

Высокомолекулярные соединения (серия С), 2020, T. 62, № 2, стр. 108-121
ПОЛИАРИЛЕНЭФИРКЕТОНЫ − ТЕРМО-, ТЕПЛО- И ХЕМОСТОЙКИЕ ТЕРМОПЛАСТЫ И ПЕРСПЕКТИВЫ СОЗДАНИЯ РАЗЛИЧНЫХ МАТЕРИАЛОВ НА ИХ ОСНОВЕ
С. Н. Салазкин 1, В. В. Шапошникова 1, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: vsh@ineos.ac.ru
Поступила в редакцию 02.03.2020
После доработки 24.03.2020
Принята к публикации 10.04.2020
Аннотация
Обобщены литературные данные о синтезе и свойствах полиариленэфиркетонов, сочетающих в себе высокую термо-, тепло- и хемостойкость с уникальными механическими и электрофизическими характеристиками. Особое внимание уделено синтезу полиариленэфиркетонов реакцией нуклеофильного замещения активированного арилдигалогенида и показана возможность регулирования разнообразных свойств полимеров.
ВВЕДЕНИЕ
Химия тепло- и термостойких термопластичных полимеров стремительно развивается, начиная с 50-х годов ХХ века. В результате было синтезировано много новых полимеров различного строения с широким набором свойств.
Интенсивное развитие техники и высокотехнологичных отраслей мировой экономики обеспечивает повышенный спрос на термо-, теплостойкие конструкционные термопласты [1–6]. Несмотря на большое разнообразие классов конструкционных термопластов, полиариленэфиркетоны (ПАЭК) (в частности, производимый в промышленном масштабе за рубежом полиэфирэфиркетон (PEEK) марки Victrex) занимают одно из ведущих мест по широте применения в технике [7], что обусловлено наличием у них комплекса ценных свойств, а именно, сочетания высокой термо-, тепло-, хемостойкости с уникальными механическими, хорошими оптическими и электрофизическими характеристиками [8–12].
МЕТОДЫ СИНТЕЗА ПОЛИАРИЛЕНЭФИРКЕТОНОВ
Традиционные, наиболее распространенные методы получения ПАЭК, имеющие большое практическое значение, основаны на реакциях электрофильного и нуклеофильного замещения, а также полимеризации с раскрытием цикла [13–17]. К недостаткам последнего метода следует отнести трудность контроля полимеризации, недостижимость 100%-ной конверсии мономеров даже при повышенной температуре и широкое ММР получаемых полимеров. В некоторых случаях полимеризация с раскрытием цикла затруднена из-за высокой температуры плавления макроциклических олигомерных ПАЭК. Катион-радикальная полимеризация диарилоксипроизводных (реакция Шолля) [18–21] и поликонденсация металлоорганических соединений с арилдигалогенидами по реакции палладий- или никель-катализируемого кросс-сочетания [22, 23] не нашли широкого применения в синтезе ПАЭК. Необходимо отметить, что реакции кросс-сочетания широко используются в органическом синтезе, поскольку осуществляются в мягких условиях и позволяют получать продукты с высоким выходом и селективностью.
Осуществление синтеза ПАЭК реакцией электрофильного замещения возможно двумя путями: гомополиконденсацией ароматических монокарбоновых кислот и их хлор- или фторангидридов, а также поликонденсацией полиядерных ароматических углеводородов, в том числе содержащих в молекуле простую эфирную связь, с ароматическими дикарбоновыми кислотами и их хлор- или фторангидридами, а также фосгеном. Реакцию проводят в среде органических растворителей (метиленхлорид, 1,2-дихлорэтан, нитробензол и другие) в присутствии катализаторов AlCl3, FeCl3, SbCl5 и т.д.; с использованием системы растворитель–катализатор HF/BF3; в сильных кислотах (полифосфорной, трифторметансульфокислоте, метансульфокислоте, а также в двух последних в присутствии P2O5 и PCl5). Подробная информация о синтезе и свойствах ПАЭК, получаемых реакцией электрофильного замещения, представлена в обзорах [24–28].
Широкий выбор и доступность исходных мономеров, низкие температуры синтеза, возможность получения ПАЭК без мостиковой эфирной связи в основной цепи макромолекулы являются несомненными достоинствами синтеза ПАЭК реакцией электрофильного замещения. В то же время имеются и недостатки, которые ограничивают возможности использования этого метода для получения ПАЭК в промышленном масштабе. К ним относятся необходимость использования больших количеств катализатора AlCl3, проблема очистки полимера от AlCl3 и регенерации катализатора, а также сложность технологической реализации синтеза ПАЭК (по окончании синтеза реакционная масса представляет собой монолит, который трудно выгрузить из реактора).
Основное преимущество синтеза ПАЭК реакцией нуклеофильного замещения – высокая селективность и возможность использования широкого ряда ароматических бисфенолов, в том числе и кардовых, что позволяет получать полимеры различного строения и регулировать их свойства в широком диапазоне. Отсутствие в России промышленного производства одного из исходных мономеров – 4,4'-дифторбензофенона, высокие температуры синтеза, необходимость удаления из полимера примесей (остатки растворителя, KF, NaF, KCl, NaCl) относятся к недостаткам синтеза ПАЭК реакцией нуклеофильного замещения. Отметим, что проблему примесей в полимере при синтезе ПАЭК реакцией нуклеофильного замещения разрешить легче, чем при синтезе ПАЭК реакцией электрофильного замещения.
Реакция нуклеофильного замещения является перспективной для синтеза новых полимеров и для разработки методов промышленного получения уже известных полимеров, в том числе ПАЭК. В связи с этим представляется целесообразным уделить значительное внимание анализу литературы по синтезу, свойствам и применению ПАЭК, получаемых с помощью данной реакции.
СИНТЕЗ ПАЭК РЕАКЦИЕЙ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
Реакция нуклеофильного замещения широко используется для синтеза ПАЭК, что нашло отражение в многочисленных литературных источниках, включая обзоры [24, 28–31], и патентах.
Известны два пути синтеза ПАЭК реакцией нуклеофильного замещения: гомополиконденсация фенолятов частично гидролизованных дигалогенидов, содержащих в молекуле карбонильную группу и поликонденсация производных ароматических бисфенолов с активированными ароматическими дигалогенидами, а также ароматическими нитросоединениями.
Впервые о синтезе ПАЭК реакцией нуклеофильного замещения галогена в активированном арилдигалогениде стало известно в 1967 г. [32, 33]. Взаимодействием 4,4'-дихлорбензофенона с динатриевым дифенолятом бисфенола А в присутствии окиси меди, используемой в качестве катализатора, в ДМСО был получен низкомолекулярный полимер [32]. В случае замены 4,4'-дихлорбензофенона на 4,4'-дифторбензофенон были синтезированы высокомолекулярные ПАЭК различного химического строения без применения катализатора [33].
Эффективность осуществления синтеза ПАЭК реакцией нуклеофильного замещения определяется строением исходных мономеров, природой растворителя, типом фенолята, протеканием побочных реакций.
Строение исходных мономеров
Реакционная способность фенолятов бисфенолов в реакции нуклеофильного замещения изменяется обратно пропорционально их кислотности. Иными словами, сильные электроотрицательные группы (–SO2–, –SO–, –CO–) в бисфеноле делокализуют отрицательный заряд на кислороде и тем самым снижают его реакционную способность, а электронодонорные группы (–C(CH3)2–, –S–, –O– и другие) увеличивают реакционную способность фенолята бисфенола.
Реакционная способность ароматического дигалогенида зависит от природы галогена и от присутствия в его молекуле электроноакцепторных групп. Галогены по реакционной способности в реакции ароматического нуклеофильного замещения располагаются в ряд F $ \gg $ Cl $ \gg $ Br $ \gg $ I. Наиболее реакционноспособными являются дигалогениды, имеющие в своем составе группы –SO2– и –NO2–. Причем группа NO2 активирует дигалогенид сильнее, чем группа SO2. Наличие в дигалогениде таких групп позволяет использовать в реакции нуклеофильного замещения не только фтор-, но и хлорпроизводные. Карбонильная (–CO–), азо-(–N=N–), сульфоксидная группа (–SO–), оксадиазольный цикл по сравнению с группой SO2 активируют дигалогенид в значительно меньшей степени. В связи с этим в данном случае достаточно активны только фторпроизводные [33]. Активация фтор- и хлорпроизводных циан группой (–CN–) [34–36], пиридиновым циклом [35], а также активация фторпроизводных хиноксалиновым, бензоксазольным, бензимидазольным, бензтиазольным [37], пираноновым [38] циклами, 3,6-дикарбонилдибензофурановым [39], карбазольным [40] фрагментами, амидной (–CO–NH–) [37, 41], перфторалкильной группой (–(–CF2–)6–) [37, 42], фенилфосфиноксидной (–PO–C6H5–) [43–46] и другими группами является достаточной для эффективного протекания реакции нуклеофильного замещения.
Реакция нуклеофильного замещения при синтезе ПАЭК осуществляется через стадию образования комплекса Мейзенгеймера (σ-комплекса). Детальное изучение механизма этой реакции для ПАЭК [47–49] показало, что в некоторых случаях наряду с реакцией нуклеофильного ароматического замещения (SNAr) активированного арилгалогенида возможна реакция по механизму радикального мономолекулярного нуклеофильного замещения (SRN1) по схеме
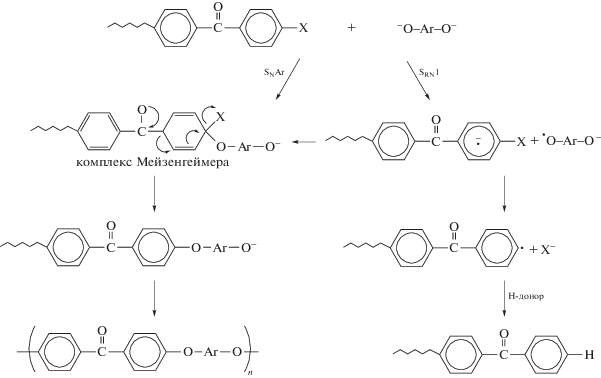
Протекание реакции по механизму SRN1 приводит к образованию нереакционноспособных бензофеноновых концевых групп и к получению полимеров с низкой ММ. Авторы [48] установили, что возможность и степень такой реакции определяется типом дигалогенидов и дифенолятов. Вероятность протекания этой реакции более высокая в случае синтеза ПАЭК на основе дихлорариленов и гидрохинона (по сравнению с получением ПАЭК путем взаимодействия дифторариленов с гидрохиноном).
Природа растворителя
Наиболее подходящими для синтеза ПАЭК реакцией нуклеофильного замещения являются диполярные апротонные растворители, поскольку в них хорошо растворяются исходные мономеры, образующиеся феноляты бисфенолов, олигомеры с концевыми фенолятными группами и высокомолекулярный полимер. В отличие от протонных апротонные диполярные растворители в случае растворенных ионных соединений сольватируют в основном катионы, оставляя анионы относительно свободными и сильно реакционноспособными. Это приводит к увеличению донорной способности нуклеофила и, следовательно, к очень сильному ускорению (на несколько порядков) процесса поликонденсации. Наиболее широко используемые диполярные апротонные растворители – ДМСО, ДМФА, ДМАА, N-метил-2-пирролидон и другие.
Для синтеза кристаллизующихся ПАЭК актуальна проблема выбора растворителя, поскольку такие полимеры при проведении реакции в обычном диполярном апротонном растворителе осаждаются из реакционной массы, не достигнув высокой или даже средней ММ. Для решения данной проблемы поликонденсацию осуществляют при высокой температуре (до 350°С) и используют растворители, которые при этом не разлагаются. К ним относятся дифенилсульфон, бензофенон [50], сульфолан [50–52]. Применение таких растворителей, как 9-ксантон, флуоренон, антрахинон позволяет осуществлять синтез ПАЭК при 325–350°С [53]. Авторами [54, 55] была предпринята попытка синтеза ПАЭК в высококипящих растворителях, таких как N,N,N',N'-тетраметилмочевина, N-ацетилпиперидин, N-бензоилпиперидин и N,N-диметилбензамид, ранее не использовавшихся для получения этих полимеров.
Необходимо отметить, что использование в синтезе ПАЭК дифенилсульфона, являющегося твердым при комнатной температуре, упрощает методику их выделения, поскольку исключает стадию переосаждения полимера, и позволяет получить высокомолекулярные полимеры. В соответствии с данными, представленными в работах [56, 57], после проведения синтеза ПАЭК реакционную массу охлаждали, экструдировали в пленку, экстрагировали из пленки растворитель, образовавшийся при реакции KF и остатки K2СO3, соответственно метанолом и водой.
Тип фенолята
В синтезе ПАЭК реакцией нуклеофильного замещения используют натриевые и калиевые дифеноляты бисфенолов, что обусловлено их растворимостью. При этом необходимо учитывать, что применение менее активного по сравнению с калиевым натриевого фенолята бисфенола на основе ароматических дихлорпроизводных неэффективно. Для получения фенолятов в реакционную массу вводят растворы NaOH и KOH, твердые карбонаты и бикарбонаты натрия и калия, карбонаты и бикарбонаты натрия и других щелочных металлов в смеси с K-, Rb-, Cs-солями органических кислот. Для предотвращения разветвления полимерной цепи в процессе синтеза ПАЭК применяют смеси карбонатов натрия и калия [58, 59]. Фенолят может быть приготовлен заранее или in situ.
Побочные реакции
Большое значение при синтезе конденсационных полимеров имеет соблюдение стехиометрии мономеров, проведение реакции в инертных условиях с целью защиты от кислорода и влаги, содержащейся в воздухе, и тщательное удаление воды из зоны реакции путем азеотропной отгонки. Преимущество использования твердых карбонатов и бикарбонатов натрия или калия по сравнению с водными растворами NaOH или KOH состоит в том, что не требуется точная дозировка концентрации щелочи и удаление больших количеств воды. Необходимость защиты от кислорода воздуха обусловлена тем, что с ростом температуры увеличивается способность фенолятов к окислению. Для более тщательного удаления воды из зоны реакции при азеотропной отгонке используют молекулярные сита [60].
Выполненное авторами [61] исследование возможности протекания побочных реакций при синтезе ПАЭК на основе 4,4'-дифторбензофенона и бисфенола А в присутствии поташа в ДМАА показало, что деструктивные процессы возможны только под воздействием калиевого дифенолята бисфенола А.
К побочным реакциям относятся также процессы ветвления и сшивания макромолекул, которые могут протекать при проведении синтеза в высококипящих растворителях при температуре более 300°С [62, 57 ].
Использование фундаментальных результатов исследования закономерностей поликонденсации [27, 59, 63–68] позволило оптимизировать условия синтеза полимеров и получить ПАЭК с регулируемой ММ от 10 до 100 × 103 [27, 66–68].
В начале 70-х годов ХХ века были синтезированы кардовые полимеры [69], свойства которых обусловливают повышенный интерес к ним до настоящего времени. Кардовые группировки могут быть введены в полимерную цепь в составе бисфенолов [69–73] и дигалогенпроизводных [74–76].
Несмотря на то, что кардовые ПАЭК впервые были синтезированы в ИНЭОС АН СССР, метод синтеза ПАЭК на основе фенолфталеина и 4,4'-дигалогенбензофенона был запатентован китайскими исследователями [77]. В настоящее время в КНР в промышленном масштабе производят ПАЭК на основе 4,4'-дифторбензофенона и фенолфталеина (PEK-C) [78].
Использование в поликонденсации активированных олигомерных гомологов 4,4'-дифторбензофенона позволило впервые получить статистические кардовые со-ПАЭК, проявляющие склонность к кристаллизации [79, 80].
Другой подход к синтезу кардовых ПАЭК, основанный на использовании в поликонденсации кардовых активированных ароматических дифтор-, дихлор- и дибромпроизводных [74, 75], обеспечил получение высокомолекулярных кардовых ПАЭК только в случае применения кардового дифторпроизводного:
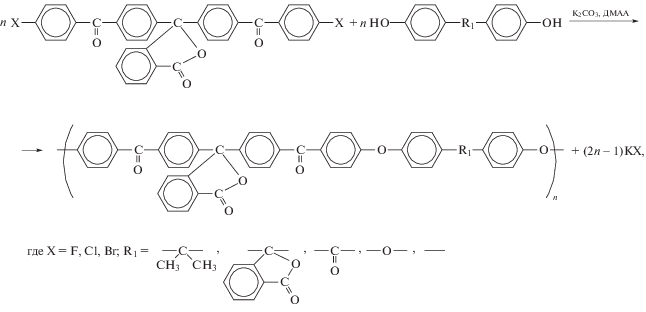
Попытка синтеза кардовых ПАЭК на основе 3,3-бис-(4-фторфенил)фталида и ряда бисфенолов [76] оказалась неудачной, так как не позволила получить высокомолекулярные полимеры даже при использовании жестких условий синтеза (растворитель N,N-диметилпропиленмочевина, температура до 220°С, продолжительность синтеза до 52 ч), что, по-видимому, обусловлено низкой активацией дифторпроизводного фталидным циклом.
Наряду с кардовыми полимерами разнообразием ценных свойств отличаются фторированные ПАЭК [60, 81–85]. Известны два подхода к синтезу таких полимеров – введение фтор-, трифторметильных групп в нефторированный ПАЭК путем химической модификации и поликонденсация мономеров, содержащих фтор-, трифторметильные группы. Традиционным является второй подход. При его реализации возможны два варианта проведения поликонденсации: на основе мономеров, содержащих гексафторизопропилиденовую группу [86], а также с использованием мономеров, содержащих фтор-, трифторметильные группы в ароматическом кольце [85, 87]. Фторированные ПАЭК могут быть получены реакцией нуклеофильного замещения, как F-, так и NO2-групп. Анализ литературных данных по синтезу и свойствам фторированных ПАЭК содержится в обзоре [83].
Как было отмечено выше, при получении полукристаллических ПАЭК актуальная проблема – преждевременная кристаллизация полимера и его осаждение из реакционной массы в процессе синтеза. Широко известный коммерческий суперконструкционный полимер PEEK получают нагреванием в токе азота эквимольных количеств 4,4'-дифторбензофенона и гидрохинона, взятого в небольшом избытке тщательно измельченного поташа в дифенилсульфоне при 200°С 1 ч, 250°С 1 ч и при 320°С 1 ч [88]. Необходимость использования столь жестких условий синтеза PEEK обусловлена высокой кристалличностью и нерастворимостью этого полимера в обычных органических растворителях. К отрицательным моментам синтеза PEEK можно отнести высокую чувствительность калиевого дифенолята гидрохинона к кислороду воздуха.
Для решения проблемы преждевременного осаждения кристаллического полимера из реакционной массы используется подход, заключающийся в синтезе аморфных, растворимых в широком ряде органических растворителей преполимеров PEEK, содержащих объемные боковые заместители (например, трет-бутильные [89], кетальные [90, 91], кетиминные [92–94], аминонитрильные [95] группы), и дальнейшем удалении этих групп путем химических превращений, что позволяет получить высокомолекулярные полукристаллические полимеры.
Авторы [89] реализовали синтез PEEK в соответствии со схемой
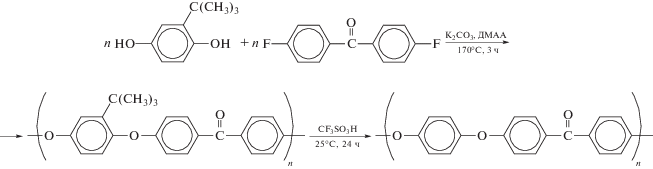
и показали, что полное удаление трет-бутильной группы возможно при использовании в качестве катализатора и растворителя трифторметансульфоновой кислоты.
Наибольшее внимание ученые уделяют химическим превращениям ПАЭК в боковых или основных цепях макромолекул, не приводящим к изменению длины цепи. В случае ПАЭК деструкция макромолекул и реакции их сшивания друг с другом исследуются преимущественно как побочные реакции [61], протекающие в процессе синтеза, переработки или эксплуатации полимеров.
Результаты исследования химических превращений ПАЭК с участием как бензольного кольца [96–104] и карбонильных групп [105–109] основной цепи, так и боковых функциональных групп (атомов галогенов [110–112], сульфокислотных [113–117], карбоксильных [69, 118–123], циано- [124–128], амино- [129] и других [112, 130–133] групп) свидетельствуют о широких возможностях модификации строения и свойств полимеров.
Осуществление синтеза ПАЭК, содержащих боковые реакционноспособные функциональные группы, возможно двумя путями: введением реакционноспособных групп в готовую макромолекулу и поликонденсацией мономеров, содержащих в своем составе наряду с двумя участвующими в реакции функциональными группами, еще одну или несколько групп, инертных в условиях синтеза полимера. Второй путь синтеза ПАЭК является более интересным, несмотря на трудности его практической реализации. Например, получение высокомолекулярных ПАЭК, содержащих боковые реакционноспособные карбоксильные группы, затруднено осаждением образующейся полимерной соли из реакционной среды и частичной деструкцией макромолекул при их взаимодействии с карбоксилатными группами. Авторы работ [73, 134] преодолели эти трудности и получили высокомолекулярные ПАЭК.
Введение в макромолекулу ПАЭК объемных боковых групп (как алифатических, так и ароматических) очень часто бывает необходимым не с целью их дальнейших химических превращений, а для придания полимерам новых функциональных свойств [135, 136], улучшения их растворимости и способности к переработке [137–140].
Подробный анализ литературных данных по химическим превращениям ПАЭК сделан в литературном обзоре диссертации [141].
Синтез ароматических ПАЭК может быть успешно осуществлен без растворителя с использованием больших избытков карбонатов и бикарбонатов щелочных металлов при нагревании от 200 до 350–370°С [142].
Перспективным методом синтеза ПАЭК является метод, основанный на использовании триметилсилильных эфиров бисфенолов, заключающийся в нагревании эквимольных количеств 4,4'-дифторбензофенона и триметилсилильного эфира бисфенола в присутствии CsF или KF при 230–270°С в течение 0.5–1 ч, 350°С – 0.5–1 ч [143, 144].
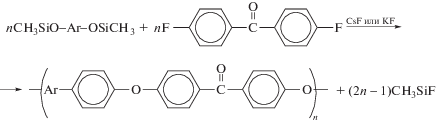
Преимущества этого метода состоят в следующем: происходит непрерывная регенерация катализатора в процессе синтеза; единственным побочным продуктом является летучий триметилсилилфторид; полученный полимер не требует дополнительной очистки. Однако полимеры, полученные данным методом, характеризуются высокой полидисперсностью (от 4 до 10). Низкая реакционная способность дихлорпроизводных не позволяет использовать их для получения ПАЭК на основе триметилсилильных эфиров бисфенолов.
Другой вариант синтеза ПАЭК без использования щелочей и карбонатов щелочных металлов – метод, основанный на применении в реакции фторида цезия. Высокомолекулярные ПАЭК на основе 4,4'-дифторбензофенона и бисфенола А могут быть получены без использования щелочей и карбонатов щелочных металлов, а именно, в присутствии фторида цезия в N-метил-2-пирролидоне при 160°С (продолжительность 3 ч) [145]. Фториды металлов могут выступать в роли промотирующих агентов при поликонденсации дихлорпроизводных с бисфенолами. Применение фторида цезия в смеси с гидридом кальция [81, 82] существенно увеличивает скорость реакции и позволяет получить высокомолекулярные ПАЭК на основе бисфенола AF и декафторбензофенона при нагревании при 60°С в течение 3 ч (в темноте и в среде аргона). К сожалению, потенциальная взрывоопасность гидрида кальция в процессе синтеза ограничивает широкое использование этого метода.
Разновидностью нуклеофильного замещения при синтезе ПАЭК является реакция с отщеплением NO2-группы [146].
Известный способ получения высокомолекулярных ПАЭК, при котором легко выдержать стехиометрию исходных мономеров, – гомополиконденсация частично гидролизованных ароматических дигалогенидов, например 4-фтор-4'-гидроксибензофенона (в присутствии K2CO3 в дифенилсульфоне при 335°С) [62, 147]. Этим способом получают коммерческий полимер зарубежной марки PEK.
Получение высокомолекулярных ПАЭК на основе дигалоген(фтор, хлор)производных без использования ароматических бисфенолов описано в работах [148–150]. Синтез ПАЭК осуществляют реакцией 4,4'-дихлорбензофенона с Na2CO3 в присутствии каталитической системы SiO2/CuCl2 в дифенилсульфоне при 280–320°С [148]:

Реакция 4,4'-дифторбензофенона с K2CO3 в присутствии SiO2 протекает быстро, и полимер достигает максимального значения ММ за короткий промежуток времени. Однако в указанных полимерах обнаружен гель. Авторы [148] предполагают, что K2CO3 способен в процессе синтеза разрывать простую эфирную связь в макромолекуле с образованием свободного фенола, который может вызывать побочные реакции, приводящие к разветвлению полимерной цепи и образованию геля. В связи с этим для синтеза ПАЭК данным методом желательно использовать Na2CO3 при мольном соотношении ароматический дигалогенид : Na2CO3 = 1 : (1–2).
Относительно новым и перспективным является синтез высокомолекулярных ПАЭК реакцией нуклеофильного замещения под воздействием СВЧ-излучения [151, 152]. С целью оптимизации условий поликонденсации авторы работы [151] исследовали влияние параметров синтеза (интенсивность СВЧ-излучения, природа растворителя, продолжительность синтеза) на ММ полимеров и показали положительное влияние СВЧ-излучения: под его воздействием ПАЭК достигают высокой ММ за более короткий промежуток времени.
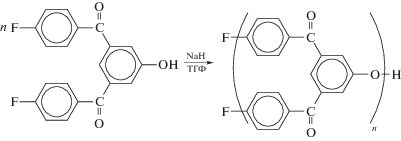
Реакцию нуклеофильного замещения успешно используют для получения гиперразветвленных ПАЭК [153–155]. Впервые такие полимеры были получены авторами [153] в присутствии гидрида натрия в ТГФ:
СВОЙСТВА И ПРИМЕНЕНИЕ ПАЭК
Анализ большого числа литературных источников показал возможность получения ПАЭК разнообразного химического строения, определяющего их функциональные свойства. Значения температуры плавления Тпл и стеклования Тс полимеров повышаются при увеличении жесткости полимерных цепей за счет введения в макромолекулу объемных алифатических [156], ароматических [137, 140, 157–160], в том числе кардовых, и полярных групп, гетероциклических [86, 135, 161, 162], фениленовых [163] и нафталиновых фрагментов [107, 164]. Например, при замещении водорода в мостиковой метиленовой группе исходного бисфенола, являющегося фрагментом основной полимерной цепи, на метильную или фенильную группы Тс увеличивается на 15–25°С, при замещении на две фенильные группы – на 40–50°С [29, 33]. ПАЭК, содержащие кардовые группы, обладают более высокой Тс (на 60–100°С) по сравнению с ПАЭК без кардовых групп [165]. Замена карбонильной группы в основной цепи макромолекулы на мостиковые простые эфирные группы приводит к уменьшению Тпл, Тс и степени кристалличности [30]. Гексафторизопропилиденовые группы фрагмента бисфенола способствуют увеличению Тс [86]. В целом фторированные ПАЭК характеризуются более высокими термическими свойствами по сравнению с нефторированными полимерами. Высокие термические свойства фторированных ПАЭК, по-видимому, объясняются более высокой прочностью связи C–F по сравнению с C–H [84].
Сведения о термоокислительной устойчивости ПАЭК позволяют сделать вывод о том, что полимеры, имеющие объемные боковые заместители, в том числе галогенсодержащие [166, 167], характеризуются высокой термоокислительной устойчивостью (>500°С) [51, 52, 136, 160, 162, 166, 168, 169]. Большой интервал между величинами температуры переработки расплава и Тразл создает значительные преимущества при переработке полимеров.
Для многих ПАЭК существует корреляция между химическим строением элементарного звена и значениями Тпл, Тс, что позволяет рассчитать их величину и спрогнозировать свойства вновь синтезируемых полимеров [170, 171].
К сожалению, данные о механических свойствах ПАЭК, не производимых в промышленном масштабе, в литературе недостаточно систематизированы. Необходимо отметить важные с практической точки зрения публикации [64, 67, 172–175], в которых представлены результаты исследования зависимости физико-механических, реологических свойств ПАЭК от их ММ. Полученные сведения позволили определить значение ММ, являющееся оптимальным для достижения желаемых свойств ПАЭК. Данные, представленные в работах [67, 172], свидетельствуют о том, что высокие механические свойства пленок (для ПАЭК на основе 4,4'-дифторбензофенона и бисфенола А прочность при разрыве ${{\bar {\sigma }}_{р}}$ составляет до 95 МПа, относительное удлинение ${{\bar {\varepsilon }}_{р}}$ – до 240%) и монолитных образцов ПАЭК (например, ПАЭК на основе 4,4'-дифторбензофенона и фенолфталеина обладает удельной ударной вязкостью с надрезом до 35 кДж/м2 [151], а со-ПАЭК на основе 4,4'-дифторбензофенона и смеси бисфенола А, анилида фенолфталеина – до 44 кДж/м2 [176]) [177] обеспечивают перспективность применения этих полимеров в качестве связующих для композиционных материалов, для получения из них пленок, волокон. Добавление ПАЭК на основе 4,4'-дифторбензофенона и фенолфталеина (например, производимого промышленностью Китая полимера PEK-C) в композицию с эпоксидным связующим при получении углепластиков позволяет существенно повысить показатель удельной ударной вязкости композиционного материала [178]. По-видимому, это обусловлено не только высокой ударной вязкостью ПАЭК, но и улучшением адгезии эпоксидного связующего к армирующему материалу [179–181].
Реакция нуклеофильного замещения позволяет получать ПАЭК разного химического строения как кристаллической, так и аморфной структуры. В основном кристаллические ПАЭК растворимы только в концентрированной серной кислоте. Аморфные ПАЭК растворимы в апротонных диполярных растворителях (ДМАА, ДМСО, ДМФА и других) и хлорсодержащих растворителях (метиленхлориде, хлороформе, 1,2-дихлорэтане и т.д.). Присутствие в макромолекуле кардовых и других объемных группировок (например, гексафторизопропилиденовых групп [86]), фтор-, трифторметильных групп, присоединенных к ароматическому кольцу, способствует улучшению растворимости ПАЭК [83, 84, 86, 167]. Наличие фтора в макромолекуле наряду с хорошей растворимостью в органических растворителях и улучшенными термическими показателями обеспечивает высокую гидрофобность, низкую диэлектрическую постоянную и слабую адгезию фторированных полимеров [83] по сравнению с не фторированными. В отличие от фторсодержащих полимеров ПАЭК, содержащие фрагменты 3,3-бис-(4'-гидроксифенил)фталимидина или 2-(β-гидроксиэтил)-3,3-бис-(4'-гидроксифенил)фталимидина, благодаря наличию водородных связей являются гидрофильными и характеризуются повышенной адгезией [141, 182].
Очень ценное качество ПАЭК – их высокая гидролитическая и химическая устойчивость при высоких температурах [183].
ПАЭК перспективны для получения на их основе мембран для топливных элементов [121, 162, 184–193]. Данные всестороннего исследования характеристик аморфного PEK-C [194–196] показывают, что наряду с высокой термо-, тепло-, хемостойкостью и уникальными механическими характеристиками этот полимер имеет хорошие газотранспортные [194, 196] и протонобменные [197] свойства, что позволяет использовать его для получения газоразделительных мембран и мембран топливных элементов. Серьезного внимания заслуживают гомо-ПАЭК на основе 4,4'-дифторбензофенона и имида фенолфталеина и его сополимеры с PEK-C, которые имеют очень хорошие газотранспортные характеристики [196]. Существует очень много публикаций, посвященных исследованию свойств мембран из ПАЭК разнообразного химического строения [87, 117, 123, 159, 169, 198–231 ], что свидетельствует о большой перспективности этого научного направления.
Среди ценных функциональных свойств ПАЭК, прежде всего кардовых ПАЭК, необходимо отметить следующие: эффект электронного переключения при внешних воздействиях (давление, температура, электрическое и магнитное поле и т.д.) [232–235], электро- и фотолюминесценция, анизотропная электропроводность [236, 237]. Наличие у ПАЭК таких свойств указывает на большие возможности использования полимеров в электронике, радиотехнике и приборостроении. Показано, что некоторые из представителей ПАЭК могут применяться в качестве оптически прозрачных пленочных адгезивов с анизотропной электропроводностью при создании солнечных элементов [236, 237].
Высокие биологическая стабильность, чистота, химическая стойкость, механические и трибологические свойства, стойкость к различным способам стерилизации, хорошая биологическая совместимость с тканями человеческого организма определяют эффективность использования PEEK в качестве материала для имплантации, обеспечивающего отсутствие трения при функционировании в экстремально агрессивной среде человеческого тела [238–240]. PEEK применяют для изготовления деталей медицинского оборудования и инструментария (насосов, диализных машин, стерилизаторов и т.д.). Известно получение ПАЭК, обладающих антигрибковым и антимикробным действием [241].
В последнее время активно развиваются аддитивные технологии – получение изделий из полимерных материалов с помощью компьютерных 3D технологий. Известно, что благодаря комплексу ценных свойств частично кристаллический полимер PEEK перспективен для применения в этих технологиях [242–246]. Преимущества 3D технологии заключаются в следующем: в отличие от традиционных способов переработки полимеров (прессование, литье под давлением и другие) данная технология позволяет получать объекты любой степени сложности и геометрии, используя цифровую модель; сокращается продолжительность цикла получения изделия. Для изготовления 3D-изделий из PEEK в основном используют селективное лазерное спекание [245, 246]. Однако образцы изделий, полученные данным методом, уступают по механическим свойствам образцам, полученным методом литья под давлением, поскольку из-за отсутствия давления при спекании частиц порошка полимера изделие имеет пористую структуру. Таким образом, для широкого внедрения 3D технологии получения изделий на основе PEEK требуются дополнительные исследования по оптимизации условий лазерного спекания, обеспечивающих производство бездефектных образцов.
ЗАКЛЮЧЕНИЕ
Анализ большого объема литературных данных по синтезу, свойствам и областям применения ПАЭК свидетельствует о значительном интересе исследователей к этому классу полимеров, что объясняется уникальным сочетанием функциональных и технологических свойств, присущих ПАЭК. Усилия многих ученых сконцентрированы на комплексных исследованиях синтеза, свойств и химических превращений гомо- и со-ПАЭК, синтезируемых реакцией нуклеофильного замещения.
Исследуемые ПАЭК имеют практическую ценность как высокотермо-, тепло- и хемостойкие конструкционные термопласты с хорошими механическими свойствами, в частности уникальной удельной ударной вязкостью. Выявленные новые функциональные свойства ПАЭК обеспечивают возможность их применения для создания ценных материалов: мембран для газоразделения и топливных элементов; материалов с эффектом обратимого электронного переключения в результате внешних воздействий, с электро- и фотолюминесценцией; оптически прозрачных пленочных адгезивов с анизотропной электропроводностью; материалов для медицины. Высокие термические (теплостойкость от 150 до 300°С и выше, термостойкость более 450°С) и механические характеристики аморфных и частично кристаллических ПАЭК, стабильные в условиях высокой влажности и температуры, стойкость к радиации, гидролизу, к химическим реагентам позволяют использовать материалы на основе ПАЭК в авиакосмической, оборонной, атомной, химической промышленности, электронике, электротехнике, машиностроении.
Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации.
Список литературы
Михайлин Ю.А. Термоустойчивые полимеры и полимерные материалы. СПб.: Профессия, 2006.
Михайлин Ю.А. Специальные полимерные композиционные материалы. СПб.: Научные основы и технологии, 2009.
Михайлин Ю.А. Конструкционные полимерные композиционные материалы. СПб.: Научные основы и технологии, 2010.
Михайлин Ю.А. Тепло-, термо- и огнестойкость полимерных материалов. СПб.: Научные основы и технологии, 2011.
Fink J.K. High Performance Polymers. Norwich; New York: William Andrew Inc., 2008.
Mittal V. High Performance Polymers and Engineering Plastics. Hoboken, New Jersey: Wiley; Salem, Massachusetts: Scrivener Publ. LLC, 2011.
Bhatnagar N., Jha S., Bhowmik S. //Inventi J. 2010, V. 1. № 1. Published on Web 18.09.2010, www.inventi.in, 2010etr004.
Goosey M.T. Plastics for Electronics. Dordrecht: Kluwer Acad. Publ., 1999.
Cousins K. Polymers for Electronic Components. Shawbury: Smithers Rapra Press, 2001.
Cousins K. Polymers in Electronics: market report. Shawbury: Smithers Rapra Press, 2006.
Sabu T., Visakh P.M. Handbook of Engineering and Speciality Thermoplastics. Hoboken, New Jersey: Wiley; Salem, Massachusetts: Scrivener Publ. LLC, 2011. V. 3.
Kemmish D. Update on the Technology and Applications of Polyaryletherketones. Shawbury: Smithers Rapra Technol., 2010.
Chan K.P., Wang Y.-F., Hay A.S., Hronovski X.L., Cotter R.J. // Macromolecules. 1995. V. 28. № 20. P. 6705.
Ben-Haida A., Colguhoun H.M., Hodge P., Williams D.J. // J. Mater. Chem. 2000. V. 10. № 9. P. 2011.
Ben-Haida A., Colguhoun H.M., Hodge P., Williams D.J. // Macromolecules. 2006. V. 36. № 19. P. 6467.
Colquhoun H.M., Sestiaa L.G., Zolotukhin M.G., Williams D.J. // Macromolecules. 2000. V. 33. № 23. P. 8907.
Colquhoun H.M., Zhu Z.X., Dudman C.C. // Macromolecules. 2005. V. 38. № 25. P. 10421.
Percec V., Nava H. // J. Polym. Sci., Polym. Chem. 1988. V. 26. № 3. P. 783.
Percec V., Wang J.H., Okita S. // J. Polym. Sci., Polym. Chem. 1991. V. 29. № 12. P. 1789.
Percec V., Wang J.H. // Makromol. Chem., Macromol. Symp. 1992. V. 54–55. № 1. P. 337.
Percec V., Wang J.H., Lisha Yu. // Polym. Bull. 1992. V. 27. № 5. P. 503.
Bochmann M., Kelly K., Lu J. // J. Polym. Sci., Polym. Chem. 1992. V. 30. № 12. P. 2511.
Deeter G.A., Moore J.S. // Macromolecules. 1993. V. 26. № 10. P. 2535.
Mullins M.J., Woo E.P. // J. Macromol. Sci. C. 1987. V. 27. № 2. P. 313.
Lakshmana R.V. // J. Macromol. Sci. C. 1995. V. 35. № 4. P. 661.
Гилева Н.Г. Дис. … канд. хим. наук. Уфа: ИХ БФ АН СССР, 1989.
Шапошникова В.В. Дис. … канд. хим. наук. М.: ИНЭОС РАН, 1993.
Shaposhnikova V.V., Salazkin S.N. // Russ. Chem. Bull. 2014. V. 63. № 10. P. 2213.
Maiti S., Mandal B.K. // Prog. Polym. Sci. 1986. V. 12. № 1–2. P. 111.
McGrail P.T. // Polym. Int. 1996. V. 41. № 2. P. 103.
Jayakannan M., Ramakrishnan S. // Macromol. Rapid Commun. 2001. V. 22. № 18. P. 1463.
Jennings B.E., Jones M.E.B., Rose J.B. // J. Polym. Sci. C. 1967. V. 16. № 2. P. 715.
Johnson R.N., Farnham A.G., Clendinning R.A., Hale W.F., Merriam C.N. // J. Polym. Sci. A-1. 1967. V. 5. № 9. P. 2375.
Kricheldorf H.R., Jahnke P. // Macromol. Chem. 1990. V. 191. № 9. P. 2027.
Ohta M., Oikawa H., Sugimoto K., Yamaguchi K., Yamaguchi A. Pat. 4803258A USA. 1989.
Maresca L.M., Farnham A.G., Schwab Th.H., Steiner U.A. Pat. 4963643 USA. 1990.
Labadie J.W., Hedrick J.L. // Macromol. Chem., Macromol. Symp. 1992. V. 54–55. P. 313.
Gomez-Valdemoro A., Martinez-Manez R., Sancenon F., Garcia F.C., Garcia J.M. // Macromolecules. 2010. V. 43. № 17. P. 7111.
Cormier L., Lucotte G., Delfort B. // Polym. Bull. 1991. V. 26. № 4. P. 395.
Paya N., Ghaffari M., Mighani H. // J. Environ. An. Chem. 2018. V. 5. № 1. P. 1000233.
Lucas M., Brock P., Hedrick J.L. // J. Polym. Sci., Polym. Chem. 1993. V. 31. № 9. P. 2179.
Labadie J.W., Hedrick J.L. // Macromolecules. 1990. V. 23. № 26. P. 5371.
Fitch J.W., Reddy V.S., Youngman P.W., Wohlfahrt G.A., Cassidy P.E. // Polymer. 2000. V. 41. № 6. P. 2301.
Chen X.T., Sun H., Tang X.D., Wang C.Y. // J. Appl. Polym. Sci. 2008. V. 110. № 3. P. 1304.
Dang T.D., Dalton M.J., Venkatasubramanian N., Johnson J.A., Cerbus C.A., Feld W.A. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 23. P. 6134.
Li K., Chen X., Sun H., Tang X. // Chin. J. Polym. Sci. 2007. V. 25. № 6. P. 651.
Percec V., Clough R.S., Rinaldi P.L., Litman V.E. // Macromolecules. 1991. V. 24. № 21. P. 5889.
Percec V., Wang J.H., Clough R.S. // Macromol. Chem., Makromol. Symp. 1992. V. 54/55. P. 275.
Percec V., Clough R.S., Rinaldi P.L., Litman V.E. // Macromolecules. 1994. V. 27. № 6. P. 1535.
Konno K., Kobayashi T., Yonetake K., Ueda M., Fitch J.W., Cassidy P.E. // Polymer. 1998. V. 39. № 3. P. 719.
Liu Y., Zhong M., Song Ch., Sheng Sh., Liu X., Hou H. // High Performance Polymers. 2019. V. 31. № 4. P. 409.
Bao F., Zong L., Li N., Song Y., Pan Y., Wang J., Jian X. // Thermochim. Acta. 2020. V. 683. P. 178184.
Jansons V. Pat. 4767837 USA. 1988.
Шарапов Д.С. Дис. … канд. хим. наук. М.: ИНЭОС РАН, 2006.
Sharapov D.S., Shaposhnikova V.V., Salazkin S.N. // Polymer Science. A. 2004. V. 46. № 4. P. 381.
Rose J.B. Pat. 4010147 USA. 1977.
Rose J.B. Pat. 1414424 Great Britain. 1975.
Fukuoka Sh., Matsuda H. Pat. 4703102 USA. 1987.
Sharapov D.S., Shaposhnikova V.V., Salazkin S.N. // Polymer Science. B. 2003. V. 45. № 1–2. P. 10.
Ding J., Liu F., Li M., Day M., Zhou M. // J. Polym. Sci., Polym. Chem. 2002. V. 40. № 23. P. 4205.
Shaposhnikova V.V., Sharapov D.S., Kaibova I.A., Gorlov V.V., Salazkin S.N., Dubrovina L.V., Bragina T.P., Kazantseva V.V., Bychko K.A., Askadskii A.A., Tkachenko A.S., Nikiforova G.G., Petrovskii P.V., Peregudov A.S. // Polymer Science. A. 2007. V. 49. № 10. P. 1071.
Attwood T.E., Dawson P.C., Freeman J.L., Hoy L.R.J., Rose J.B., Staniland P.A. // Polymer. 1981. V. 22. № 8. P. 1096.
Percec V., Clough R.S., Rinaldi P.L., Litman V.E. // Polym. Prepr. Am. Chem. Soc. 1991. V. 32. № 1. P. 353.
Jensen B.J., Hergenrother P.M. // High Performance Polymers. 1989. V. 1. № 1. P. 31.
Percec V., Grigoras M., Clough R.S., Fanjul J. // J. Polym. Sci., Polym. Chem. 1995. V. 33. № 2. P. 331.
Shaposhnikova V.V., Salazkin S.N., Sergeev V.A., Blagodatskikh I.V., Dubrovina L.V., Sakunts A.A., Pavlova S.-S.A. // Russ. Chem. Bull. 1996. V. 45. № 10. P. 2397.
Shaposhnikova V.V., Salazkin S.N., Donetskii K.I., Gorshkov G.V., Askadskii A.A., Bychko K.A., Kazantseva V.V., Samoryadov A.V., Krasnov A.P., Lioznov B.S., Afonicheva O.V., Svetlova N.A., Kogan A.S., Tkachenko A.A. // Polymer Science. A. 1999. V. 41. № 2. P. 124.
Bender T.P., Burt R.A., Hamer G.K., DeVisser C., Smith P.F., Saban M. // Org. Proc. Res. Development. 2002. V. 6. № 5. P. 714.
Виноградова С.В., Коршак В.В., Салазкин С.Н., Кульков А.А. // Высокомолек. соед. А. 1972. Т. 14. № 12. С. 2545.
Vinogradova S.V., Vasnev V.A., Vygodskii Ya.S. // Russ. Chem. Revs. 1996. V. 65. № 3. P. 249.
Hergenrother P.M. // Polym. J. 1987. V. 19. № 1. P. 73.
Kim M.K., Kwon K.J., Han Y.K. // Bull. Korean Chem. Soc. 2011. V. 32. № 9. P. 3311.
Донецкий К.И. Дис. … канд. хим. наук. М.: ИНЭОС РАН, 2000.
Salazkin S.N., Shaposhnikova V.V., Donetskii K.I., Petrovskii P.V. // Mendeleev Commun. 1997. № 5. P. 210.
Salazkin S.N., Shaposhnikova V.V., Donetskii K.I., Gorshkov G.V., Petrovskii P.V., Komarova L.I., Genina M.M., Tkachenko A.S. // Russ. Chem. Bull. 2000. V. 49. № 6. P. 1093.
Knauss D.M., Bender J.T. // J. Polym. Sci., Polym. Chem. 2002. V. 40. № 17. P. 3046.
Zhang H.C., Chen T.L., Yuan Y.G. Pat. CN85108751 China. 1987.
Han Y., Li B., Yang Y. // J. Appl. Polym. Sci. 1996. V. 62. № 10. P. 1739.
Salazkin S.N., Donetskii K.I., Gorshkov G.V., Shaposhnikova V.V. // Dokl. Chem. 1996. V. 348. № 1–3. P. 115.
Salazkin S.N., Donetskii K.I., Gorshkov G.V., Shaposhnikova V.V., Genin Ya.V., Genina M.M. // Polymer Science A. 1997. V. 39. № 9. P. 960.
Qi Y., Ding J., Day M., Jiang J., Callender C.L. // Chem. Mater. 2005. V. 17. № 3. P. 676.
Qi Y., Ding J., Day M., Jiang J., Callender C.L. // Polymer. 2006. V. 47. № 25. P. 8263.
Dhara M.G., Banerjee S. // Prog. Polym. Sci. 2010. V. 35. № 8. P. 1022.
Xie J., Peng W., Li G., Jiang J. // Polym. Bull. 2011. V. 67. № 1. P. 45.
Kimura K., Nishichi A., Yamashita Y. // Polym. Adv. Technol. 2004. V. 15. № 6. P. 313.
Hamciuc C., Hamciuc E., Bruma M., Klapper M., Pakula T. // Polym. Bull. 2001. V. 47. № 1. P. 1.
Liu B., Hu W., Chen Ch., Jiang Zh., Zhang W., Wu Zh., Matsumoto T. // Polymer. 2004. V. 45. № 10. P. 3241.
Rose J.B., Staniland P.A. Pat. 4320224 USA. 1982.
Risse W., Sogah D.Y. // Macromolecules. 1990. V. 23. № 18. P. 4029.
Kelsey D.R. Eur. Pat. 0148633. 1985.
Kelsey D.R., Robeson L.M., Clendinning R.A., Blackwell C.S. // Macromolecules. 1987. V. 20. № 5. P. 1204.
Lindfors B.E., Mani R.S., McGrath J.E., Mohanty D.K. // Macromol. Chem., Rapid. Commun. 1991. V. 12. № 12. P. 337.
Brink A.E., Gutzeit S., Lin T., Marand H., Lyon K., Hua T., Davis R., Riffle J.S. // Polymer. 1993. V. 34. P. 825.
Bourgeois Y., Devaux J., Legras R. // Polymer. 1996. V. 37. № 14. P. 3171.
Yang J., Tyberg C.S., Gibson H.W. // Macromolecules. 1999. V. 32. № 25. P. 8259.
Huang R.Y.M., Shao P., Burns C.M., Feng X. // J. Appl. Polym. Sci. 2001. V. 82. № 11. P. 2651.
Daoust D., Devaux J., Godard P. // Polym. Int. 2001. V. 50. № 8. P. 917.
Daoust D., Devaux J., Godard P. // Polym. Int. 2001. V. 50. № 8. P. 925.
Daoust D., Devaux J., Godard P. // Polym. Int. 2001. V. 50. № 8. P. 932.
Lakshmi M.R.T.S., Choudhary V., Varma I.K. // J. Mater. Sci. 2005. V. 40. № 3. P. 629.
Hamciuc C., Bruma M., Klapper M. // J. Macromol. Sci. A. 2001. V. 38. № 7. P. 659.
Shang X., Li X., Xiao M., Meng Y. // Polymer. 2006. V. 47. № 11. P. 3807.
Di Vona M.L., Marani D., D’Epifanio A., Traversa E., Trombetta M., Licoccia S. // Polymer. 2005. V. 46. № 5. P. 1754.
Trotta F., Drioli E., Gordano A. // J. Appl. Polym. Sci. 2001. V. 80. № 7. P. 1037.
Gao Y., Jian X., Xuan Y., Xu Z., Lu Q. // J. Appl. Polym. Sci. 2002. V. 84. № 10. P. 1866.
Gao Y., Jian X., Xuan Y., Xiang S., Liang P., Guiver M.D. // J. Polym. Sci., Polym. Chem. 2002. V. 40. № 20. P. 3449.
Kang N., Hlil A.R., Hay A.S. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 22. P. 5745.
Henneuse-Boxus C., De Ro A., Bertrand P., Marchand-Brynaert J. // Polymer. 2000. V. 41. № 7. P. 2339.
Henneuse-Boxus C., Duliere E., Marchand-Brynaert J. // Eur. Polym. J. 2001. V. 37. № 1. P. 9.
Ma W., Zhao Ch., Lin H., Zhang G., Na H. // J. Appl. Polym. Sci. 2011. V. 120. № 6. P. 3477.
Lu Z., Liu Y., Qin J., Yang Z., Ye C. // React. Funct. Polym. 2004. V. 61. № 3. P. 379.
Zhang W., Qiu X., Ueda M., Sui Y., Hu H., Zhang X., Wang L. // Solid State Ionics. 2018. V. 314. P. 187.
Wang F., Chen T., Xu J., Liu T., Jiang H., Qi Y., Liu S., Li X. // Polymer. 2006. V. 47. № 11. P. 4148.
Gao Y., Robertson G.P., Guiver M.D., Mikhailenko S.D., Li X., Kaliaguine S. // Macromolecules. 2004. V. 37. № 18. P. 6748.
Xiao G., Sun G., Yan D. // Polym. Bull. 2002. V. 48. № 4–5. P. 309.
Li X., Zhao C., Lu H., Wang Z., Na H. // Polymer. 2005. V. 46. № 15. P. 5820.
Xing P., Robertson G.P., Guiver M.D., Mikhailenko S.D., Kaliaguine S. // Polymer. 2005. V. 46. № 10. P. 3257.
Xuan Y., Gao Y., Huang Y., Jian X. // J. Appl. Polym. Sci. 2003. V. 88. № 5. P. 1111.
Hunter R.A., Turner P.D., Rimmer S. // J. Mater. Chem. 2001. V. 11. № 3. P. 736.
Zolotukhin M.G., Colguhoun H.M., Sestiaa L.G., Rueda D.R., Flot D. // Macromolecules. 2003. V. 36. № 13. P. 4766.
Kim S., Yang S., Kim D. // Int. J. Hydrogen Energy. 2017. V. 42. № 17. P. 12496.
Zhao Zh., Gu Y., Chao D., Liu X. // Eur. Polym. J. 2019. V. 116. P. 336.
Le Mong A., Kim D. // Electrochim. Acta. 2018. V. 290. P. 544.
Hill A.R., Meng Y., Hay A.S., Abu-Yousef I.A. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 8. P. 1318.
Saxena A., Sadhana R., Rao V.L., Kanakavel M., Ninan R.N. // Polym. Bull. 2003. V. 50. № 4. P. 219.
Zhang Y., Sun X., Niu Y., Xu R., Wang G., Jiang Z. // Polymer. 2006. V. 47. № 5. P. 1569.
Yu Y.K., Xiao F., Cai M.Z. // J. Appl. Polym. Sci. 2007. V. 104. № 6. P. 3601.
Zhang Y., Sun X., Niu Y., Xu R., Wang G., Jiang Zh. // Polymer. 2006. V. 47. № 5. P. 1569.
Yang S., Ahn Y., Kim D. // J. Mater. Chem. A. 2017. V. 5. P. 2261.
Yonezawa N., Mori S., Miyata S., Ueha-Anyashiki Y., Maeyama K. // React. Funct. Polym. 2002. V. 53. № 1. P. 11.
Zhang S., Fu L., Li J., Yang D., Gao Z., Jia M., Zheng Y., Wu Z. // Macromol. Chem. Phys. 2000. V. 201. № 6. P. 649.
Chen J., Yang D. // J. Polym. Sci., Polym. Phys. 2007. V. 45. № 22. P. 3011.
Fritsch D., Vakhtangishvili L., Kricheldorf H.R. // J. Macromol. Sci. A. 2002. V. 39. № 11. P. 1335.
Donetskii K.I., Salazkin S.N., Gorshkov G.V., Shaposhnikova V.V. // Dokl. Chem. 1996. V. 350. № 1–3. P. 216.
Feng J., Xu Y., Sun Y., Wen Sh., Lei Y., Zhang L., Huo J. // J. Polym. Res. 2016. V. 23. № 12. P. 247.
Liu B., Chen Zh., Lin L., Han Y., Pang J., Jiang Zh. // High Performance Polymers. 2017. V. 29. № 5. P. 575.
Yildiz E., Inan T.Y., Yildirum H., Kuyulu A., Giingor A. // Macromol. Mater. Eng. 2001. V. 286. № 10. P. 634.
Cassidy P.E., Fitch J.W., Gronewald S.D., St. Clair A.K., Stoakley D.M. Pat. 6372877 USA. 2002.
More A.S., Pasale S.K., Honkhambe P.N., Wadgaonkar P.P. // J. Appl. Polym. Sci. 2011. V. 121. № 6. P. 3689.
Lu J., Yan G., Zhang G., Yang J. // J. Polym. Res. 2017. V. 24. № 10. P. 152.
Рябев А.Н. Дис. … канд. хим. наук. М.: ИНЭОС РАН, 2009.
Staniland P.A., Wilde C.J., Bottino F.A., Di Pasquale G., Pollicino A., Recca A. // Polymer. 1992. V. 33. № 9. P. 1976.
Kricheldorf H.R., Bier G. // Polymer. 1984. V. 25. № 8. P. 1151.
Ellzey K.A., Farris R.J., Emrick T. // Polym. Bull. 2003. V. 50. № 4. P. 235.
Imai Y., Ishikawa H., Park K., Kakimoto M. // J. Polym. Sci., Polym. Chem. 1997. V. 35. № 10. P. 2055.
Radlmann E., Schmidt W., Nischk G. // Macromol. Chem. Phys. 1969. V. 130. № 1. P. 45.
Attwood T.E., Dawson P.C., Freeman J.L., Hoy L.R.J., Rose J.B., Staniland P.A. // Polym. Prepr. Am. Chem. Soc. 1979. V. 20. № 1. P. 191.
Fukawa I., Tanabe T., Dozono T. // Macromolecules. 1991. V. 24. № 13. P. 3838.
Fukawa I., Tanabe T. // J. Polym. Sci., Polym. Chem. 1992. V. 30. № 9. P. 1977.
Fukawa I., Tanabe T. // J. Polym. Sci., Polym. Chem. 1993. V. 31. № 2. P. 535.
Brunel R., Marestin C., Martin V., Mercier R., Schiets F. // High Performance Polymers. 2008. V. 20. № 2. P. 185.
Li M., Zhang A., Yin J., Liew K.Y. // Polym. Eng. Sci. 2011. V. 51. № 1. P. 23.
Miller T.M., Neenan T.X., Kwock E.W., Stein S.M. // J. Am. Chem. Soc. 1993. V. 115. № 1. P. 356.
Morikawa A., Ono K. // Macromolecules. 1999. V. 32. № 4. P. 1062.
Sennet L., Fossum E., Tan L.S. // Polymer. 2008. V. 49. № 17. P. 3731.
Mohanty D.K., Sachdeva Y., Hedrick J.L., Wolfe J.F., McGrath J.E. // Polym. Prepr. Am. Chem. Soc. 1984. V. 25. № 2. P. 19.
Liu B., Dai Y., Robertson G.P., Guiver M.D., Hu W., Jiang Z. // Polymer. 2005. V. 46. № 25. P. 11279.
Gao Y., Jian X., Dai Y., Xue J., Peng S., Liu S. // J. Appl. Polym. Sci. 2000. V. 78. № 1. P. 20.
Zheng Y., Yang X., Yuan M., Luo J. // High Performance Polymers. 2019. V. 31. № 9–10. C. 1173.
Han X., Xie Y., Liu D., Chen Zh., Zhang H., Pang J., Jiang Zh. // J. Membr. Sci. 2019. V. 589. P. 117230.
Xu Y., Feng J., Ren H., Bi Y., Zhu J., Sun Y., Wen Sh., Huo J., Zhang L. // J. Polym. Res. 2017. V. 24. № 6. P. 90.
Liang J., Ge J., Wu K., Zhang Q., Wang J., Ye Zh. // J. Membr. Sci. 2020. V. 597. P. 117626.
Kawaguchi S., Morikawa A. // High Performance Polymers. 2018. V. 30. №. 1. P. 67.
Sun S., Guo M., Yi X., Zhang Z. // Polym. Bull. 2017. V. 74. № 5. P. 1519.
Салазкин С.Н. Дис. … д-ра хим. наук. М.: ИНЭОС РАН, 1979.
Li L., Liu S., Liu Zh., Jing L., Guan Sh., Jiang Zh., Liu B., Matsumoto T. // High Performance Polymers. 2012. V. 24. № 5. P. 425.
Niu Y., Zhu X., Liu L., Zhang Y., Wang G., Jiang Z. // React. Funct. Polym. 2006. V. 66. № 5. P. 559.
Wang Z., Chen T., Xu J. // J. Macromol. Sci. A. 2000. V. 37. № 12. P. 1571.
Hou L., Wang Z., Xu J., Chen Zh. // J. Polym. Res. 2019. T. 26. № 10. P. 243.
Аскадский А.А., Кондращенко В.И. Компьютерное материаловедение полимеров. М.: Научный мир, 1999. Т. 1.
Carlier V., Devaux J., Legras R., McGrail P.T. // Macromolecules. 1992. V. 25. № 24. P. 6646.
Shaposhnikova V.V., Askadskii A.A., Salazkin S.N., Sergeev V.A., Samoryadov A.V., Krasnov A.P., Bychko K.A., Kazantseva V.V., Lioznov B.S. // Polymer Science. A. 1997. V. 39. № 4. P. 499.
Hou T.H., Jensen B.J., Bai J.M. // High Performance Polymers. 1989. V. 1. № 1. P. 41.
Hinkley J.A., Crook R.A., Davis J.R.J. // High Performance Polymers. 1989. V. 1. № 1. P. 61.
Chivers R.A., Moore D.R. // Polymer. 1994. V. 35. № 1. P. 110.
Shaposhnikova V.V., Salazkin S.N., Donetskii K.I., Gorshkov G.V., Sharapov D.S., Mamedova I.A., Petrovskii P.V., Askadskii A.A., Bychko K.A., Kazantseva V.V., Krasnov A.P., Afonicheva O.V., Tkachenko A.S., Genina M.M. // Polymer Science. A. 2002. V. 44. № 6. P. 563.
Richter A., Lohkaemper H.G. Pat. 0292073 USA. 2009.
Xuefeng A.N., Shuagying J.I., Bangming T., Zilong Zh., Xiao-Su Y. // J. Mater. Sci. Lett. 2002. V. 21. № 22. P. 1763.
Kalaev D.V., Brantseva T.V., Gorbatkina Yu.A., Kerber M.L., Kravchenko T.P., Salazkin S.N., Shaposhnikova V.V. // Polymer Science. A. 2003. V. 45. № 5. P. 465.
Volkov A.S., Gorbatkina Yu.A., Gorbunova I.Yu., Kerber M.L., Salazkin S.N., Shaposhnikova V.V. // Polymer Science. A. 2007. V. 49. № 5. P. 558.
Горбаткина Ю.А., Иванова-Мумжиева В.Г. Адгезия модифицированных эпоксидов к волокнам. М.: ТОРУС ПРЕСС, 2018.
Салазкин С.Н., Донецкий К.И. // Докл. РАН. 1998. Т. 362. № 6. С. 789.
Кульков А.А. Дис. … канд. хим. наук. М.: МХТИ им. Д.И. Менделеева, 1972.
Jansen J.C., Drioli E. // Polymer Science. A. 2009. V. 51. № 11–12. P. 1355.
Jagur-Grodzinski J. // Polym. Adv. Technol. 2007. V. 18. № 10. P. 785.
Nagarale R.K., Shina W., Singh P.K. // Polym. Chem. 2010. V. 1. № 4. P. 388.
Dobrovolsky Yu.A., Jannasch P., Lafitte B., Belomoina N.M., Rusanov A.L., Likhachev D.Yu. // Russ. J. Electrochem. 2007. V. 43. № 5. P. 489.
Bose S., Kuila T., Nguyen T.X.H., Kim N.H., Lau K., Lee J.H. // Prog. Polym. Sci. 2011. V. 36. № 6. P. 813.
Awasthi S., Kiran V., Gaur B. // Int. J. Hydrogen Energy. 2017. V. 42. № 16. P. 11710.
Le Mong A., Yang S., Kim D. // J. Membr. Sci. 2017. V. 543. P. 133.
Lin L., Chen Zh., Zhang Zh., Feng S., Liu B., Zhang H., Pang J., Jiang Zh. // Polymer. 2016. V. 96. P. 188.
Wang Y., Xu J., Zang H., Wang Zh. // Int. J. Hydrogen Energy. 2019. V. 44. № 12. P. 6136.
Xu J., Zhenguo Zh., Yang K., Zhang H., Wang Zh. // Renewable Energy. 2019. V. 138. P. 1104.
Wang Zh., Chen T., Xu J. // Macromolecules. 2000. V. 33. № 15. P. 5672.
Li L., Wang B., Tan H., Chen T., Xu J. // J. Membr. Sci. 2006. V. 269. № 1–2. P. 84.
Wang Zh., Chen T., Xu J. // Macromolecules. 2007. V. 40. № 9. P. 3238.
Drioli E., Regina A., Casciola M., Oliveti A., Trotta F., Massari T. // J. Membr. Sci. 2004. V. 228. № 2. P. 139.
Dang H.S., Kim D. // J. Power Sources. 2013. V. 222. P. 103.
Xing P., Robertson G.P., Guiver M.D., Mikhailenko S.D., Kaliaguine S. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 12. P. 2866.
Xing P., Robertson G.P., Guiver M.D., Mikhailenko S.D., Kaliaguine S. // Macromolecules. 2004. V. 37. № 21. P. 7960.
Chen Y., Meng Y.Z., Wang S.J., Tian Sh., Chen Y., Hay A.S. // J. Membr. Sci. 2006. V. 280. № 1–2. P. 433.
Wootthikanokkhan J., Seeponkai N. // J. Appl. Polym. Sci. 2006. V. 102. № 6. P. 5941.
Li Y.X., Roy A., Badami A.S., Hill M., Yang J., Dunn S., McGrath J.E. // J. Power Sources. 2007. V. 172. № 1. P. 30.
Shang X.Y., Fang S.M., Meng Y.Z. // J. Membr. Sci. 2007. V. 297. № 1–2. P. 90.
Jeong M.H., Lee K.S., Hong Y.T., Lee J.S. // J. Membr. Sci. 2008. V. 314. № 1–2. P. 212.
Pang J.H., Zhang H.B., Li X.F., Wang L.F., Liu B.J., Jiang Zh. // J. Membr. Sci. 2008. V. 318. № 1–2. P. 271.
Zhao C., Li X., Lin H., Shao K., Na H. // J. Appl. Polym. Sci. 2008. V. 108. № 1. P. 671.
Jeong M.H., Lee K.S., Lee J.S. // J. Membr. Sci. 2009. V. 337. № 1–2. P. 145.
Jeong M.H., Lee K.S., Lee J.S. // Macromolecules. 2009. V. 42. № 5. P. 1652.
Lin H.D., Zhao C.J., Cui Z.M., Ma W.J., Fu T.Z., Na H., Xing W. // J. Power Sources. 2009. V. 193. № 2. P. 507.
Shao K., Zhu J., Zhao C.J., Li X., Cui Z., Zhang Y., Li H., Xu D., Zhang G., Fu T.Z., Wu J., Na H., Xing W. // J. Polym. Sci., Polym. Chem. 2009. V. 47. № 21. P. 5772.
Zhang G., Fu T.Z., Shao K., Li X.F., Zhao C.J., Na H., Zhang H. // J. Power Sources. 2009. V. 189. № 2. P. 875.
Zhang Y., Cui Z.M., Zhao C.J., Shao K., Li H.T., Fu T.Z., Na H., Xing W. // J. Power Sources. 2009. V. 191. № 2. P. 253.
Zhang Y., Wan Y., Zhao C.J., Shao K., Zhang G., Li H.T., Lin H.D., Na H. // Polymer. 2009. V. 50. № 19. P. 4471.
Luo J.J., Wang S.J., Xiao M., Han D.M., Meng Y.Z. // Eur. Polym. J. 2010. V. 46. № 8. P. 1736.
Li Zh., Liu X., Chao D., Lu X., He L., Yang Y., Zhang W. // J. Appl. Polym. Sci. 2010. V. 118. № 6. P. 3318.
Mikami T., Miyatake K., Watanabe M. // ACS Appl. Mater. Interfaces. 2010. V. 2. № 6. P. 1714.
Zhao C.J., Lin H.D., Na H. // Int. J. Hydrogen Energy. 2010. V. 35. № 5. P. 2176.
Zhao C.J., Lin H.D., Zhang Q.A., Na H. // Int. J. Hydrogen Energy. 2010. V. 35. № 19. P. 10482.
Zhao C.J., Lin H.D., Han M.M., Na H. // J. Membr. Sci. 2010. V. 353. № 1–2. P. 10.
Feng S.G., Shang Y.M, Wang Y.W., Liu G.S., Xie X.F., Dong W.Q., Xu J.M., Mathur V.K. // J. Membr. Sci. 2010. V. 352. № 1–2. P. 14.
Fu L.C., Xiao G.Y., Yan D.Y. // J. Membr. Sci. 2010. V. 362. № 1–2. P. 509.
Guo M.M., Liu B.J., Guan S.W., Liu C., Qin H.Y., Jiang Z.H. // J. Membr. Sci. 2010. V. 362. № 1–2. P. 38.
Zhang Y., Wan Y., Zhang G., Shao K., Zhao C.J., Li H.T., Na H. // J. Membr. Sci. 2010. V. 348. № 1–2. P. 353.
Chen L.K., Wu Ch.Sh., Chen M.C., Hsu K.L., Li H.Ch., Hsieh Ch.H., Hsiao M.H., Chang C.L., Chu P.P.J. // J. Membr. Sci. 2010. V. 361. № 1–2. P. 143.
De Bonis C., D’Epifanio A., Di Vona M.L., Mecheri B., Traversa E., Trombetta M., Licoccia S. // J. Polym. Sci. Part A-1. 2010. V. 48. № 10. P. 2178.
Zhang Y., Zhang G., Wan Y., Zhao C., Shao K., Li H., Han M., Zhu J., Xu S., Liu Z., Na H. // J. Polym. Sci., Polym. Chem. 2010. V. 48. № 24. P. 5824.
Lin H.D., Zhao C.J., Jiang Y.N., Ma W.J., Na H. // J. Power Sources. 2011. V. 196. № 4. P. 1744.
Mikami T., Miyatake K., Watanabe M. // J. Polym. Sci., Polym. Chem. 2011. V. 49. № 2. P. 452.
Wang J., Zhao C.J., Lin H.D., Zhang G., Zhang Y., Ni J., Ma W.J., Na H. // J. Power Sources. 2011. V. 196. № 13. P. 5432.
Wang L., Zhu G.M., Li J.Q., Gao C.M. // Polym. Bull. 2011. V. 66. № 7. P. 925.
Salazkin S.N. // Polymer Science. B. 2004. V. 46. № 7–8. P. 203.
Ponomarev A.F., Moshelev A.V., Il’yasov V. Kh., Lachinov A.N., Salazkin S.N., Shaposhnikova V.V., Sharapov D.S., Kornilov V.M. // Polymer Science. C. 2009. V. 51. № 1. P. 46.
Shaposhnikova V.V., Tkachenko A.S., Zvukova N.D., Peregudov A.S., Klemenkova Z.S., Ponomarev A.F., Il’yasov V.Kh., Lachinov A.N., Salazkin S.N. // Russ. Chem. Bull. 2016. V. 65. № 2. P. 502.
Салазкин С.Н., Шапошникова В.В., Лачинов А.Н. Пат. 2573009 Россия. 2016.
Chebotareva A.B., Untila G.G., Kost T.N., Stepanov A.S., Salazkin S.N., Shaposhnikova V.V. // Solar Energy Mater. Solar Cells. 2017. V. 165. P. 1.
Chebotareva A.B., Untila G.G., Kost T.N., Stepanov A.S., Salazkin S.N., Shaposhnikova V.V. // Solar Energy. 2019. V. 193. P. 828.
Kurtz S.M., Devine J.N. // Biomaterials. 2007. V. 28. № 32. P. 4845.
Kurtz S.M. PEEK biomaterials handbook. New York: World Plastics Library, Elsevier, 2011.
Geringer J., Tatkiewicz W., Rouchouse G. // Wear. 2011. V. 271. № 11–12. P. 2793.
Patel S.A., Patel M.V., Ray A., Patel R.M. // J. Polym. Sci., Polym. Chem. 2003. V. 41. № 15. P. 2335.
Wu W.Z., Geng P., Zhao J., Zhang Y., Rosen D.W., Zhang H.B. // Materials Research Innovations. 2014. V. 18. № 5 (suppl). P. S5-12.
Wu W., Geng P., Li G., Zhao D., Zhang H., Zhao J. // Materials. 2015. V. 8. № 9. P. 5834.
Yang C., Tian X., Li D., Cao Y., Zhao F., Shi C. // J. Mater. Proc. Tech. 2017. V. 248. P. 1.
Berretta S., Ghita O., Evans K.E. // Eur. Polym. J. 2014. V. 59. P. 218.
Berretta S., Evans K.E., Ghita O. // Eur. Polym. J. 2015. V. 68. P. 243.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)