut G., Veronese F.M. // J. Control. Release. 2012. V. 161. № 2. P. 461.
Bailon P., Won C.Y. // Exp. Opin. Drug. Deliv. 2009. V. 6. № 1. P. 1.
Veronese F.M. // Biomaterials. 2001. V. 22. № 5. P. 405.
Roberts M.J., Harris J.M. // J. Pharm. Sci. 1998. V. 87. № 11. P. 1440.
Fee C.J. // Biotechnol. Bioeng. 2007. V. 98. № 4. P. 725.
Kusterle M., Jevševar S., Porekar V.G. // Acta Chim. Slov. 2008. V. 55. № 3. P. 594.
Veronese F.M., Caliceti P., Schiavon O. // J. Bioact. Compat. Polym. 1997. V. 12. № 3. P. 196.
Abuchowski A., van Es T., Palczuk N.C., Davis F.F. // J. Biol. Chem. 1977. V. 252. № 11. P. 3578.
Zalipsky S., Lee C. // Poly(Ethylene Glycol) Chemistry. Topics in Applied Chemistry / Ed. by J.M. Harris, S. Zalipsky. Boston: Springer, 1992. P. 347.
Bentley M.D., Harris J.M., Kozlowski A. Pat. US6448369B1 USA. 1999.
Harris J.M., Herati R.M. Pat. US5252714A USA. 1993.
Kinstler O.B., Brems D.N., Lauren S.L., Paige A.G., Hamburger J.B., Treuheit M.J. // Pharm. Res. 1996. V. 13. № 7. P. 996.
Kinstler O., Molineux G., Treuheit M., Ladd D., Gegg C. // Adv. Drug. Deliv. Rev. 2002. V. 54. № 4. P. 477.
Bell S.J., Fam C.M., Chlipala E.A., Carlson S.J., Lee J.I., Rosendahl M.S., Doherty D.H., Cox G.N. // Bioconjug. Chem. 2008. V. 19. № 1. P. 299.
Wang J., Hu T., Liu Y., Zhang G., Ma G., Su Z. // Anal. Biochem. 2011. V. 412. № 1. P. 114.
Lee H., Jang I.H., Ryu S.H., Park T.G. // Pharm. Res. 2003. V. 20. № 5. P. 818.
Tong Y., Zhong K., Tian H., Gao X., Xu X., Yin X., Yao W. // Int. J. Biol. Macromol. 2010. V. 46. № 3. P. 331.
Goodson R.J., Katre N.V. // Biotechnology. 1990. V. 8. № 4. P. 343.
Harris J.M., Chess R.B. // Nat. Rev. Drug. Discov. 2003. V. 2. № 3. P. 214.
EMEA. Cimzia ‘assesment report’. www.ema.europa.eu/docs/en_GB/document_library/ EPAR_-_Public_assessment_report/human/001037/ WC500069735.pdf.
Zalipsky S. // Poly(ethylene glycol) Chemistry and Biological Applications / Ed. by J.M. Harris, S. Zalipsky. Washington: ACS Books, 1997. P. 318.
Turecek P.L., Bossard M.J., Schoetens F., Ivens I.A. // J. Pharm. Sci. 2016. V. 105. № 2. P. 460.
Banci L., Bertini I., Scolaro L.M., Caliceti P., Schiavon O., Veronese F.M. // J. Inorg. Biochem. 1990. V. 39. № 2. P. 149.
Veronese F.M., Sartore L., Schiavon O., Caliceti P. // Ann. Acad. Sci. 1990. V. 613. P. 468.
Digilio G., Barbero L., Bracco C., Corpillo D., Esposito P., Piquet G., Traversa S., Aime S. // J. Am. Chem. Soc. 2003. V. 125. № 12. P. 3458.
Hinds K.D., Kim S.W. // Adv. Drug. Deliv. Rev. 2002. V. 54. № 4. P. 505.
Meng F., Manjula B.N., Smith P.K., Acharya S.A. // Bioconjug. Chem. 2008. V. 19. № 7. P. 1352.
Veronese F.M., Mero A., Caboi F., Sergi M., Marongiu C., Pasut G. // Bioconjug. Chem. 2007. V. 18. № 6. P. 1824.
Hayashi Y., Harris J.M., Hoffman A.S. // React. Funct. Polym. 2007. V. 67. № 11. P. 1330.
Yamaoka T., Tabata Y., Ikada Y. // J. Pharm. Sci. 1994. V. 83. № 4. P. 601.
Garay R.P., Labaune J.R. // Open Conf. Proc. J. 2011. V. 2. P. 104.
Armstrong J.K., Hempel G., Koling S., Chan L.S., Fisher T., Meiselman H.J., Garratty G. // Cancer. 2007. V. 110. № 1. P. 103.
Ganson N.J., Kelly S.J., Scarlett E., Sundy J.S., Hershfield M.S. // Arthritis Res. Ther. 2006. V. 8. № 1. P. R12.
Yoo J.W., Chambers E., Mitragotri S. // Curr. Pharm. Des. 2010. V. 16. № 21. P. 2298.
Nance E., Timbie K., Miller G.W., Song J., Louttit C., Klibanov A.L., Shih T.Y., Swaminathan G., Tamargo R.J., Woodworth G.F., Hanes J., Price R.J. // J. Control. Release. 2014. V. 189. P. 123.
Gref R., Lück M., Quellec P., Marchand M., Dellacherie E., Harnisch S., Blunk T., Müller R.H. // Colloids Surf. B. 2000. V. 18. № 3–4. P. 301.
Miteva M., Kirkbride K.C., Kilchrist K.V., Werfel T.A., Li H., Nelson C.E., Gupta M.K., Giorgio T.D., Duvall C.L. // Biomaterials. 2015. V. 38. P. 97.
Moghimi S.M., Hedeman H., Muir I.S., Illum L., Davis S.S. // Biochim. Biophys. Acta. 1993. V. 1157. № 3. P. 233.
Owens D.E., Peppas N.A. // Int. J. Pharm. 2006. V. 307. № 1. P. 93.
Porter C.J., Moghimi S.M., Illum L., Davis S.S. // FEBS Lett. 1992. V. 305. № 1. P. 62.
Leu D., Manthey B., Kreuter J., Speiser P., Delucax P.P. // J. Pharm. Sci. 1984. V. 73. № 10. P. 1433.
Tröster S.D., Wallis K.H., Müller R.H., Kreuter J. // J. Control. Release. 1992. V. 20. № 3. P. 247.
Gref R., Minamitake Y., Peracchia M.T., Trubetskoy V., Torchilin V., Langer R. // Science. 1994. V. 263. № 5153. P. 1600.
Bazile D., Prud’homme C., Bassoullet M.T., Marlard M., Spenlehauer G., Veillard M. // J. Pharm. Sci. 1995. V. 84. № 4. P. 493.
De Jaeghere F., Allemann E., Feijen J., Kissel T., Doelker E., Gurny R. // J. Drug Target. 2000. V. 8. № 3. P. 143.
Jaulin N., Appel M., Passirani C., Barratt G., Labarre D. // J. Drug. Target. 2000. V. 8. № 3. P. 165.
Jiang X., Qu W., Pan D., Ren Y., Williford J.M., Cui H., Luijten E., Mao H.Q. // Adv. Mater. 2013. V. 25. № 2. P. 227.
Alexis F., Pridgen E., Molnar L.K., Farokhzad O.C. // Mol. Pharm. 2008. V. 5. № 4. P. 505.
Kim J., Kim P.H., Kim S.W., Yun C.O. // Biomaterials. 2012. V. 33. № 6. P. 1838.
Ensign L.M., Schneider C., Suk J.S., Cone R., Hanes J. // Adv. Mater. 2012. V. 24. № 28. P. 3887.
Lai S.K., Hida K., Shukair S., Wang Y.Y., Figueiredo A., Cone R., Hope T.J., Hanes J. // J. Virol. 2009. V. 83. № 21. P. 11196.
McAuley J.L., Linden S.K., Png C.W., King R.M., Pennington H.L., Gendler S.J., Florin T.H., Hill G.R., Korolik V., McGuckin M.A. // J. Clin. Invest. 2007. V. 117. № 8. P. 2313.
Jachak A., Lai S.K., Hida K., Suk J.S., Markovic N., Biswal S., Breysse P.N., Hanes J. // Nanotoxicology. 2012. V. 6. № 6. P. 614.
Lai S.K., O’Hanlon D.E., Harrold S., Man S.T., Wang Y.Y., Cone R., Hanes J. // Proc. Natl. Acad. Sci. 2007. V. 104. № 5. P. 1482.
Olmsted S.S., Padgett J.L., Yudin A.I., Whaley K.J., Moench T.R., Cone R.A. // Biophys. J. 2001. V. 81. № 4. P. 1930.
Lai S.K., Wang Y.Y., Hanes J. // Adv. Drug. Deliv. Rev. 2009. V. 61. № 2. P. 158.
Cu Y., Saltzman W.M. // Mol. Pharm. 2009. V. 6. № 1. P. 173.
Agafonov M., Volkova T., Kumeev R., Chibunova E., Terekhova I. // J. Mol. Liq. 2019. V. 274. P. 770.
Lai S.K., Suk J.S., Pace A., Wang Y.Y., Yang M., Mert O., Chen J., Kim J., Hanes J. // Biomaterials. 2011. V. 32. № 26. P. 6285.
Suk J.S., Lai S.K., Wang Y.Y., Ensign L.M., Zeitlin P.L., Boyle M.P., Hanes J. // Biomaterials. 2009. V. 30. № 13. P. 2591.
Mun E.A., Morrison P.W.J., Williams A.C., Khutoryanskiy V.V. // Mol. Pharm. 2014. V. 11. № 10. P. 3556.
Irmukhametova G.S., Mun G.A., Khutoryanskiy V. V. // Langmuir. 2011. V. 27. № 15. P. 9551.
Xu Q., Boylan N.J., Suk J.S., Wang Y.Y., Nance E.A., Yang J.C., McDonnell P.J., Cone R.A., Duh E.J., Hanes J. // J. Control. Release. 2013. V. 167. № 1. P. 76.
Lautenschläger C., Schmidt C., Lehr C.M., Fischer D., Stallmach A. // Eur. J. Pharm. Biopharm. A. 2013. V. 85. № 3. P. 578.
Casettari L., Vllasaliu D., Castagnino E., Stolnik S., Howdle S., Illum L. // Prog. Polym. Sci. 2012. V. 37. № 5. P. 659.
Sogias I.A., Khutoryanskiy V.V., Williams A.C. // Macromol. Chem. Phys. 2010. V. 211. № 4. P. 426.
Ways T.M.M. PhD Thesis. Reading: University of Reading, 2019.
Bae K.H., Moon C.W., Lee Y., Park T.G. // Pharm. Res. 2009. V. 26. № 1. P. 93.
Yang X., Zhang Q., Wang Y., Chen H., Zhang H., Gao F., Liu L. // Colloids Surf. B. 2008. V. 61. № 2. P. 125.
Jiang X., Dai H., Leong K.W., Goh S.H., Mao H.Q., Yang Y.Y. // J. Gene Med. 2006. V. 8. № 4. P. 477.
Merkel O.M., Librizzi D., Pfestroff A., Schurrat T., Buyens K., Sanders N.N., De Smedt S.C., Béhé M., Kissel T. // J. Control. Release. 2009. V. 138. № 2. P. 148.
Itaka K., Yamauchi K., Harada A., Nakamura K., Kawaguchi H., Kataoka K. // Biomaterials. 2003. V. 24. № 24. P. 4495.
Alibolandi M., Abnous K., Anvari S., Mohammadi M., Ramezani M., Taghdisi S.M. // Artif. Cells. Nanomed. Biotechnol. 2018. V. 46. № 1. P. 1159.
Shin J.M., Oh S.J., Kwon S., Deepagan V.G., Lee M., Song S.H., Lee H.J., Kim S., Song K.H., Kim T.W., Park J.H. // J. Control. Release. 2017. V. 267. P. 181.
Lele B.S., Hoffman A.S. // J. Control. Release. 2000. V. 69. № 2. P. 237.
Greenwald R.B., Gilbert C.W., Pendri A., Conover C.D., Xia J., Martinez A. // J. Med. Chem. 1996. V. 39. № 2. P. 424.
Zalipsky S. // Bioconjug. Chem. 1995. V. 6. № 2. P. 150.
Greenwald R.B., Pendri A., Conover C., Gilbert C., Yang R., Xia J. // J. Med. Chem. 1996. V. 39. № 10. P. 1938.
Li C., Wallace S. // Adv. Drug. Deliv. Rev. 2008. V. 60. № 8. P. 886.
Posey J.A. // Clin. Cancer Res. 2005. V. 11. № 21. P. 7866.
Kelland L. // Exp. Opin. Inv. Drugs. 2006. V. 15. № 11. P. 1309.
Li W., Zhan P., De Clercq E., Lou H., Liu X. // Prog. Polym. Sci. 2013. V. 38. № 3–4. P. 421.
Ahuja A., Khar R.K., Ali J. // Drug. Dev. Ind. Pharm. 1997. V. 23. № 5. P. 489.
Heath F., Newman A., Clementi C., Pasut G., Lin H., Stephens G.J., Whalley B.J., Osborn H.M.I., Greco F. // Polym. Chem. 2016. V. 7. № 47. P. 7204.
Naloxegol (Movantik) for Opioid-Induced Constipation. J. Am. Medical Association. 2016. V. 315. № 2. P. 194.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)