

Высокомолекулярные соединения (серия С), 2020, T. 62, № 1, стр. 55-65
Межфазный слой в полимерных нанокомпозитах
В. И. Иржак *
Институт проблем химической физики Российской академии наук
142432 Московской области, Черноголовка, просп. Академика Семенова, 1, Россия
* E-mail: irzhak@icp.ac.ru
Поступила в редакцию 03.10.2019
После доработки 13.11.2019
Принята к публикации 25.12.2019
Аннотация
Рассмотрены особенности формирования и структуры межфазного слоя в полимерных нанокомпозитах. Показано взаимное влияние наполнителей и матрицы на структуру и свойства межфазного слоя. Учтена зависимость характеристик межфазного слоя от размерности наночастиц: одномерных (нанотрубки), двумерных (графен и слоистые силикатные глины), трехмерных (металлсодержащие наночастицы) и 0-размерных квантовых точек.
ВВЕДЕНИЕ
Межфазный слой между матрицей и наполнителем рассматривается как ключевая область взаимодействия, которая в значительной степени определяет свойства конечного материала [1–4]. Как было показано во многих работах, в основном с использованием метода наноиндентирования [4–8], независимо от природы волокна и матрицы толщина межфазного слоя в полимерных волокнистых композитах достигает нескольких микрометров. Моделирование методом молекулярной динамики [9–11] приводит к таким же величинам. Очевидно, в случае полимерных нанокомпозитов следует ожидать аналогичного результата. Более того, у наноразмерных частиц имеется огромная удельная поверхность, которая на три порядка больше, чем у микрочастиц. Вследствие этого даже при низком наполнении межфазная область весьма велика и ее вклад в свойства композита может быть существенным. Прямые расчеты [12] демонстрируют влияние, которое даже небольшая объемная доля наполнителя оказывает на окружающую матрицу. Например, всего 1 об. % наносфер (радиус 2 нм) при толщине межфазного слоя 6 нм дает объемную долю межфазной области до 63%. В работе [13], исходя из кинетики отверждения диглицидилового эфира дифенилолпропана (бисфенола А – ДГЭБА) в присутствии окиси графена, показано, что доля матрицы, которая находится в межфазной области, составляет около 30% от общего объема.
Поверхностная энергия наночастиц довольно велика, поэтому они способны адсорбировать различные молекулы [14–19]. Следует иметь в виду, что структура адсорбционных слоев различна для одномерных углеродных нанотрубок (УНТ) и галлуазитных (каолиновых) многослойных трубок, двумерных графена и слоистых силикатных глин типа монтмориллонита (MMT), трехмерных металлсодержащих наночастиц, наноалмазов и фуллеренов, хотя последние иногда считаются 0-мерными. Истинно 0-мерными являются квантовые точки [20].
УНТ и графен могут быть нековалентно функционализированы без искажения морфологической структуры как путем образования π–π-связей с ароматическими соединениями, так и в результате ионных взаимодействий с соответствующими полимерами и поверхностно-активными веществами [14–21]. Так, адсорбция ведет к образованию межфазного слоя.
Другим фактором, способствующим стабильному межфазному взаимодействию наночастиц с матрицей, является их ковалентная функционализация [21–25]. В отличие от нековалентной ковалентная модификация может вносить дефекты в структуру наночастиц. Например, введение поверхностных групп СООН путем окисления УНТ приводит к изгибу трубки и таким образом ухудшает ее механические и проводящие свойства [25].
Присутствие реакционноспособных групп на поверхности наночастиц позволяет использовать их для введения соответствующих лигандов методом grafting-from, в котором используется полимеризация, инициируемая с поверхности наночастиц присоединенными инициирующими группами [26], или методом grafting-to [27], согласно которому полимерные молекулы реагируют своей концевой группой с функциональными группами, расположенными на поверхности исходных или предварительно функционализированных наночастиц.
Наконец, следует учитывать особенность нанокомпозитов в тех случаях, когда синтез матрицы происходит в присутствии наночастиц при повышенных температурах, а полученный материал используется при комнатной температуре (например, эпоксиды). Из-за разницы в коэффициентах теплового расширения наполнителя и матрицы межфазный слой может быть в определенной степени деформирован.
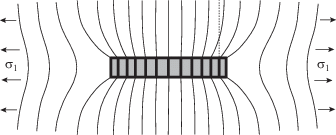
Важность межфазного слоя иллюстрирует рис. 1. Видно, что именно этот структурный элемент определяет механические свойства композита: напряжение концентрируется на межфазной границе [28]. Для электропроводящих наномпозитов важнейшее значение имеет слой матрицы, который обеспечивает туннельную передачу заряда [29]. Фактически его следует рассматривать как межфазный слой.
Рис. 1.
Схема деформации полимерной матрицы и частицы наполнителя под действием растягивающего напряжения σ1, действующего на матрицу [28].
Температура стеклования Tg характеризует матрицу, поскольку она является функцией химической природы и молекулярной архитектуры и зависит от таких параметров, как функциональность олигомера и отвердителя, а также степень превращения.
Изучение процессов формирования матрицы в присутствии наночастиц показывает, что их влияние на Tg вряд ли однозначно [30]. Это связано с наличием межфазного слоя. Эффект межфазной энергии полимер–наночастица исследован на примере нанокомпозитов с полистирольной матрицей и низкой концентрацией наночастиц с привитым полистиролом [31]. Установлено, что межфазная энергия этой системы коррелирует с плотностью прививки и молекулярно-массовым соотношением матрицы и привитых полимеров. Изменения Tg варьируют в зависимости от обоих параметров и количественно согласуются с величиной межфазной энергии.
Задача настоящего обзора – обсуждение особенностей формирования межфазного слоя в полимерных нанокомпозитах и взаимного влияния наполнителей и матрицы на структуру и свойства межфазного слоя.
ОДНОМЕРНЫЕ НАПОЛНИТЕЛИ
Очевидно, что способ синтеза нанокомпозита накладывает отпечаток на его морфологическую структуру. Типичная методика приготовления реакционной смеси изложена ниже [32], в других работах она может отличаться только в деталях.
Смесь эпоксидной смолы с предварительно очищенными многослойными УНТ обрабатывали ультразвуком в течение 2 ч, затем ее помещали в масляную баню при температуре 120°С и медленно добавляли стехиометрическое количество отвердителя при непрерывном механическом перемешивании до получения однородной смеси, что занимало около 10 мин. Далее происходило отверждение по тому или иному режиму.
В работе [33] показано, что влияние многослойных УНТ в концентрации 0.25 и 0.5 мас. % на Tg зависит от плотности сетки матрицы: Tg растет в слабо сшитых и уменьшается в плотно сшитых композитах. Предполагается, что в этих случаях взаимодействие в межфазных слоях имеет решающее значение. В рыхлых сетках наполнитель ограничивает подвижность кинетических фрагментов в результате их адсорбции, тогда как в плотных сетках многослойные УНT препятствуют образованию узлов. В связи с этим влияние наночастиц на Tg за счет межфазной области с более низкой плотностью сшивки является значительным.
Образование межфазных слоев при наличии ковалентной функционализации доказано в работе [34]. Однослойные и многослойные УНТ, а также углеродные нановолокна были обработаны смесью азотной и серной кислот, в результате чего на поверхности появились карбоксильные группы. Последние были превращены в сложноэфирные группы по реакции с фенилглицидиловым эфиром. Матрица была получена путем отверждения ДГЭБА полиэфиртриамином. Различие процедуры синтеза отражается на структуре межфазного слоя, что проявляется в релаксационных спектрах. На рис. 2 приведены кривые tgδ(Т), полученные при частоте 0.1 Гц. Как видно, высокотемпературный переход, типичный для матрицы (кривые 1), зарегистрирован для композитов на основе однослойных и многослойных УНТ, а также нановолокон. Но в случае немодифицированных (кривые 2) и СООН-модифицированных (кривые 3) наночастиц наблюдается дополнительный переход при более низкой температуре.
Рис. 2.
Температурная зависимость tgδ для эпоксидных матриц и эпоксидных нанокомпозитов, содержащих 1 мас. % однослойных (а), и многослойных УНТ (б), а также нановолокон (в). 1 – чистая матрица, 2–4 – эпоксидный композит с немодифицированными наночастицами (2), наночастицами, модифицированными группами СООН (3) и привитыми фрагментами сложного эфира (4). Данные работы [34].
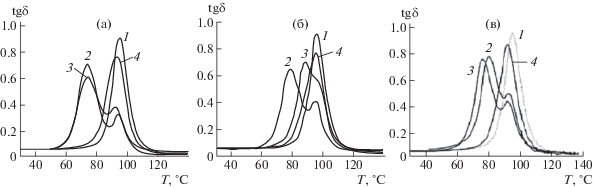
Этот результат указывает на наличие структуры, которая менее совершенна, чем структура матрицы, и, по-видимому, локализована в межфазном слое. Модификация наночастиц привитыми фрагментами сложного эфира (кривые 4) не ведет к сдвигу температуры, что может трактоваться как наличие межфазного слоя с плотностью упаковки не меньшей, чем у матрицы. Авторы [34] считают, что наблюдаемый эффект связан с селективной адсорбцией эпоксидного олигомера на наночастицы, что вряд ли возможно при появлении на поверхности фрагментов сложных эфиров (кривые 4).
Не исключено другое объяснение данного эффекта: эфирные хвосты способствуют релаксации напряжений межфазного слоя. Обращает на себя внимание то, что для однослойных УНТ эффект СООН более выражен, чем для многослойных. Это является следствием того, что однослойные УНТ модифицированы в большей степени (8.6% против 4.9% по отношению к карбоксильным группам и 21.1% против 13.7% по отношению к эфирным фрагментам).
В работе [35] оценена толщина межфазного слоя b в композите УНТ/ПВС в зависимости от диаметра трубки d.
| d, нм | 25 ± 5 | 16 ± 5 | 14 ± 5 | 15 ± 5 | 2.5 ± 1 |
| 2b/d | 0.81 ± 0.01 | 1.47 ± 0.04 | 2.27 ± 0.10 | 2.83 ± 0.10 | 4.87 ± 0.80 |
Как видно, толщина межфазного слоя соизмерима с диаметром УНТ, а отношение b/d тем больше, чем тоньше трубки.
S. Wang и соавторами [23] оценили прочность на сдвиг τ границы раздела однослойная УНТ–эпоксидная матрица в 306 МПа, предполагая, что трубки не кластеризованы и что прививка диэтилтолуолдиамина приводит в среднем к образованию одной связи на 25 атомов углерода. В соответствии с теми же расчетами в случае немодифицированных однослойных УНТ τ составляет ∼50 МПа. Однако следует учитывать, что τ зависит от радиуса УНТ. Например, согласно данным [36], полученным методом вытягивания (pull-out) из полиэтиленбутиленовой матрицы, τ ≈ 85 МПа для многослойной УНТ с радиусом 10–20 нм, и она уменьшается до ∼15 МПа при радиусе УНТ 60–70 нм.
Межфазную связь УНT/ДГЭБA изучали методом молекулярной динамики при комнатной температуре в работе [37]. В исследовании сравнивали характеристики зигзагообразных УНТ с различными диаметрами, легирующими добавками, функциональными группами и дефектами. Зигзагообразные (n, 0) УНТ с n = 5, 7, 9, 11, 13, 15 были рассмотрены в категории диаметров (в этом случае диаметр пропорционален n); B-, N- и Si-легированные УНТ – в категории легирующих веществ; кислород, гидроксил, амин, карбоксил и комбинация кислорода и гидроксила были включены в число функциональных групп; УНТ с моновакансиями, дефектами Стоун–Уэльса и более сложными моновакансиями и дивакансиями рассматривались в группе дефектов. Анализ взаимодействия между УНТ и ДГЭБА привел к выводу о ковалентном сшивании, хотя не исключил взаимодействие Ван-дер-Ваальса. Было установлено, что ковалентное сшивание с большей вероятностью улучшится при меньшем диаметре УНТ, когда кремний находится в положении прямого легирования, с комбинацией кислородных и гидроксильных функциональных групп, и с моновакансиями.
Результаты исследований методом молекулярной динамики [9] показывают, что диаметр УНТ влияет на толщину области раздела УНТ–ДГЭБA, но этот эффект зависит от степени сшивания эпоксидной смолы. Тип отвердителя также сказывается в определенной степени на толщине межфазного слоя.
Метод вытягивания [4, 36–41] позволяет оценить межфазное напряжение сдвига – важный параметр, характеризующий межфазное взаимодействие (рис. 3).
Рис. 3.
Зависимость усилия отрыва от времени: а – УНТ встроена в полимерную матрицу; б – по мере вытягивания УНТ из полимера изгибается консоль; в – при максимальной силе достигается максимальный изгиб консоли; г – отрывание, д – полное отделение УНТ от полимера. Данные работы [37].
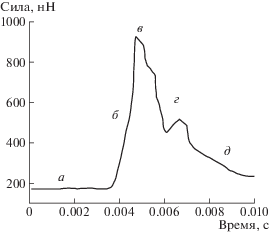
Анализ опубликованных экспериментальных данных и различных вычислительных результатов, выполненный в работе [38], показал, что как первые, так и вторые не дают оснований для однозначных выводов. Эксперименты по вытягиванию нанотрубок привели к выводу, что такие методы могут преувеличивать значения межфазного напряжения сдвига полимерных композитов.
Например, в работе [37], в которой был использован сканирующий зондовый микроскоп при вытягивании нанотрубки из матрицы, было обнаружено, что межфазное напряжение сдвига между многослойной УНT и эпоксидной матрицей находится в диапазоне 35–376 МПа. Однако применение в аналогичных экспериментах атомно-силового микроскопа [38] показало, что в системе многослойная УНТ–полиэтилен-бутеновая матрица среднее значение межфазного напряжения сдвига равно примерно 47 МПа.
Атомистическое описание проблемы [39] позволило оценить влияние на межфазное напряжение сдвига таких факторов, как длина внедрения УНТ, число ван-дер-ваальсовых взаимодействий, толщина границы раздела, диаметр УНТ. Некоторые результаты приведены в табл. 1.
Таблица 1.
Результаты извлечения УНТ разного диаметра [39]
| Хиральность УНТ | Диаметр УНТ, нм | Усилие вытягивания, нН | Межфазное напряжение сдвига, МПа |
|---|---|---|---|
| (5, 5) | 0.678 | 0.310 | 34.7 |
| (8, 8) | 1.085 | 0.439 | 30.6 |
| (12, 12) | 1.628 | 0.590 | 27.5 |
| (15, 15) | 2.035 | 0.696 | 25.9 |
| (18, 18) | 2.443 | 0.835 | 25.9 |
В работе [41] использован метод моделирования вытягивания для детального анализа влияния волнистости, аспектного отношения, диаметра УНТ, объемной доли, коэффициента Пуассона и модуля матрицы на межфазное напряжение сдвига. Показано, что с увеличением длины волны волнистости и объемной доли УНТ максимальное значение межфазного напряжения сдвига возрастает.
Заметим, что в обзоре [4] содержится обстоятельный анализ современных методов исследования межфазных слоев в композитах, содержащих УНТ.
Особенностью химической структуры галлуазитовых нанотрубок является то, что их внутренняя поверхность покрыта группами Al–OH, а внешняя поверхность в основном состоит из силоксановых групп Si–O–Si. Следовательно, стратегия модификации нанотрубок галлуазита, нацеленная на создание оптимального межфазного слоя, заключается в прививке силанов [42]. Р. Sun с сотрудниками [43] успешно привили 3-аминопропилтриэтоксисилан на поверхность нанотрубок галлуазита, предварительно обработанных смесью H2SO4 и H2O2, чтобы увеличить концентрацию гидроксильных групп, обладающих более высокой активностью в отношении силанизации.
ДВУМЕРНЫЕ НАНОЧАСТИЦЫ
В качестве исходного материала для синтеза графена используют коммерчески доступный расширенный графит. В результате термического воздействия образуются сгустки графена толщиной от десятков до ста нанометров. Длительная обработка ультразвуком может расщепить их в более тонкие листы толщиной до нескольких нанометров [44].
Методы функционализации графена, которые в принципе не отличаются от применявшихся в предыдущих случаях, описаны в работах [13, 25, 45] и других. Например, дисперсию графена в смеси метилсульфоновой кислоты и Р2О5 ацилировали дикарбоновой кислотой (4,4'-дикарбоксидифениловый эфир), а затем проводили реакцию поликонденсации с 3,3'-диамино-бензидином [25].
Оксид графена, который получается, в частности, путем окисления графита, часто используют в качестве исходного реагента при ковалентной функционализации. Например, амино- и эпокси-функционализированные образцы оксида графена были синтезированы с помощью 3-аминопропилтриэтоксисилана и 3-глицидоксипропилтриметоксисилана [46].
Алкиламины (гексил, гексадецил и октадецил амины), состоящие из длинноцепочечных гидрофобных алкильных групп и гидрофильных аминогрупп, прививаются на поверхность окиси графена через присутствующие на ней два типа реакционноспособных групп, а именно, карбоксильные и эпоксидные. Первые группы участвуют в реакции амидирования, а вторые – в реакции нуклеофильного замещения [47]. Бифункциональный поли(эфирдиамин) позволяет прививать аминосодержащие фрагменты, способные реагировать с эпоксидными группами связующего [48]. Были использованы молекулы с различной длиной цепи (D230 и D2000). Предполагалось, что будут созданы различные структуры межфазного слоя. Прививка цепей D230 на поверхность окиси графена приводит к ограниченной подвижности сегментов и соответственно к минимальной деформируемости. Цепи D2000 обладают гораздо большей деформируемостью благодаря большей длине. Это помогает увеличить подвижность наполнителя в матрице и вызывает рассеяние энергии при деформации. Указанное различие в свойствах межфазной среды, полученной с помощью D230 и D2000, определяет разницу в механических свойствах нанокомпозитов.
Y. Wang и соавторами [49] показали, что методы ковалентной и нековалентной функционализации могут значительно снизить тепловое сопротивление между графеном и эпоксидной смолой в нанокомпозитaх. Оказалось, что бутильная функциональная группа более эффективна в снижении межфазного термического сопротивления, чем карбоксильная и гидроксильная. Различные нековалентно присоединенные функциональные молекулы, включая 1-пиренбутил, 1-пирен-бутириновую кислоту и 1-пиренбутиламин, создают одинаковый тепловой эффект. Хотя ковалентная функционализация более эффективна для усиления межфазного теплового транспорта, метод нековалентной функционализации включает в себя менее сложные процессы синтеза, а также не вносит дефектов в базисную плоскость графена. Ковалентная функционализация вносит дефекты, что снижает характеристики теплопереноса графена.
Авторы работы [50], используя соотношение
оценили объемную долю интерфазы ${{{v}}_{{int}}}$ в эпоксидных нанокомпозитах, в которых в качестве наполнителя была использована окись графена в исходном состоянии и с привитым сополимером акрилонитрила и бутадиена. В формуле (1) tgδ и tgδm – значения тангенса угла потерь нанокомпозита и матрицы с объемной долей наполнителя φ, В – параметр, по существу характеризующий объем межфазного слоя. Расчеты показали, что для концентрации наполнителя 0.6 мас. % соответствующие значения B равны 3.12 и 8.84, ${{{v}}_{{int}}}$ составляют 0.0040 и 0.0114.Для получения графена с привитыми молекулами важным требованием является дезагрегация графеновых листов [51], причем следует учитывать, что последние получают из интеркалированного графита путем термической обработки [44, 51, 52].
Возможна интеркаляция различных соединений в слоистые силикаты (рис. 4), типичным представителем которых является ММТ. После расслоения наполнителя, как и в предыдущем случае графена, молекулы интеркалята фактически принимают участие в образовании межфазного слоя [53].
Рис. 4.
Схема взаимодействия слоистых силикатов с полимерной матрицей: а – разделение на фазы – образование микрокомпозита, б – интеркалирование, в – расслоение – образование нанокомпозита. Данные работы [53].
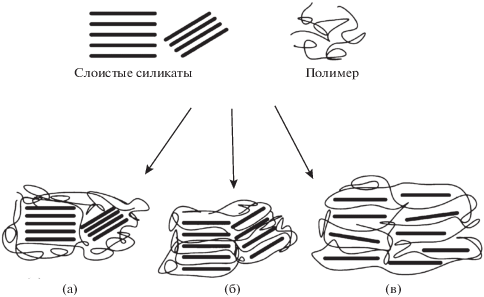
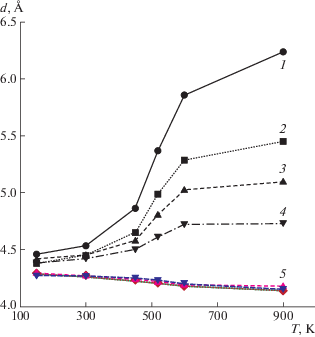
D. Kong и С. Park [54] методом рентгеновской дифракции проследили за процессом расслоения ММТ, интеркалированного октадециламмонием, в ходе изотермического отверждения ДГЭБА 4,4′-диаминодифенилсульфоном и нашли, что его можно разбить на три этапа (рис. 5). Первый этап связан с проникновением ДГЭБА в межпакетное пространство ММТ. На втором этапе происходит катионная полимеризация эпоксидной смолы, катализируемая аммонием, на третьем этапе – отверждение амином ДГЭБА, сорбированного ММТ.
Рис. 5.
Изменение расстояния d между пакетами ММТ в ходе изотермического отверждения ДГЭБА при 140 (1), 130 (2) и 120°С (3). Римскими цифрами обозначены этапы процесса расслоения. Стрелки указывают ожидаемую тенденцию расслоения. Данные работы [54].

С целью идентификации внутриполостной полимеризации, ведущей в конце концов к расслоению, смесь ММТ и ДГЭБА выдерживали при разной температуре в течение десятков дней в отсутствие аминного отвердителя. Затем, добавив 4,4′-диаминодифенилсульфон, вели отверждение в неизотермическом режиме. Аналогичный прием использовали для системы ДГЭБА–ММТ–полиэфир–диамин. На первом этапе измеряли рост эпоксидного эквивалента и температуры стеклования. Такая технология позволяет улучшить как степень дисперсности ММТ в эпоксидной смоле, так и последующее расслоение глины при формировании нанокомпозита. Период внутриполостной полимеризации удается сократить до десятков минут за счет применения в качестве катализатора комплекса BF3⋅C2H5NH2.
Силикатную поверхность наночастиц ММТ обычно модифицируют органическими соединениями, включая амины и силаны [55–59]. В данном случае могут возникать ионные связи. С этой целью в работе [57] использовали 2-(3,4-дигидроксифенил)-этиламин, который был вовлечен в окислительную полимеризацию, и поверхность ММТ была покрыта образовавшимся полимером. Расстояние между слоями увеличилось во время полимеризации.
В работе [60] были получены эпоксидные нанокомпозиты на основе бентонита, модифицированного дифениламин-4-диазонием посредством ионного обмена. Межфазный слой образован окислительной полимеризацией анилина. Арильные группы диазония оказывают сильное влияние на межфазное взаимодействие.
ТРЕХМЕРНЫЕ НАНОЧАСТИЦЫ
Если углеродные наночастицы, фуллерены, наноалмазы вводятся в реакционную смесь и отверждение происходит в их присутствии, то металлсодержащие нередко образуются в ходе реакции in situ [61]. Особенность формирования наночастиц в отверждающейся эпоксидной матрице заключается в том, что данный процесс протекает в условиях нарастающей вязкости и, в конечном счете, стеклования системы. В принципе это может перевести из кинетической в диффузионную область любую стадию процесса: зародышеобразование, рост наночастиц вследствие присоединения нуль-валентных атомов, вторичные реакции типа коалесценции или оствальдовского созревания. Действительно, как показано в работе [62], в изотермическом режиме при 70°C процесс тормозится из-за стеклования. Повышение температуры до 130°C ведет к его “реанимации”. Вместе с тем доказана возможность получения наночастиц золота в ПММА-матрице, содержащей HAuCl4, при воздействии УФ-излучения с последующим прогреванием при разной температуре, не превышающей температуру стеклования ПММА [63].
Наноалмазы – это разновидность наночастиц на основе углерода, которая привносит свойства алмаза в мир наноразмеров [64]. Его сферическая форма со средним диаметром 4–6 нм приводит к очень большой удельной поверхности, обычно около 400 м2/г, т.е. много больше по сравнению с пластинчатыми и стержнеобразными наночастицами. Большое отношение поверхности к объему дает больше места для присоединения функциональных групп к поверхности наноалмазов. Отличительной особенностью наноалмазов [65, 66] по сравнению с УНТ и другими углеродными наночастицами является наличие огромного числа различных функциональных групп, которые покрывают поверхность частицы наноалмаза. Слой поверхностных функциональных групп является неотъемлемым свойством этого материала. Химия поверхности наноалмазов богата, и функционализация их поверхности может быть достигнута в любой степени без ущерба для свойств алмазного ядра [67]. В частности, аминирование наноалмазов (НА) позволило при синтезе композита эпоксид–наноалмазы обойтись без специального отверждающего агента [68]:

Влияние высокой концентрации и малых размеров наноалмазов на прочность эпоксидного нанокомпозита обусловлено тем, что межфазные слои перекрываются, образуя сеть, которая останавливает распространение трещин [69].
Среди ковалентных производных фуллеренов следует упомянуть так называемые фуллеренолы. Полигидроксилированные фуллеренолы содержат различное количество гидроксильных групп [70]. Качество дисперсии фуллерена C60 в эпоксидном композите обеспечивается химической реакцией поверхности наполнителя с матрицей. Исследования показали, что именно межфазная адгезия имеет решающее значение [71, 72]. Наночастицы фуллерена подавляют разрушение межфазного слоя в волокнистых композитах по механизму рассеивания энергии [73]. По-видимому, этим же можно объяснить синергетическое влияние фуллерена и оксида графена на механические свойства сшитых полиуретанов [74].
Что касается металлических наночастиц, то адсорбция часто приводит к образованию химических связей [75]. Например,
Зависимость конформации лиганда от размера наночастиц металла (рис. 6), полученная в работе [77], позволяет рассчитать расстояние от поверхности наночастиц Au до функциональной (S) и “хвостовой” (CH3) групп молекулы алкилтиола. Видно, что первая величина не зависит от размера частиц и незначительно уменьшается с температурой. Вторая величина, отражающая конформацию лигандов, увеличивается с повышением температуры, и тем сильнее, чем меньше диаметр наночастиц Au.
Рис. 6.
Зависимость конформации лиганда СН3(СН2)13SН от температуры и размера наночастиц Au. Диаметр наночастиц 3 (1), 5 (2), 7 (3) и 11 нм (4); 5 – длина связи S–Au. Данные работы [77].
С одной стороны, слой лиганда защищает наночастицу и тем самым стабилизирует ее, но, с другой стороны, он делает ее метастабильной, что проявляется в снижении температуры плавления, причем основным фактором этого является энергия взаимодействия между цепями лигандов [75].
Для понимания структуры межфазного слоя в полимерных нанокомпозитах, нужно ответить на фундаментальный вопрос – каково изменение размеров цепи при добавлении наночастиц к полимеру и что контролирует это изменение [79]. Полимерные нанокомпозиты можно рассматривать как аналог концентрированных растворов полимеров. Теория и эксперименты сходятся во мнении, что полимерные цепи могут менять конформацию в зависимости от взаимодействия их с растворителем и концентрации полимера: наночастицы в этом случае играют роль растворителя.
Влияние наночастиц на размер полимерных цепей изучено в работе [80]. Радиус инерции Rg дейтерированных цепей полистирола в хорошо диспергированной смеси протонированного полистирола и химически идентичных наночастиц (сшитый ПС) определяли методом рассеяния нейтронов. Было обнаружено, что Rg растет на 10–20%, если только Rg больше, чем радиус наночастицы Rp. Аналогичные результаты получены для полисиликатных наночастиц, смешанных с ПДМС [81]. Главное, что следует иметь в виду, смешиваемость наночастиц и набухание полимерной цепи напрямую связаны, и управляющим параметром является соотношение Rg/Rp [79].
Вместе с тем было обнаружено, что наночастицы кремнезема в ПС не влияют на радиус инерции макромолекул [82]. В работе [83] исследовали два основных аспекта проблемы раствора полимер–фуллерен: влияние C60 на конформацию цепей ПС и влияние ПС на смешиваемость и агрегацию C60. Радиус инерции Rg и гидродинамический радиус Rh полимера остаются неизменными при добавлении C60, хотя Rg/Rp $ \gg $ 1 (Rp фуллерена 0.35 нм). Однако смешиваемость C60 в толуоле уменьшается: при добавлении полистирола образуются агрегаты с радиусом порядка 100 нм, и этот эффект усиливается с увеличением Mw полимера. Работы [82, 83] указывают на определяющую роль межфазного взаимодействия, которое является аналогом качества растворителя.
КВАНТОВЫЕ ТОЧКИ
Квантовые точки – синтезированные коллоидным методом флуоресцирующие полупроводниковые нанокристаллы, состоящие из металла второй или четвертой групп и халькогенида шестой группы периодической системы (CdSe, CdS, ZnS, PbSe), имеющие размеры от 2 до 7 нм [20, 84]. Как видно, квантовые точки соизмеримы с другими наночастицами, однако их рассматривают как особый тип наночастиц: вследствие специфической электронной структуры они имеют высокий квантовый выход люминесценции.
Обычно квантовые точки получают путем высокотемпературного коллоидного органического синтеза в среде инертного газа. Неорганическое ядро покрывают длинной цепью органического стабилизатора [85]. Данная оболочка обеспечивает образование полимерных нанокомпозитов [86, 87].
На рубеже XXI века был открыт процесс дижестивного созревания, который представляет собой “перекачку” атомов от крупных металлических наночастиц в более мелкие и является удобным способом сужения функции распределения частиц по размерам [88]. Кипячением порошкообразных веществ в трет-бутилтолуоле в присутствии оксида триоктилфосфина, гексадециламина и додекантиола в результате дижестивного созревания получали наночастицы ZnS, CdS, CdSe, CdTe, SnS, PbS, PbSе [89, 90]. Данный метод позволяет наращивать одну или несколько оболочек, также содержащих металл второй группы и халькогенид шестой группы (CdSe/ZnSe, CdSe/ZnSe/ZnS, CdS/ZnS, CdSe/CdS, CdSe/CdTe/ZnSe). При этом определяющую роль играют стабилизирующие агенты, образующие лигандный слой на поверхности наночастиц.
В последнее время все большее внимание привлекают углеродные квантовые точки [91–93], которые получают путем деструкции углеродных материалов, таких как графит, УНТ, сажа, или гидротермального синтеза из углеводов (глюкозы, сахарозы, крахмала) в присутствии HCl или NaOH. Углеродные квантовые точки содержат большое число связей С–С графенового типа с sp2 гибридными орбиталями атомов углерода, а также некоторое число sp3 гибридизованных атомов, что приводит к более неупорядоченной структуре. Эти наночастицы не имеют определенной формы, но фрагменты графена содержат, помимо гексагональных, дефектные участки, влияющие на люминесцентные свойства. По сравнению с другими квантовыми точками размер углеродных частиц меньше, несколько нанометров, молекулярная масса составляет от тысяч до десятков тысяч. На их поверхности имеется большое количество групп –ОН, –СООН, –NH2 и других, которые обеспечивают, с одной стороны, хорошую растворимость в воде, с другой, возможность получения путем полимеризации различных неорганических, органических или биологически активных покрытий [93]. К сожалению, для таких композитов прямых данных о структуре и свойствах межфазных слоев не найдено.
ЗАКЛЮЧЕНИЕ
Результаты комплексного исследования образцов эпоксидных полимеров, отвержденных в присутствии УНТ [94], указывают на структурирование матрицы их поверхностью. Полимерные матрицы другой природы также принимают непосредственное участие в формировании межфазного слоя [95, 96]. Роль сорбатов с ковалентным и/или нековалентным взаимодействием, концентрация которых на поверхности УНТ к тому же невелика, сводится, во-первых, к предотвращению образования жгутов из трубок (кластеров с нематической структурой), во-вторых, к модификации межфазного слоя. Итак, межфазный слой одномерных наночастиц по молекулярной структуре идентичен матрице.
Поскольку непременной стадией формирования полимерных нанокомпозитов с двумерными наночастицами является расслоение последних путем их интеркалирования [53], именно интеркаляты определяют состав и структуру межфазного слоя.
Для стабилизации полярных трехмерных наночастиц и квантовых точек в неполярной матрице необходим лигандный слой, который и формирует межфазную границу в нанокомпозите.
В этом собственно заключаются особенности образования и структуры межфазных слоев в полимерных нанокомпозитах.
Работа выполнена в рамках Госзадания ИПХФ РАН (рег. номер темы АААА-А19-119032690060-9).
Список литературы
Козлов Г.В., Яновский Ю.Г., Карнет Ю.Н. Структура и свойства дисперсно-наполненных полимерных композитов. Фрактальный анализ. М.: Альянстрансатом. 2008.
Филиппов Р.А. // Вест. ННГУ. 2011. № 4. Ч. 2. С. 545.
Chen J., Liu B., Gao X., Xu D. // RSC Adv. 2018. V. 8. № 49. P. 28048.
Chen J., Gao X., Xu D. // Adv. Mater. Sci. Eng. 2019. V. 2019. P. 5268267.
Downing T.D., Kumar R., Cross W.M., Kjerengtroen L., Kellar J.J. // J. Adhes. Sci. Technol. 2000. V. 14. № 14. P. 1801.
Diez-Pascual A.M., Gómez-Fatou M.A., Ania F., Flores A. // Prog. Mater. Sci. 2015. V. 67. P. 1.
Gibson R.F. // Compos. Sci. Technol. 2014. V. 105. P. 51.
Enrique-Jimenez P., Quiles-Díaz S., Salavagione H.J., Fernández-Blázquez J.P., Monclús M.A., Guzman de Villoria R., Gómez-Fatou M.A., Ania F., Flores A. // Compos. Sci. Technol. 2019. V. 169. P. 151.
Sáenz Ezquerro C., Laspalas M., Chiminelli A., Serrano F., Valero C. // Fibers. 2018. V. 6. № 3. P. 54.
Chawla R., Sharma S. // Compos. Sci. Technol. 2017. V. 144. P. 169.
Zaminpayma E., Mirabbaszadeh K. // Comput. Mater. Sci. 2012. V. 58. P. 7.
Winey K.I., Vaia R.A. // MRS Bull. 2007. V. 32. № 4. P. 314.
Galpaya D.G.D., Fernando J.F.S., Rintoul L., Motta N., Waclawik E.R., Yan C., George G.A. // Polymer. 2015. V. 71. P. 122.
Georgakilas V., Otyepka M., Bourlinos A.B., Chandra V., Kim N., Kemp K.C., Hobza P., Zboril R., Kim K.S. // Chem. Rev. 2012. V. 112. № 11. P. 6156.
Britz D.A., Khlobystov A.N. // Chem. Soc. Rev. 2006. V. 35. № 7. P. 637.
Zhao Y.-L., Stoddart J.F. // Acc. Chem. Res. 2009. V. 42. № 8. P. 1161.
Liu L., Etika K.C., Liao K.-S., Hess L.A., Bergbreiter D.E., Grunlan J.C. // Macromol. Rapid. Commun. 2009. V. 30. № 8. P. 627.
Terrones M., Martin O., González M., Pozuelo J., Serrano B., Cabanelas J.C., Vega-Díaz S.M., Baselga J. // Adv. Mater. 2011. V. 23. № 44. P. 5302.
Zhao S., Song Z., Cui J., Li C., Yan Y. // J. Polym. Sci., Polym Chem. 2012. V. 50. № 21. P. 4548.
Бричкин С.Б., Разумов В.Ф. // Успехи химии. 2016. Т. 85. № 12. С. 1297.
Бадамшина Э.Р., Гафурова М.П., Эстрин Я.И. // Успехи химии. 2010. Т. 79. № 11. С. 1027.
Seong M., Kim D.S. // J. Appl. Polym. Sci. 2015. V. 132. № 28. P. 42269.
Wang S., Liang R., Wang B., Sun S. // Polym. Compos. 2009. V. 30. № 8. P. 1050.
Zhang Y., Wang Y., Yu J., Chen L., Zhu J., Hu Z. // Polymer. 2014. V. 55. P. 4990.
Sahoo N.G., Cheng H.K.F., Li L., Chan S.H., Judeh Z., Zhao J. // Adv. Funct. Mater. 2009. V. 19. № 24. P. 3962.
Rubio N., Au H., Leese H.S., Hu S., Clancy A.J., Shaffer M.S.P. // Macromolecules. 2017. V. 50. № 18. P. 7070.
Zdyrko B., Luzinov I. // Macromol. Rapid. Commun. 2011. V. 32. № 12. P. 859.
Hull D., Clyne T.W. An Introduction to Composite Materials. Cambridge Solid State Science Series. Cambridge: Univ. Press, 1996.
Li J., Kim J.-K. // Compos. Sci. Technol. 2007. V. 67. № 10. P. 2114.
Allaoui A., El Bounia N. // Exp. Polym. Lett. 2009. V. 3. № 9. P. 588.
Chen F., Clough A., Reinhard B.M., Grinstaff M.W., Jiang N., Koga T., Tsui O.K.C. // Macromolecules. 2013. V. 46. № 11. P. 4663.
Xie H., Liu B., Yuan Z., Shen J., Cheng R. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 20. P. 3701.
Putz K.W., Palmeri M.J., Cohn R.B., Andrews R., Brinson L.C. // Macromolecules. 2008. V. 41. № 18. P. 6752.
Auad M.L., Mosiewicki M.A., Uzunpinar C., Williams R.J.J. // Polym. Eng. Sci. 2010. V. 50. № 1. P. 183.
Coleman J.N., Cadek M., Ryan K.P., Fonseca A., Nagy J.B., Blau W.J., Ferreira M.S. // Polymer. 2006. V. 47. № 26. P. 8556.
Tsafack T., Alred J.M., Wise K.E., Jensen B., Siochi E., Yakobson B.I. // Carbon. 2016. V. 105. P. 600.
Barber A.H., Cohen S.R., Wagner H.D. // Appl. Phys. Lett. 2003. V. 82. № 23. P. 4140.
Wernik J.M., Cornwell-Mott B.J., Meguid S.A. // Int. J. Solids Struct. 2012. V. 49. № 13. P. 1852.
Li Y., Liu Y., Peng X., Yan C., Liu S., Hu N. // Comput. Mater. Sci. 2011. V. 50. № 6. P. 1854.
Cooper C.A., Cohen S.R., Barber A.H., Wagner H.D. // Appl. Phys. Lett. 2002. V. 81. P. 3873.
Yazdchi K., Salehi M. // Compos. Part A. 2011. V. 42. № 10. P. 1301.
Prashantha K., Lacrampe M.-F., Krawczak P. // Adv. Mater. Manufact. Charact. 2013. V. 3. № 1. P. 11.
Sun P., Liu G., Lv D., Dong X., Wu J., Wang D. // J. Appl. Polym. Sci. 2016. V. 133. № 13. P. 43249.
Veca L.M., Meziani M.J., Wang W., Wang X., Lu F., Zhang P., Sun Y.-P. // Adv. Mater. 2009. V. 21. № 20. P. 2088.
Wan Y.-J., Tang L.-C., Gong L.-X., Yan D., Li Y.-B., Wu L.-B., Jiang J.-X., Lai G.-Q. // Carbon. 2014. V. 69. P. 467.
Li Z., Wang R., Young R.J., Yang F., Hao L., Jiao W., Liu W. // Polymer. 2013. V. 54. P. 6437.
Shanmugharaj A.M., Yoon J.H., Yang W.J., Ryu S.H. // J. Colloid. Interface Sci. 2013. V. 401. P. 148.
Guan L.-Z., Wan Y.-J., Gong L.-X., Yan D., Tang L.-C., Wu L.-B., Jiang J.-X., Lai G.-Q. // J. Mater. Chem. A. 2014. V. 2. № 25. P. 15058.
Wang Y., Yang C., Pei Q.-X., Zhang Y. // ACS Appl. Mater. Interface. 2016. V. 8. № 12. P. 8272.
Konnola R., Joji J., Parameswaranpillai J., Joseph K. // RSC Adv. 2015. V. 5. № 11. P. 61775.
Deng H., Wu F., Chen L., Xu Z., Liu L., Yang C., Mai W., Cheng B. // J. Appl. Polym. Sci. 2014. V. 131. № 23. P. 41164.
Соловьев М.Е., Раухваргер А.Б., Савинский Н.Г., Иржак В.И. // Журн. общ. химии 2017. Т. 87. № 4. С. 677.
Fu S.-Y., Sun Z., Huang P., Li Y.-Q., Hu N. // Nano Mater. Sci. 2019. V. 1. № 1. P. 2.
Kong D., Park C.E. // Chem. Mater. 2003. V. 15. № 2. P. 419.
Becker O., Simon G.P. // Adv. Polym. Sci. 2005. V. 179. P. 29.
Paiva L.B., de Morales A.R., Valenzuela Diaz F.R. // Appl. Clay Sci. 2008. V. 42. № 1–2. P. 8.
Yang L., Phua S.L., Teo J.K.H., Toh C.-L., Lau S.K., Ma J., Lu X. // ACS Appl. Mater. Interface. 2011. V. 3. № 8. P. 3026.
Azeez A.A., Rhee K.Y., Park S.J., Hui D. // Compos. Part B. 2013. V. 45. № 1. P. 308.
Jagtap S.B., Rao V.S., Barman S., Ratna D. // Polymer. 2015. V. 63. P. 41.
Jlassi K., Chandran S., Poothanari M.A., Benna-Zayani M., Thomas S., Chehimi M.M. // Langmuir. 2016. V. 32. № 14. P. 3514.
Bogdanova L., Kuzub L., Dzhavadjan E., Rabenok E., Novikov G., Pomogailo A.D. // Macromol. Sympos. 2012. V. 317–318. P. 117.
Богданова Л.М., Кузуб Л.И., Спирин М.Г., Джардималиева Г.И., Иржак В.И., Дебердеев Р.Я. // Вест. КГТУ. 2015. Т. 18. № 16. С. 10.
Агарева Н.А., Александров А.П., Смирнова Л.А., Битюрин Н.М. // Перспектив. матер. 2009. № 1. С. 5.
Krueger A. // J. Mater. Chem. 2008. V. 18. № 4. P. 1485.
Долматов В.Ю. // Успехи химии. 2007. Т. 76. № 4. С. 375.
Jabeen S., Kausar A., Muhammad B., Gulb S., Farooq M. // Polym.-Plast. Technol. Eng. 2015. V. 54. № 13. P. 1379.
Krueger A., Lang D. // Adv. Funct. Mater. 2012. V. 22. № 5. P. 890.
Mochalin V.N., Neitzel I., Etzold B.J.M., Peterson A., Palmese G., Gogotsi Yu. // ACS Nano. 2011. V. 5. № 9. P. 7494.
Haleem Y., Song P., Liu D., Wang C., Gan W., Saleem M.F., Song L. // Mater. 2016. V. 9. № 7. P. 507.
Пенькова А.В., Acquah S.F.A., Пиотровский Л.Б., Маркелов Д.А., Семисалова А.С., Kroto H.W. // Успехи химии. 2017. Т. 86. № 6. С. 530.
Rafiee M., Yavari F., Rafiee J., Rafiee J., Koratkar N. // J. Nanopart. Res. 2011. V. 13. № 2. P. 733.
Pikhurov D.V., Zuev V.V. // Nanosystem. 2013. V. 4. № 6. P. 834.
Jiang Z., Zhang H., Zhang Z., Murayama H., Okamoto K. // Compos. A. 2008. V. 39. № 11. P. 1762.
Tarasov A.E., Anokhin D.V., Propad Y.V., Bersenev E.A., Razorenov S.V., Garkushin G.V., Badamshina E.R. // J. Compos. Mater. 2019. V. 53. № 26–27. P. 3797.
Henz B.J., Hawa T., Zachariah M.R. // Langmuir. 2008. V. 24. № 15. P. 773.
Gooding J.J., Ciampi S. // Chem. Soc. Rev. 2011. V. 40. № 10. P. 2704.
Ghorai P.K., Glotzer S.C. // J. Phys. Chem. C. 2007. V. 111. № 43. P. 15857.
Kaushik A.P., Clancy P. // J. Chem. Phys. 2012. V. 136. № 11. P. 114702.
Crawford M.K., Smalley R.J., Cohen G., Hogan B., Wood B., Kumar S.K., Melnichenko Y.B., He L., Guise W., Hammouda B. // Phys. Rev. Lett. 2013. V. 110. № 19. P. 196001.
Tuteja A., Duxbury P.M., Mackay M.E. // Phys. Rev. Lett. 2008. V. 100. № 7. P. 077801.
Nakatani A.I., Chen W., Schmidt R.G., Gordon G.V., Han C.C. // Polymer. 2001. V. 42. № 8. P. 3713.
Sen S., Xie Y., Kumar S., Yang H., Bansal A., Ho D.L., Schweizer K.S. // Phys. Rev. Lett. 2007. V. 98. № 12. P. 128302.
Dattani R., Michels R., Nedoma A.J., Schweins R., Westacott P., Huber K., Cabral J.T. // Macromolecules. 2014. V. 47. № 17. P. 6113.
Waldron D.L., Preske A., Zawodny J.M., Krauss T.D., Gupta M.C. // Nanotechnol. 2015. V. 26. № 7. P. 075705.
Vasiliev R.B., Dorofeev S.G., Dirin D.N., Belov D.A., Kuznetsova T.A. // Mendeleev Commun. 2004. V. 14. № 4. P. 169.
Vasilets V.N., Savenkov G.N., Merekalov A.S., Shandryuk G.A., Satalova A.M., Talroze R.V. // Polymer Science A. 2011. V. 53. № 6. P. 521.
Tselikov G.I., Shandryuk G.A., Kutergina I.Y., Satalova A.M., Merekalov A.S., Timoshenko V.Yu., Talroze R.V. // Polymer Science A. 2014. V. 56. № 6. P. 781.
Иржак В.И. // Журн. физ. химии. 2017. Т. 91. № 8. С. 1359.
Heroux D., Ponce A., Cingarapu S., Klabunde K.J. // Adv. Funct. Mater. 2007. V. 17. № 17. P. 3562.
Cingarapu S., Yang Z., Sorensen C.M., Klabunde K.J. // Inorg. Chem. 2011. V. 50. № 11. P. 5000.
Zuo J., Jiang T., Zhao X., Xiong X., Xiao S., Zhu Z. // J. Nanomater. 2015. V. 2015. P. 787862.
Кокорина А.А., Прихожденко E.C., Сухоруков Г.Б., Сапелкин А.В., Горячева И.Ю. // Успехи xимии. 2017. Т. 86. № 11. С. 1157.
Kandasamy G. // J. Carbon Res. 2019. V. 5. № 2. P. 24.
Cao L., Wang X., Meziani M.J., Wang H., Luo P.G., Lin Y., Harruff B.A., Veca L.M., Murray D., Xie S.-Y., Sun Y.-P. // J. Am. Chem. Soc. 2007. V. 129. № 37. P. 11318.
Kondrashov S.V., Grachev V.P., Akatenkov R.V., Aleksashin V.N., Deev I.S., Anoshkin I.V., Rakov E.G., Irzhak V.I. // Polymer Science A. 2014. V. 56. № 3. P. 330.
Mikitaev A.K., Kozlov G.V. // Polymer Science D. 2016. V. 9. № 1. P. 110.
Козлов Г.В., Долбин И.В. // Физика и химия стекла. 2019. Т. 45. № 4. С. 255
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)