

Высокомолекулярные соединения (серия С), 2021, T. 63, № 1, стр. 3-13
ПОЛИМЕРНЫЕ ЗАГАДКИ ХИМИЧЕСКОЙ ФИЗИКИ
a Федеральный исследовательский центр химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: berlin@chph.ras.ru
Поступила в редакцию 17.12.2020
После доработки 12.01.2021
Принята к публикации 26.01.2021
Аннотация
Рассмотрены проблемы, возникшие в процессе исследовательской работы при решении разнообразных прикладных и научных задач. В основном, это касалось синтеза и различных свойств полимеров. Особое внимание уделено явлениям, на первый взгляд, загадочным, требующим нетривиального подхода для их объяснения. Только понимание сути самого явления позволило получить ответ и найти адекватное качественное и количественное описание очередной загадки природы.
Что такое наука?.. Каждый человек имеет свой ответ на этот вопрос. Одни считают, наука – это установление новых законов природы, но такое происходит чрезвычайно редко. Некоторые полагают, что наука – это создание моделей, в том числе математических, описывающих различные природные явления. Для меня наука состоит из загадок, преподносимых нам природой, которые необходимо разгадать. Причем, когда наконец удается решить проблему, т.е. отгадать загадку (или думать так), получаешь несказанное удовольствие – главный стимул занятий наукой, во всяком случае для меня. Это и есть фундаментальная наука. Но существует и прикладная наука, цель которой – создание новых материалов, веществ, процессов. Везде присутствует творчество, но оно разное. В первом случае нами движет любопытство, во втором – некоторое задание извне. Впрочем, разделения науки в жизни не происходит в том смысле, что часто люди, занимающиеся фундаментальной наукой, одновременно делают и какие-то прикладные работы, поскольку фундаментальный результат приводит к необходимости довести его до практики. Иначе, выполнение прикладной задачи иногда приводит к возникновению фундаментальной проблемы. Так, изучение практически важных проблем горения для борьбы с пожарами, создания двигателей и прочего привело к теории горения, распространения пламени и т.д. Напротив, чисто фундаментальные ядерные исследования вылились в атомную бомбу. Причем занимались фундаментальными и прикладными исследованиями одни и те же люди. Конечно, в прикладные работы вовлечено гораздо больше людей, ресурсов, и таких примеров множество.
Конструирование приборов или создание процессов по известным принципам – здесь уже выступает инженерия, тоже, конечно, требующая творческого подхода. На всем этом зиждется развитие промышленности и вообще цивилизации, прогресс человечества.
Приведу несколько примеров таких загадок, встретившихся на моем пути и касающихся, главным образом, природных и искусственных полимерных материалов.
ПОЧЕМУ АТМОСФЕРА ЗЕМЛИ СОДЕРЖИТ 21% КИСЛОРОДА?
Возможным ключом к ответу является процесс горения лесов. Леса увеличивают концентрацию кислорода, а пожары снижают. Существенным моментом в этом динамическом равновесии оказываются критические условия горения древесины [1–3]. При исследовании и сравнении горючести различных материалов пользуются такой характеристикой, как кислородный индекс. Это минимальная концентрация кислорода в подаваемой смеси азот–кислород, при которой поддерживается самостоятельное горение образца материала в определенных стандартных условиях. Удивительным образом для древесины такая величина (~21%) совпадает с составом атмосферы Земли.
ОТКУДА В ЖИВЫХ ОРГАНИЗМАХ ВЫДЕЛЕННАЯ ХИРАЛЬНОСТЬ?
Биологические молекулы (аминокислоты в белках, сахара в ДНК и РНК) во всех живых организмах имеют одну и ту же хиральность и лишь один из двух возможных стереоизомеров, причем во всех случаях они закручены в одну и ту же сторону. Это общее свойство, присущее живым организмам, – использовать аминокислоты только одного типа, впервые обнаружил великий французский ученый XIX века Луи Пастер. Как же возникла в живых организмах выделенная хиральность?
В работах японских авторов [4–8] продемонстрирован следующий удивительный эффект. Для определенных систем – макромолекулы и смеси оптических изомеров низкомолекулярного хирального соединения – полимерная цепь при комплексообразовании с одним изомером сворачивается в правую (условно) спираль, а с другим – в левую:

Самым необычным оказалось то, что при использовании смеси хиральных изомеров цепь образует только один тип спирали, соответствующий преобладающему изомеру, даже если его избыток очень невелик. Так, на рис. 1 можно видеть резкий переход двулучепреломления, т.е. переход спирали одного типа в другой при незначительном отклонении от рацемической смеси11.
Рис. 1.
Зависимость интенсивности двулучепреломления от соотношения L- и D-изомеров при двух температурах [4]; на вставке – данные около стехиометрии в увеличенном масштабе.
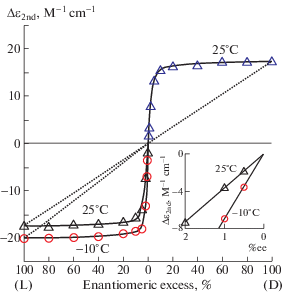
Возможным объяснением данного эффекта является необходимость наличия зародыша спирали большой длины для сворачивания всей макромолекулы [9]. Соотношение вероятностей образования L- и D-зародышей зависит от их длины по степенному закону. Поэтому небольшое различие в количестве изомеров приводит к значительной разнице вероятностей образования зародыша спирали, тем бóльшей, чем длиннее зародыш. Такие рассуждения объясняют возможный механизм усиления незначительной разницы в хиральности низкомолекулярных соединений, возникшей, например, случайным образом при их синтезе. Главная роль в таком усиливающим эффекте отведена полимерному состоянию, ведь не зря белки, ДНК, РНК и другие элементы живого считаются высокомолекулярными соединениями.
ХИМИЧЕСКИЕ РЕАКЦИИ В ТВЕРДОЙ ФАЗЕ
Авторы, изучающие химические реакции в твердой фазе при высоких значениях давления в условиях деформации сдвига на наковальнях Бриджмена (рис. 2a), обратили внимание на тот факт, что выход продукта определяется углом поворота деформации, а не ее скоростью, т.е. практически не зависит от времени [10, 11]. Иными словами, при построении кинетических кривых следует использовать координаты выход–сдвиговая деформация вместо привычных координат выход–время. При сдвиговой деформации смеси двух твердых порошков поверхность раздела, вблизи которой идет их взаимодействие и образуется продукт реакции, однозначно связана с величиной деформации (рис. 2б) [12].
Рис. 2.
Наковальни Бриджмена (а) и зависимость выхода стеарата кальция от угла поворота наковален при реакции стеариновой кислоты с оксидом кальция (б) [11].
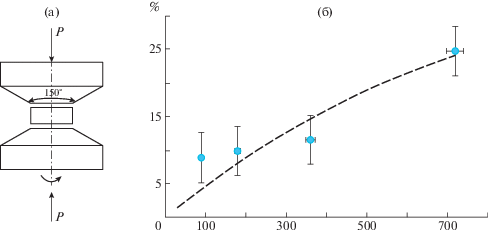
Поэтому выход продукта зависит только от деформаций в отсутствие диффузионных процессов, затрудненных в твердой фазе. Очевидно, что при проведении твердофазных химических реакций в других смесительных аппаратах (экструдеры, смеситель Брабендера и другие) ситуация будет аналогичной, хотя и более сложной из-за неоднородности сдвиговых деформаций.
ПЛАВЛЕНИЕ И СТЕКЛОВАНИЕ – РАЗЛИЧНОЕ И ОБЩЕЕ
Следующие две загадки касаются стеклообразного состояния вещества (в том числе полимеров), которое, несмотря на отсутствие дальнего порядка, демонстрирует часто те же макроскопические свойства, что и кристаллы, хотя объяснение этим свойствам для кристаллов дается с учетом существования в них дальнего порядка. Остановлюсь на двух явлениях: переход твердое тело–жидкость и пластическая деформация.
Молекулярно-динамическое моделирование простых геометрических объектов – дисков и сфер позволяет наблюдать постепенный переход от кристаллического состояния к стеклообразному [13–17]. Для этого достаточно рассмотреть однородную смесь частиц двух диаметров. Когда все частицы одинаковые, система представляет собой полностью упорядоченный кристалл. При изменении отношения диаметров смесь постепенно переходит в неупорядоченное стеклообразное состояние при низкой температуре. Нагревая эти системы, можно наблюдать переход из твердого состояния в жидкое. Для одинаковых частиц при небольшой разнице диаметров такой переход сопровождается скачком плотности (фазовый переход первого рода, плавление), а когда разница диаметров превышает некоторое критическое значение, скачка плотности не происходит (размягчение стекла, стеклование при охлаждении). Важно отметить, что с ростом разницы в размере частиц температура перехода из твердого состояния в жидкое непрерывно снижается (рис. 3).
Рис. 3.
Зависимости скачка плотности (а) и температуры перехода (б) от разницы радиусов сфер двух типов, случайно расположенных в пространстве [17].
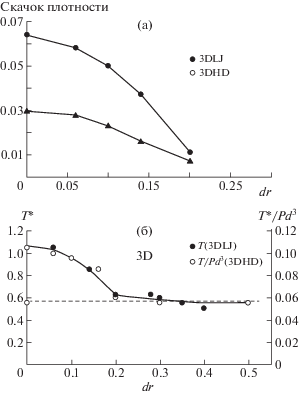
Молекулярно-динамическое моделирование показало, что и плавление кристаллов, и размягчение стекла при увеличении температуры, а соответственно, и объема происходят тогда, когда становится возможным обмен соседними частицами. Простая модель из семи дисков на плоскости, демонстрирующая переход твердого тела (1, 2) в жидкость (3) при увеличении температуры:
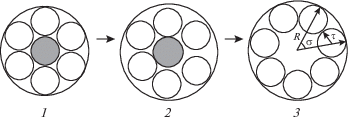
Так, при дальнейшем увеличении температуры начинается диффузия частиц, в результате чего сдвиговая деформация может возникать между переходами кристалл–жидкость и стекло–жидкость, при этом различие переходов заключается в том, что в точке перехода при плавлении кристалла теряется дальний порядок и, следовательно, наблюдается скачок энтропии, а значит, и энтальпии, что характерно для фазового перехода первого рода. При размягчении стекла упорядоченность не меняется, так как в твердом стекле нет дальнего порядка, и поэтому скачков энтропии и энтальпии не возникает.
Пластическая деформация кристаллических тел происходит с увеличением числа дислокаций и их перемещением. В стекле нет дальнего порядка и нет дислокаций, но макроскопические проявления деформации, такие как предел текучести и полосы сдвига, те же, что и в кристаллах. Молекулярно-динамическое моделирование показало [18–22], что пластическая деформация в стеклах всегда (в том числе и при одноосном сжатии) сопровождается увеличением объема, т.е. снижением плотности. Аналогичные результаты демонстрирует эксперимент. Заметим, что при деформации вблизи температуры стеклования наблюдается релаксация объема, и в эксперименте уменьшения плотности можно не заметить. Красивое доказательство снижения плотности было получено при моделировании процесса деформации твердых дисков и эллипсов на плоскости. В процессе деформации при переходе к пластическому течению (максимум на диаграмме сжатия, напряжение – деформация, σ–ε) появляются полосы сдвига (рис. 4). При этом увеличивается объем, снижается плотность, а также уменьшается число контактов между твердыми частицами, и именно в области полос сдвига частицы имеют малое количество контактов друг с другом и, соответственно, пониженную плотность.
Рис. 4.
Пространственное распределение локальной деформации (а), частиц с малым числом контактов, отмеченных красным цветом (б), и направлений контактов (в) на пластической стадии деформации (г, показано стрелкой) при одноосном сжатии по вертикальной оси неупорядоченной системы твердых эллипсов [19].
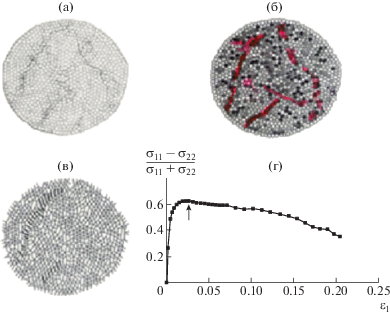
Таким образом, в стеклах при сдвиге формируются зоны пониженной плотности, в которых облегчена деформация, точно так же, как в областях скопления дислокаций в кристаллических телах.
ВСЕ ОБЫЧНЫЕ ИЗОТРОПНЫЕ ТЕЛА ИМЕЮТ ПОЛОЖИТЕЛЬНЫЙ КОЭФФИЦИЕНТ ПУАССОНА
Обычные изотропные тела расширяются в поперечном направлении при одноосном сжатии. Однако теория дает возможность коэффициенту Пуассона μ изменяться в пределах от 0.5 до –1.0. Тело сохраняет свой объем при μ = 0.5 и форму при μ = –1.0. Моделирование системы мягких сфер с двумя независимыми коэффициентами – нормальной и тангенциальной жесткостью, характеризующими контакт между частицами, показало, что величина μ = –1.0 может быть достигнута при условии значительного преобладания тангенциальной жесткости над нормальной [23, 24]:

Все обычные потенциалы взаимодействия атомов друг с другом – центральные (тангенциальная жесткость существенно меньше нормальной) или близки к ним. Это могло бы оказаться абстрактным рассуждением, если бы не экспериментальная работа американских авторов [25], в которой специальным образом был синтезирован новый класс полимерных и металлических пен с отрицательным коэффициентом Пуассона (около –1.0). Метод получения такой пены состоял в следующем: обычную очень легкую полимерную или металлическую пену подвергали всестороннему сжатию и фиксировали в таком состоянии. При этом внутренние перегородки теряли устойчивость и изгибались (рис. 5). Такая изогнутая перегородка имеет тангенциальную жесткость больше, чем нормальная жесткость. Следовательно, для получения отрицательного коэффициента Пуассона достаточно создать структуру, узлы которой связаны потенциалом или элементами, имеющими большую тангенциальную и малую нормальную жесткость.
Рис. 5.
Электронные микрофотографии пены из полиэфира (a, б) и меди (в, г) до (a, в) и после (б, г) всестороннего сжатия [25].

КИНЕТИЧЕСКИЙ ИЛИ ДИФФУЗИОННЫЙ РЕЖИМ ПРОВЕДЕНИЯ ХИМИЧЕСКИХ РЕАКЦИЙ
Принято считать, что при организации промышленных процессов следует стремиться к проведению химических реакций в кинетическом режиме. Приведем примеры, когда с точки зрения улучшения качества продукта и технологических параметров процесса выгоднее диффузионный режим.
Представим полимеризацию газообразного формальдегида в открытой системе [26, 27]. Газообразный формальдегид подается в реактор с раствором катализатора, растворяется и полимеризуется в нем. В зависимости от условий (скорость подачи и растворения, концентрация катализатора и другие) возможны различные режимы химического процесса. Предельными режимами являются кинетический, когда скорость лимитируется скоростью самой химической реакции полимеризации, и диффузионный, когда процесс лимитируется скоростью подвода мономера, т.е. скоростью его подачи в реактор или растворения в жидкой фазе. Важно, что критическим, устанавливающим качество полимера, параметром в данном случае является его молекулярная масса. Она, в свою очередь, определяется реакцией передачи цепи на примеси воды и метанола в формальдегиде. Поэтому необходимой и важной стадией производства полиформальдегида можно назвать его очистку от примесей. Рассмотрим два предельных случая.
При кинетическом режиме значения концентрации формальдегида [Ф]ж и примеси [Пр]ж равны их растворимости, а средняя степень (DP) полимеризации, пропорциональная молекулярной массе полимера, равна отношению скорости роста и передачи цепи:
При диффузионном режиме в реакцию полностью вступают поступающие в раствор из газовой фазы формальдегид и примесь. Тогда DP = $\frac{{{{\beta }_{{\text{Ф}}}}{{{\left[ {\text{Ф}} \right]}}_{{\text{Г}}}}}}{{{{\beta }_{{{\text{Пр}}}}}{{{\left[ {{\text{Пр}}} \right]}}_{{\text{Г}}}}}}$, где βФ и βПр – коэффициенты массопередачи для формальдегида и примеси. Отношения $\frac{{{{K}_{{\text{Ф}}}}}}{{{{K}_{{{\text{Пр}}}}}}}$ и $\frac{{{{{{\beta }}}_{{\text{Ф}}}}}}{{{{{{\beta }}}_{{{\text{Пр}}}}}}}$ мало отличаются от единицы, в то время как для катионной полимеризации формальдегида $\frac{{{{k}_{p}}}}{{{{k}_{п}}}} \ll 1$. Именно поэтому ММ полимера оказывается выше при проведении реакции в диффузионном режиме.
Еще один интересный и неожиданный результат состоит в том, что ММ полимера возрастает при увеличении концентрации катализатора, поскольку при этом происходит переход от кинетического к диффузионному режиму. Количественным критерием перехода служат соотношения $А{\kern 1pt} * \ll \frac{{{{{{\beta }}}_{{\text{Ф}}}}S}}{{{{k}_{P}}V{{K}_{{\text{Ф}}}}}}$ $\left( {А{\kern 1pt} * \ll \frac{{{{{{\beta }}}_{{{\text{Пр}}}}}S}}{{{{k}_{P}}V{{K}_{{{\text{Пр}}}}}}}} \right)$ для кинетического режима и $А{\kern 1pt} * \gg \frac{{{{{{\beta }}}_{{\text{Ф}}}}S}}{{{{k}_{P}}V{{K}_{{\text{Ф}}}}}}$ $\left( {А{\kern 1pt} * \gg \frac{{{{{{\beta }}}_{{{\text{Пр}}}}}S}}{{{{k}_{P}}V{{K}_{{{\text{Пр}}}}}}}} \right)$ – для диффузионного. Здесь V – объем жидкости в реакторе, а S – площадь поверхности, через которую происходит растворение газов. Предполагается, что концентрация реагентов в жидкости постоянна по объему, т.е. перемешивание осуществляется достаточно эффективно.
Промышленное крупнотоннажное производство – получение высокооктанового бензина путем сернокислотного алкилирования изобутилена бутиленами является важным производственным процессом. В таком случае диффузионный режим оказывается полезным [28]:

Общепринято, что каталитическое алкилирование изоалканов осуществляется через промежуточное образование ионов карбония:
Вторичный ион карбония менее устойчив, чем третичный, вследствие чего происходит быстрый обмен гидрид-ионом с изопарафином:
Образующийся третичный карбоний-катион реагирует с исходным олефином:
Получившийся ион карбония склонен к внутримолекулярным перегруппировкам, сопровождающимися миграцией водорода и метильных групп. Ионы карбония взаимодействуют с изобутаном, в результате чего образуются С8Н18 и третичный бутил-катион:
Основным и наиболее ценным продуктом являются изомерные углеводороды C8H18. Кроме того, процесс сопровождается побочными реакциями, например
Использование избытка изоалканов подавляет все побочные реакции, положительно влияет на выход и качество алкилата, повышает его октановое число, снижает расход катализатора и себестоимость продукта. Оказалось, однако, что если подавать вначале серную кислоту с изобутаном, создавая эмульсию, а только потом бутилены, то существенно уменьшается количество побочных продуктов и расход кислоты (образуется меньше кислого гудрона) и улучшается качество продукта (значительно повышается октановое число):
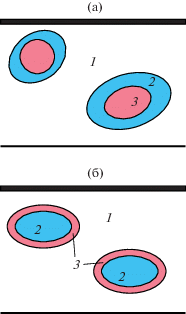
Какова же причина наблюдаемого явления? Объяснение может заключаться в следующем: после смешения кислоты с изобутаном образуется эмульсия кислоты в изобутане либо изобутана в кислоте (рис. 6). Эта эмульсия попадает в среду бутиленов, и они начинают растворяться в изобутане и (или) в кислоте. Диффузия в первом случае идет с поверхности мицеллы, и концентрация бутиленов у поверхности кислоты мала. Таким образом, реакция протекает в избытке изобутана, что и требуется для получения качественного продукта – алкилата и снижения расхода кислоты (уменьшения количества кислого гудрона). В этих условиях нет необходимости создавать большой избыток изобутана. Необходимым условием низкой концентрации бутиленов в зоне химической реакции является их медленная диффузия в изобутане по сравнению со скоростью самой реакции алкилирования:
Рис. 6.
Схема эмульсии кислоты в изобутане (а) и изобутана в кислоте (б): 1 – бутилены, 2 – изобутан, 3 – кислота.
Здесь $\frac{{{{{{\delta }}}^{2}}}}{D}$ – характерное время диффузии, где δ – толщина пленки изобутана, D – коэффициент диффузии бутилена в изобутане; $\frac{1}{{k\left[ {{{{\text{C}}}_{4}}{{{\text{H}}}_{{10}}}} \right]}}$ – характерное время химической реакции, где k – эффективная константа скорости бимолекулярного процесса превращения бутилена на границе раздела фаз кислота–изобутан.
Следовательно, переход в диффузионную область позволяет обеспечить в месте протекания химической реакции (вблизи поверхности серной кислоты) большой избыток изобутана, хотя в среднем по объему это может быть и не так. Таким образом, при полимеризации формальдегида диффузионный режим позволяет увеличить молекулярную массу продукта, а при сернокислотном алкилировании изобутана бутиленами в диффузионном режиме повышается выход продукта, октановое число и уменьшается расход катализатора.
ПОЛУЧЕНИЕ ПОЛИФОРМАЛЬДЕГИДА ПОЛИМЕРИЗАЦИЕЙ ЦИКЛИЧЕСКОГО ТРИМЕРА ФОРМАЛЬДЕГИДА ТРИОКСАНА
История началась с загадки [29–36]. Был обнаружен высокий (вплоть до семи) порядок скорости полимеризации в зависимости от концентрации триоксана, не связанный со значением диэлектрической постоянной или другими физико-химическими свойствами растворителя (таблица 1).
Таблица 1.
Порядок n скорости полимеризации триоксана по мономеру в различных растворителях, W ~ [M]n
| Катализатор | Растворитель | ε | Т, °С | Порядок по мономеру |
|---|---|---|---|---|
| BF3 · O(C2H5)2 | C6H6 | 2.0 | 55 | 3 |
| BF3 · O(C2H5)2 | C6H12 | 2.0 | 55 | 7 |
| SnCl4 | CHCl3 | 4.8 | 20 | 6 |
| BF3 · O(C2H5)2 | C6H5Cl | 5.6 | 30 | 3 |
| BF3 · O(C2H5)2 | C6H5Cl | 5.6 | 50 | 4 |
| SnCl4 | CH2Cl2 | 9.0 | 20 | 4 |
| SnCl4 | C2H4Cl2 | 10.3 | 30 | 3 |
| SnCl4 | C6H5NO2 | 35.0 | 30 | 3 |
В ряде случаев (например, в н-гептане и циклогексане) прослеживалась аномальная зависимость выхода полимера от концентрации мономера с максимумом и минимумом на кривой (рис. 7). Оказалось, что эти особенности обусловлены гетерогенным и обратимым характером полимеризации триоксана при большой (1) и малой (2) концентрации мономера:
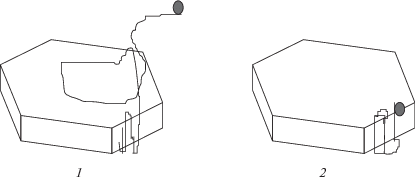
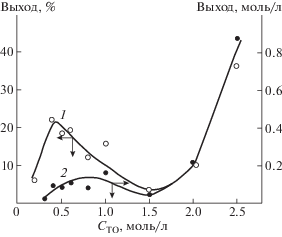
Наличием двух типов активных центров, находящихся в растворе и на поверхности твердого полимера, их превращением друг в друга при кристаллизации полимера и различием по термодинамическим и кинетическим параметрам удалось объяснить все удивительные особенности кинетики. Кроме того, структура образующегося полимера существенно зависит от условий полимеризации [37, 38]. Так, при низкой концентрации мономера, когда процесс термодинамически возможен только благодаря выпадению в твердую фазу и реакция протекает на активных центрах, расположенных на твердой поверхности полимера, формируются крупные полимерные кристаллы. При высокой концентрации мономера полимеризация идет на активных центрах в растворе, а полученный полимер выпадает в осадок в виде мелких частиц случайной формы (рис. 8).
Рис. 8.
Микрофотографии полимеров триоксана, полученных в нитробензоле (а, б) и хлористом метилене (в, г) при высокой (а, в) и низкой (б, г) концентрации мономера.

В этой связи, возникла идея термодинамического регулирования структуры (в том числе и молекулярной) полимера [39–41]. В частности, было показано, что состав сополимера триоксана и диоксалана совершенно различен при полимеризации, протекающей при концентрации триоксана выше и ниже ее предельного равновесного значения по отношению к растворенному полимеру. В первом случае наблюдается обычная зависимость состава полимера от состава мономерной смеси при примерно равных константах сополимеризации. Во втором случае независимо от состава мономерной смеси образуются крупные кристаллы сополимера одного и того же состава.
КАК ДОБИТЬСЯ ВЫСОКОЙ ПРОЧНОСТИ?
Почему проволока или полимерная расческа легко ломаются при нескольких изгибах, а деревья гнутся под ветром, но не ломаются?
Прочность – величина, характеризующая способность материала или конструкции сопротивляться разрушению под действием внешней нагрузки [42]. Есть как минимум две проблемы, которые усложняют однозначную интерпретацию этого определения, особенно для композиционных материалов. Во-первых, множество различных механизмов разрушения и их конкуренция, а во-вторых, изменение внешней нагрузки. Несколько конкретных примеров. Рассмотрим ориентированный волокнистый композит. При растяжении вдоль волокон работают три основных механизма разрушения: накопление разрывов волокон, распространение трещины поперек приложенной внешней силы растяжения и размотка, т.е. расслоение материала вдоль волокон. При сжатии вдоль волокон механизмы другие: потеря устойчивости волокон, расслоение вдоль волокон с последующей потерей устойчивости фрагментов материала и резкий изгиб или разрушение (исчерпание несущей способности) волокон с образованием складки под углом ~45° по отношению к приложенной силе сжатия. Для каждого механизма свои критические условия и требования к свойствам матрицы (связующего), границе раздела волокно–матрица и другие. Поскольку механизмы разрушения могут быть различными для конструкции и вырезанного для испытаний образца, не исключены ошибки при перенесении результатов стандартных испытаний на расчет конструкций.
Помимо упомянутых двух простых способов статического нагружения ориентированного волокнистого композита еще существует динамическое (или ударное) нагружение, циклическое приложение растягивающих, сжимающих или изгибных внешних сил и многое другое.
Часто улучшение прочности при одном виде нагрузки приводит к ее ухудшению при другом. Так, обычное оконное стекло – довольно прочный материал при статическом медленном нагружении, но оно легко бьется при ударе. В механике существует понятие статической прочности и вязкости разрушения, характеризующее сопротивление росту трещины в материале. Стекло имеет очень низкую вязкость разрушения, в нем легко распространяется трещина. Металлы могут иметь как высокую прочность, так и высокую вязкость разрушения. Эти показатели во многом определяются возможностью движения дислокаций [43]. Чем легче двигаются дислокации, тем ниже статическая прочность, но выше вязкость разрушения, и тем труднее распространяться трещине. Поэтому введение специальных добавок, затрудняющих движение дислокаций (например, углерода в железо) приводит не только к увеличению статической прочности, но и к увеличению хрупкости, легкости распространения трещин.
Природа и человек вслед за ней нашли выход из этого противоречия, создав волокнистые (армированные) композиционные материалы, такие как дерево или стеклопластик. В этом случае трещина, возникнув, допустим, при разрыве волокна, наталкивается на соседнее волокно, поворачивает и начинает двигаться вдоль него из-за большой разницы в прочности волокон и связующего или раздела фаз. Таким образом, трещина, исходно распространяющаяся поперек нагрузки, после поворота становится значительно менее опасной. Так происходит при нагружении ориентированного волокнистого материала вдоль волокон. Выбрав нужную схему армирования, расположения различных слоев в зависимости от внешней нагрузки, можно получить равнопрочный материал, чего не удается достичь для изотропного материала. Рассмотрим для примера трубу, работающую при большом внутреннем давлении, когда радиальное напряжение примерно вдвое превышает осевое. Это означает, что труба из изотропного материала (металл, полимер, керамика) всегда лопается вдоль оси от радиального напряжения, поскольку мы как бы слишком много материала положили вдоль оси. Из однонаправленного анизотропного материала можно сконструировать равнопрочную конструкцию, положив вдоль оси в 2 раза меньше слоев, чем по радиусу. Аналогичным образом удается конструировать и другие, более сложные детали, формируя равнопрочные изделия. Данный пример показывает, как можно примирить две различные характеристики прочности – статическую прочность и вязкость разрушения.
Еще одна характеристика прочности материала – усталостная прочность при циклическом изгибе. Хорошо известно, что металлическую проволоку достаточно несколько раз согнуть, чтобы она сломалась [44, 45]. То же самое происходит, например, с пластмассовой расческой. Дело в том, что в кристаллических и стеклообразных твердых телах при большой пластической деформации накапливаются дефекты (дислокации в кристаллических телах, дилатации в стеклах, крейзы в полимерах), которые превращаются в трещины, приводящие, в конце концов, к разрушению материала. В жидкостях, напротив, при деформации (течении) дефекты не накапливаются или быстро релаксируют, и тело приходит в равновесное состояние. Поэтому природа придумала для больших изгибных деформаций твердых тел, например костей, специальное устройство – сустав, содержащий жидкую смазку, в которой и происходят большие деформации. Аналогичным образом работает смазка в подшипниках.
Особенно интересно устроено дерево. В нем, живом, есть жидкий сок, который, по-видимому, и принимает на себя большие изгибные деформации [46–48]. Если дерево или ветка высыхают, то они становятся хрупкими и легко ломаются при изгибе. При низких значениях температуры жидкий сок, казалось бы, должен замерзнуть и дерево станет хрупким. Но в таких деревьях, как лиственница, которые хорошо себя чувствуют даже в сибирские морозы, в качестве сока выступает водный раствор арабиногалактана – полисахарида, замерзающего при очень низкой температуре [46, 47]. Еще один материал, в котором дефекты при деформации не накапливаются, – эластомеры и резины. Дело в том, что при обычных условиях они также находятся в жидком состоянии, и времена релаксации структуры у них очень небольшие. Так устроены мышцы у животных и человека, а также шины и другие резиновые изделия, созданные человеком [44, 45].
Работа выполнена в рамках Госзадания (гоcрегистрация № АААА-А20-120030590042-8).
Список литературы
Берлин А.А., Халтуринский Н.А., Шаулов А.Ю. // Горение и плазмохимия. 2006. Т. 4. № 2. С. 79.
Берлин Ал.Ал. // Соросовский образовательный журн. 1996. № 9. С. 57.
Марголин А.Д. // Докл. АН СССР. 1982. Т. 264. № 4. С. 888.
Nonokawa R., Yashima E. // J. Am. Chem. Soc. 2003. V. 125. P. 1278.
Yashima E., Maeda K., Okamoto Y. // Nature. 1999. V. 399. P. 449.
Ishikawa M., Maeda K., Yashima E. // J. Am. Chem. Soc. 2002. V. 124. P. 7448.
Brunsveld L., Folmer B.J.B., Meijer E.W., Sijbesma R.P. // Chem. Rev. 2001. V. 101. P. 4071.
Green M.M., Peterson N.C., Sato T., Teramoto A., Cook R., Lifson S. // Science. 1995. V. 268. P. 1860.
Аветисов В.А., Берлин А.А., Иванов В.В. // Докл. РАН. 2004. Т. 395. № 4. С. 3.
Zharov A.A. // Polymer Science B. 2004. V. 46. № 9–10. P. 268.
Момзяков А.А., Жаров А.А., Фадеева К.С., Дебердеев Т.Р., Берлин А.А., Дебердеев Р.Я. // Хим. физика. 2019. Т. 38. № 2. С. 9.
Берлин Ал.Ал. // Хим. физика. 2018. Т. 37. № 9. С. 1.
Berlin Al.Al., Gendelman O.V., Sinelnikov N.N., Mazo M.A., Manevich L.I. // J. Phys.: Condens. Matter. 1999. V. 11. № 24. P. 4583.
Berlin Al.Al., Gendel’man O.V., Sinel’nikov N.N., Mazo M.A., Manevich L.I. // Russ. J. Phys. Chem. 2000. V. 74. suppl. 1. (only in English) P. 46.
Берлин Ал.Ал., Гендельман О.В., Мазо М.А., Маневич Л.И., Балабаев Н.К. // Докл. АН СССР. 2002. Т. 382. № 6. С. 798.
Берлин Ал.Ал., Мазо М.А. // Все материалы. Энциклопедический справочник. 2012. № 8. С. 2.
Berlin Al.Al., Mazo M.A. // Polymer Science D. 2013. V. 6. № 3. P. 228.
Берлин Ал.Ал., Ротенбург Л., Басэрст Р. // Хим. физика. 1991. Т. 10. № 9. С. 1284.
Берлин Ал.Ал., Ротенбург Л., Басэрст Р. // Высокомолек. соед. А. 1992. Т. 34. № 7. С. 6.
Берлин Ал.Ал., Мазо М.А., Балабаев Н.К. // Все материалы. Энциклопедический справочник. 2012. № 10. С. 10.
Berlin Al.Al., Mazo M.A., Strel’nikov I.A., Balabaev N.K. // Polymer Science D. 2015. V. 8. № 2. P. 85.
Berlin Al.Al., Rothenburg L., Bathurst R.J. // 3-rd Japan–USSR Joint Symposium on Advanced Composite Materials. Moscow. 1991. P. 319.
Rothenburg L., Berlin Al.Al., Bathurst R.J. // Nature. 1991. V. 354. № 6353. P. 470.
Берлин Ал.Ал., Ротенбург Л., Басэрст Р. // Высокомолек. соед. Б. 1991. Т. 33. № 8. С. 619.
Fris E.A., Lakes R.S., Park J.B. // J. Mater. Sci. 1988. V. 23. P. 4406.
Берлин Ал.Ал., Вольфсон С.А. Кинетический метод в синтезе полимеров. М.: Химия, 1973.
Берлин Ал.Ал., Вольфсон С.А., Ениколопян Н.С. Кинетика полимеризационных процессов. М.: Химия, 1978.
Берлин А.А., Прочухан К.Ю., Прочухан Д.Ю., Прочухан Ю.А. // Хим. физика. 2019. Т. 38. № 3. С. 12.
Берлин Ал.Ал., Ениколопян Н.С. // Высокомолек. соед. А. 1969. Т. 11. № 12. С. 2671.
Берлин Ал.Ал., Богданова К.А., Кравчук И.П., Ракова Г.В., Ениколопян Н.С. // Докл. АН СССР. 1969. Т. 184. № 5. С. 1128.
Берлин Ал.Ал., Кравчук И.П., Ракова Г.В., Розенберг Б.А., Ениколопян Н.С. // Высокомолек. соед. А. 1973. Т. 15. № 3. С. 554.
Берлин Ал.Ал., Вольфсон С.А., Олейник Э.Ф., Ениколопян Н.С. // Высокомолек. соед. А. 1970. Т. 12. № 2. С. 443.
Богданова К.А., Ракова Г.В., Берлин Ал.Ал., Ениколопян Н.С. // Высокомолек. соед. А. 1972. Т. 14. № 9. С. 1976.
Берлин Ал.Ал., Богданова К.А., Ракова Г.В., Ениколопян Н.С. // Высокомолек. соед. А. 1975. Т. 17. № 3. С. 643.
Берлин Ал.Ал., Богданова К.А., Маркевич М.А., Ракова Г.В., Ениколопян Н.С. // Докл. АН СССР. 1973. Т. 211. № 4. С. 874.
Богданова К.А., Берлин Ал.Ал., Компаниец В.З., Ракова Г.В., Мирошниченко Е.А., Лебедев Ю.А., Ениколопян Н.С. // Высокомолек. соед. А. 1975. Т. 17. № 3. С. 658.
Берлин Ал.Ал., Карюхина Г.А., Трофимова Г.М., Ениколопян Н.С. // Докл. АН СССР. 1970. Т. 195. № 5. С. 1147.
Берлин Ал.Ал., Воробьева Г.А., Трофимова Г.М., Ениколопян Н.С. // Докл. АН СССР. 1974. Т. 214. № 2. С. 373.
Берлин Ал.Ал., Ениколопян Н.С. // Докл. АН СССР. 1971. Т. 196. № 5. С. 1111.
Берлин Ал.Ал., Дебердеев Р.Я., Перухин Ю.В., Гарипов Р.М. // Клеи. Герметики. Технологии. 2009. № 2. С. 2.
Воробьева Г.А., Трофимова Г.М., Берлин Ал.Ал., Ениколопян Н.С. // Высокомолек. соед. А. 1974. Т. 16. № 7. С. 1493.
Баженов С.Л., Берлин Ал.Ал., Кульков А.А., Ошмян В.Г. Полимерные композиционные материалы. Прочность и технология. Долгопрудный: Издательский Дом “Интеллект”, 2009.
Коттрел А. Теория дислокаций. М.: Мир, 1969.
Berlin A.A. // Polymer Science D. 2020. V. 13. № 1. P. 57.
Berlin A. // Phys. Chem. Ind J. 2019. V. 14. № 1. P. 125.
Аким Э.Л., Роговина С.З., Берлин А.А. // Докл. РАН Сер. Химия, наука о материалах. 2020. Т. 491. С. 73.
Akim E.L., Rogovina S.Z., Berlin Al.Al. // Dokl. Phys. Chem. 2020. V. 491. № 1. P. 33.
Аким Э.Л., Пекарец А.А., Роговина С.З., Берлин Ал.Ал. // Все материалы. Энциклопедический справочник. 2020. № 9. С. 3.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)