

Высокомолекулярные соединения (серия С), 2021, T. 63, № 1, стр. 14-32
КИНЕТИЧЕСКИЕ ОСОБЕННОСТИ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ МЕТИЛМЕТАКРИЛАТА В УСЛОВИЯХ ОБРАТИМОГО ИНГИБИРОВАНИЯ НИТРОКСИЛАМИ
М. Ю. Заремский a, *, В. В. Одинцова a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: zaremski@mail.ru
Поступила в редакцию 05.09.2020
После доработки 06.12.2020
Принята к публикации 28.01.2021
Аннотация
В обзоре обобщены наши и литературные данные по кинетике и механизму контролируемой радикальной полимеризации метакрилатов с участием нитроксильных радикалов. Проанализированы причины, влияющие на возможность или невозможность реализации радикальной полимеризации метилметакрилата в режиме обратимого ингибирования. Основными факторами, обусловливающими специфику полимеризации метакрилатов в присутствии нитроксильных радикалов, являются взаимодействие нитроксилов с радикалами роста по механизму диспропорционирования, “гипер”-эффект Фишера, высокая скорость реинициирования и аномально низкая скорость обратимого обрыва.
ВВЕДЕНИЕ
Известно, что не все виниловые мономеры способны участвовать в радикальной полимеризации в условиях обратимого ингибирования нитроксильными радикалами. К их числу относится ММА и его производные. Периодически появляются сведения о “первом” осуществлении контролируемой полимеризации ММА под действием нитроксилов, однако на практике область контроля обычно ограничивается 30–50% конверсии мономера [1–7]. Исследователи выдвигали различные гипотезы для объяснения данного явления. Вначале это связывали с отсутствием спонтанной полимеризации у ММА [8, 9]. В дальнейшем общепринятым является мнение, что причиной является реакция диспропорционирования радикалов роста ММА и его аналогов и нитроксилов [3, 10, 11]. Однако оказалось, что существует ряд нитроксилов, для которых реакция обрыва с радикалами ММА не происходит по механизму диспропорционирования. Тем не менее, проблема контролируемой полимеризации метакриловых мономеров в условиях обратимого ингибирования нитроксилами до сих пор не решена.
В настоящей работе мы попытались обобщить наши новые и литературные данные о кинетике и механизме полимеризации ММА под действием нитроксилов. Это позволило сформулировать отличительные особенности процесса и предложить другие возможные причины неспособности ММА к контролируемой полимеризации с участием нитроксилов. Ниже мы рассмотрим основные особенности кинетики и механизма полимеризации метакрилатов с участием нитроксилов.
РЕАКЦИЯ ДИСПРОПОРЦИОНИРОВАНИЯ РАДИКАЛОВ РОСТА С НИТРОКСИЛАМИ
Первые результаты полимеризации ММА с участием нитроксильных радикалов были получены в конце 1980-х годов в нашей стране и затем в конце 1990-х в Австралии [12, 13]. Оказалось, что полимеризация ММА в массе при 90°С с участием ТЕМПО и других нитроксилов различной природы

заканчивается в течение 1 ч и конверсия мономера не превышает 10–40%. Тем не менее среднечисловая молекулярная масса Mn ПММА близка к теоретическим значениям, свойственным для “живых” процессов. Таким образом, контролируемая полимеризация “затухала” во времени. При этом очищенные полимеры содержали двойную связь на конце цепи, что объясняли реакцией диспропорционирования нитроксилов с радикалами роста, приводящей также к образованию гидроксиламина.
Для определения количества концевых двойных связей и гидроксиламина применяли ЯМР-спектроскопию и масс-спектрометрию. Оказалось, что в полибутилметакрилате, полученном под действием ТЕМПО, содержание концевых двойных связей близко к 100% [14]. Такое не может быть результатом обычной реакции диспропорционирования между двумя радикалами роста (где эта доля не может превышать 50% молекул), а только следствием переноса атома β-водорода от радикала роста к TEMПO.
Считается, что именно реакция диспропорционирования является основной причиной необратимого ингибирования полимеризации ММА. В связи с этим основные попытки осуществления контролируемого синтеза ПММА направлены на поиск новых нитроксилов, не склонных к диспропорционированию с метакрилатными радикалами роста.
Вместе с тем к одним и тем же продуктам могут привести две реакции [11]:
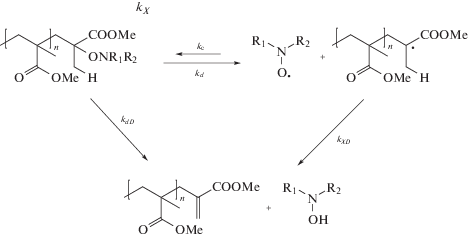
Кроме диспропорционирования возможна и внутримолекулярная перегруппировка на стадии распада концевой группы. Первая конкурирует с реакцией рекомбинации радикала роста и нитроксила и характеризуется константой скорости второго порядка kХD, а вторая – с реакцией реинициирования и описывается константой скорости первого порядка kdD.
На практике эти два механизма почти неразличимы [15], и в обоих случаях для их описания используют понятие “доли” диспропорциони-рования fD = kХD/(kХD + kХ) или fD = kdD/(kd + kdD) соответственно. Считается, что внутримолекулярная реакция переноса атома Н в алкоксиаминах определяется энергией активации гомолитической диссоциации, но не природой нитроксила и не геометрией переходного состояния [16]. Такая реакция весьма вероятна для алкоксиаминов с прочной связью C–ON (Ea > 125 кДж/моль) и маловероятна для соединений со слабой связью (Ea < 115 кДж/моль).
Чтобы определить механизм диспропорционирования в Томографическом центре Сибирского отделения Российской академии наук (Новосибирск) предложили оригинальный подход. Алкоксиаминовые инициаторы – модельные для концевой группы ПММА-нитроксил разлагали термически в присутствии и в отсутствие активного агента передачи цепи тиофенола [17]. Если реакция диспророцинирования – это внутримолекулярный процесс, то на ее скорость наличие тиофенола влиять не будет (Iб). Но если это реакция между радикалами роста и нитроксила (Iа), то скорость образования концевых двойных связей в ПММА в присутствии тиофенола резко снизится, так как последний станет конкурентом нитроксила.


Оказалось, что алкоксиамин 4-нитрофенил-2-(2,2,6,6-тетраметилпиперидин-1-илокси)-2-метилпропионат диспропорционирует исключительно по бимолекулярному механизму с fD = 3.5%. Такой же механизм реакции предполагается и для реакции макрорадикала ПММА с ТЕМПО на основании данных для алкилизобутиратных низкомолекулярных радикалов [18].
Для алкоксиаминов на основе пятичленных циклических нитроксилов механизм диспропорционирования зависит от природы заместителей в 2 и 5-положениях нитроксила [18]. Ниже приведены структуры нитроксилов на основе имидазолина, имидазолидина и пирролидина, для которых характерны меж- и внутримолекулярное диспропорционирование

только межмолекулярное диспропорционирование

Внутримолекулярный механизм требует циклического переходного состояния с хорошим перекрытием азотной орбитали nσ и разрыхляющей орбитали σ* связи C–H. Это возможно в случае менее стерически затрудненных соединений – например, нитроксила с четырьмя метильными группами в положении 2 и 5. Температурная зависимость этой реакции подчиняется уравнению Аррениуса для kdD с предэкспоненциальным множителем A = 5.2 × 1015 с–1, и высокой энергией активации (Ea ~ 140 кДж/моль). Напротив, для имидазолиновых нитроксилов с объемными 2 и 5-заместителями типична реакция межмолекулярного диспоропорцинирования.
Обычно доля диспропорционирования в полимеризации виниловых мономеров с нитроксилами мала, и этим побочным процессом пренебрегают. Так, например, при полимеризации стирола с участием ТЕМПО она составляет всего 0.4–0.6% [19, 20], а с участием нитроксила SG1 – вообще стремится к нулю. Однако она резко повышается при полимеризации метакрилатов.
Теоретический расчет [15] предсказывает полное ингибирование полимеризации, даже если доля диспропорционирования составляет всего несколько процентов. Предельно достижимая конверсия qmax связана с вероятностью диспропорционирования уравнением
(1)
${{q}_{{max}}} \approx 1 - {\text{exp}}\left\{ { - \frac{3}{2}{{k}_{p}}{{{\left( {\frac{{{{{[{\text{RX}}]}}_{0}}}}{{3f_{D}^{2}{{k}_{d}}({{k}_{X}} + {{k}_{{XD}}}){{k}_{t}}}}} \right)}}^{{1/3}}}} \right\},$Так, в типичных условиях полимеризации с участием нитроксилов с набором значений элементарных констант kd = 4.5 × 10–3 c–1, kt = 108 (моль/л)–1c–1, kX = 2.2 × 107 (моль/л)–1c–1, kp = 5 × 103 (моль/л)–1c–1 при [RX]0 = 0.1 моль/л максимальная степень превращения мономера составляет 93, 81 и 59%, при fD = 1, 2 и 5% соответственно. Обойти этот запрет не удается, даже если повысить константу роста или концентрацию инициатора. При увеличении значения константы роста в 4 раза (kp = 2 × × 104 (моль/л)–1c–1) полимеризация до глубоких конверсий становится возможной, только если вероятность диспропорционирования меньше 7%. Если при этом одновременно увеличить на порядок концентрацию инициатора, то и тогда вероятность диспропорционирования должна быть менее 22%.
Следовательно, для реализации “живого” роста цепей ПММА во всей области конверсий мономера, необходимы такие нитроксилы и/или такие условия реакции, которые бы обеспечивали вероятность диспропорционирования менее 1%.
Из уравнения (1) не сложно получить [15] упрощенную формулу для решения обратной задачи – оценки доли диспропорционирования по времени прекращения полимеризации tp ≈ 2/fDkd. Именно так была оценена величина fD = 20% для полимеризации ММА в присутствии ТЕМПО при 120°С [15]. По другим косвенным данным она равна 10% [21]. Используя такой подход, мы оценили величину fD как 11 и 9% при 90 и 80°С соответственно. Прямое измерение вероятности диспропорционирования по данным ЯМР дает оценку fD < 2% при 60°С [13]. Отсюда следует, что вероятность диспропорционирования существенно понижается с уменьшением температуры.
Это обстоятельство позволило нам повысить выход ПММА до 60% при 80°С. Более того, в таких условиях удается наблюдать рост ММ полимера при увеличении конверсии мономера (табл. 1). Те же особенности характерны и для полимеризации ММА с участием ди-трет-бутилнитроксила. Однако и в указанных условиях полимеризация затухает и не доходит до полного превращения мономера в полимер (рис. 1).
Таблица 1.
Молекулярно-массовые характеристики ПММА, полученных при полимеризации ММА, инициированной ДАК под действием 4-гидрокси-ТЕМПО и ди-трет-бутилнитроксила (ДТБН) в массе ([4-гидрокси-ТЕМПО] = [ДТБН] = [ДАК] = 0.01 моль/л)
| Нитроксил | Т, °С | Время, мин | Конверсия, % | Mn × 10–3 | Ð |
|---|---|---|---|---|---|
| 4-гидрокси-ТЕМПО | 120 | 3 | 8 | 10.1 | 1.6 |
| 10 | 12 | 12.5 | 2.7 | ||
| 160 | 17 | 11.8 | 2.7 | ||
| 4-гидрокси-ТЕМПО | 90 | 35 | 5 | 8.3 | 2.1 |
| 55 | 19 | 16.1 | 2.4 | ||
| 120 | 49 | 20.7 | 3.7 | ||
| ДТБН | 90 | 40 | 2 | 1.0 | 2.3 |
| 60 | 31 | 7.8 | 2.3 | ||
| 80 | 60 | 21.3 | 2.4 |
Рис. 1.
Интегральные (а) и дифференциальные (б) кинетические кривые полимеризации ММА. [ММА] = 9.4, [4-гидрокси-ТЕМПО] = 0.01, [ДАК] = 0.01 моль/л. Т = 80 (1), 84 (2), 90 (3) и 96°С (4). Цветные рисунки можно посмотреть в электронной версии.

Очевидно, природа заместителей в 2- и 6-положении пиперидинового нитроксила должна влиять на вероятность диспропорционирования. Однако соответствующих экспериментальных данных крайне мало. По нашим данным аналог ТЕМПО с 2,6-циклогексильными заместителями диспропорционирует еще быстрее, чем ТЕМПО: полное превращение нитроксила в гидроксиламин (а соответственно и прекращение полимеризации) происходит при 80°С всего за 15 мин. Конверсия ММА при этом не превышает 4%.
Описаны результаты исследований влияния природы заместителей в имидазолиновых нитроксилах на диспропорционирование с низкомолекулярными радикалами, модельными для радикала роста ММА – трет-бутилизобутиратным и нитрофенилизобутиратным [2, 3, 17, 18, 22–24]. Вероятность диспропорционирования понижается от 23 до 0.3% в ряду

Как видно, выполняется такое правило: вероятность диспоропорцинирования уменьшается с увеличением размера заместителей в положениях 2 и 5.
Природа алкильного заместителя в алкилметакрилате, по мнению авторов [18], не оказывает заметного влияния на скорость диспропорционирования. Тем не менее, как показали сами авторы, при переходе от трет-бутилизобутиратного радикала к п-нитрофенилизобутиратному вероятность диспропорционирования с ТЕМПО увеличивается в 1.5 раза (с 2.2 до 3.5%).
В последние годы синтезированы новые нитроксилы, которые практически не диспропорционируют с низкомолекулярными радикалами (fD ≤ 1%), имитирующими радикалы роста ММА, к которым относятся следующие [3, 23]:

Именно они и рассматриваются сейчас как потенциальные агенты контролируемого синтеза ПММА [24].
Однако следует иметь ввиду, что даже в том случае, когда продукты распада алкоксиаминового инициатора сами не могут диспропорционировать в инертной среде, такая возможность появляется, как только в системе образуются радикалы роста ММА. Например, нитроксил N2 действительно не диспропорционирует с этилизобутриатным радикалом в бензоле и хлорбензоле. Однако при замене инертной среды реакции на мономер ММА уже через 5 мин после начала полимеризации система содержит 3% гидроксиламина, а через 20 мин его концентрация повышается до ~15% и далее не меняется [25].
Аналогичный результат наблюдали при исследовании поведения нитроксила

В инертной среде вероятность диспропорционирования была невелика. Однако в среде мономера уже через 15 мин весь нитроксил превращался в гидроксиламин [25].
Это означает, что высокомолекулярные радикалы диспропорционируют намного лучше, чем их низкомолекулярные аналоги.
Та же закономерность характерна и для незамещенного ТЕМПО. Выше указывалось, что его доля диспропорционирования при полимеризации ММА составляет 20%. Однако если заменить ПММА-радикалы на их низкомолекулярные аналоги, она понижается до 3–4% [17, 26].
По поводу диспропорционирования радикалов ПММА с нитроксилом SG1 данные весьма противоречивы. Ранее считалось, что такая реакция вообще не протекает, поскольку ПММА не содержит концевых двойных связей [1, 27]. Кинетическое моделирование этого процесса в предположении бимолекулярного диспропорционирования дало величину kХD = 1.7 × 103 (моль/л)–1c–1, что отвечает значению fD = 8.5% [28]. Прямой эксперимент на модельной реакции между метилизобутиратным радикалом и SG1 дал то же самое значение [16]. Однако в работе [29] доля продуктов диспропорционирования в ПММА, полученных с SG1, составляла от 64 до 100%. Наконец, в работе [30] предполагается иной внутримолекулярный механизм диспропорционирования, продуктами которого являются ПММА с концевой двойной связью и N-оксид SG1. Было высказано предположение, что эти разногласия вызваны наличием остаточного кислорода в полимеризате [2]. Действительно, в менее дегазированных растворах, т.е. в условиях обычной дегазации с помощью вакуумирования масляным насосом или барботирования инертного газа, содержание алкена в продуктах реакции составляет 25% против 90% для дегазированных в глубоком вакууме.
Отметим, что и для нитроксила SG1 наблюдается резкое увеличение доли диспропорционирования при переходе от реакции с низкомолекулярным радикалом к реакции с полиметилметакрилатным радикалом [2].
Таким образом, во всех описанных системах реакция диспропорционирования в мономере идет намного быстрее, чем в инертном растворителе. Иными словами, вероятность диспропорционирования для высокомолекулярного радикала роста существенно выше, чем для низкомолекулярного.
В последние годы большие надежды в нитроксильной полимеризации ММА возлагают на нитроксил DPAIO.

В его присутствии удается осуществить контролируемую полимеризацию ММА до конверсии 60% [4]. Это связывают с тем, что для данного нитроксила не характерна реакция диспропорционирования с радикалами ММА. Действительно экспериментально было показано, что такая реакция между DPAIO и радикалом

отсутствует [17]. Для радикала ПММА, насколько нам известно, такой информации пока нет. Отметим, что радикальная полимеризация ММА затухает и в присутствии этого нитроксила [4].
Интересные данные были получены совсем недавно для полимеризации ММА с алкоксиаминовым инициатором, который авторы назвали Dispolreg 007 [31, 32]

В отличие от SG1 и DPAIO, нитроксил этого соединения содержит два атома H в α-положениях по отношению к группе N–O, что, по мнению авторов, препятствует протеканию диспропорционирования. Действительно, величина fD при полимеризации ММА, инициированной Dispolreg 007 при 90°С, составляет всего 0.2%, а доля продуктов диспропорционирования в конечном полимере не превышает 1%. Выход полимера достигает 90%. При этом Mn практически не увеличивается в ходе полимеризации, но близка к теоретической при конверсии 100%.
Таким образом, диспропорционирование – важная, но не единственная причина, препятствующая реализации механизма обратимого ингибирования в полимеризации ММА.
ГИПЕР-ЭФФЕКТ НАКОПЛЕНИЯ СТАБИЛЬНЫХ РАДИКАЛОВ ИНГОЛЬДА–ФИШЕРА
Хорошо известно, что полимеризация виниловых мономеров протекает на фоне высокой концентрации свободного нитроксила (на несколько порядков выше концентрации радикалов роста). Это является следствием реализации эффекта накопления стабильных радикалов – эффекта Ингольда–Фишера [33–35].
Природу этого эффекта легко понять из простейшей схемы полимеризации, инициированной алкоксиаминовым инициатором RX. При распаде RX образуются нитроксил Х. и алкильный радикал R., запускающий рост цепи
Для первичного радикала роста ${\text{P}}_{n}^{ \bullet }$ равновероятна как реакция рекомбинации с нитроксильным, так и с другим растущим радикалом
В последнем случае каждый акт квадратичного обрыва макрорадикалов приводит к образованию “мертвой” цепи и двух свободных нитроксильных радикалов Х.. Именно поэтому в начале полимеризации происходит интенсивное накопление нитроксильных радикалов в системе.
Обычно это происходит в начальный короткий период времени (от 10 мин до 2–3 ч), а затем концентрация нитроксилов практически не меняется. По относительной величине доля высвободившегося нитроксила составляет от 0.1 до 1% от исходного инициатора. С одной стороны, такой концентрации не хватает для прекращения процесса; с другой, ее вполне достаточно для практически полного подавления дальнейшей необратимой гибели макрорадикалов. В данном случае реализуется режим обратимого ингибирования, характеризующийся установлением равновесия между реакцией реинициирования и обратимого обрыва растущих цепей на нитроксилах. Хотя полностью подавить квадратичный обрыв и другие побочные реакции не удается, но доля “живых” макромолекул остается преобладающей вплоть до предельных конверсий. Такая картина наблюдается при полимеризации стирола с участием ТЕМПО, SG1, имидазолиновых, индолиновых и других нитроксилов. При полимеризации алкилакрилатов и других акриловых производных концентрация свободного нитроксила, накопленная по эффекту Фишера, более низкая и недостаточная для того, чтобы подавить квадратичный обрыв макрорадикалов. В связи с этим в указанные системы изначально добавляют дополнительное количество свободных нитроксилов.
Отличительной особенностью полимеризации ММА служит гипер-эффект Фишера. Как показали наши исследования, в короткий начальный промежуток времени концентрация свободного нитроксила достигает 5–20% от концентрации исходного инициатора.
Рассмотрим эту особенность для полимеризации ММА, инициированной алкоксиаминами, на примере инициаторов А1 и А2 на основе нитроксилов N1 и N2

На кинетических кривых изменения концентрации нитроксила, как и при полимеризации стирола [33], можно выделить два участка (рис. 2). На первом наблюдается увеличение концентрации нитроксила, что связано квадратичным обрывом первичных радикалов. На втором участке концентрация ТЕМПО выходит на постоянное значение. Чем выше исходная концентрация инициатора, тем больше свободного нитроксила высвобождается.
Рис. 2.
Зависимости концентрации N1 (а) и N2 (б) от времени при полимеризации ММА. [A1] = [А2] = = 0.03 (1), 0.01 (2) и 0.003 моль/л (3). Т = 80°С. Здесь и ниже приведены данные, полученные при полимеризации в массе в вакууме.

Для инициатора пиперидиновой природы А1 доля нитроксила на втором участке составляет 10–20% от исходной концентрации А1 (рис. 2а). При этом накопление происходит в короткий начальный период 10–15 мин. Практически такая же закономерность характерна и для инициатора имидазолиновой природы А2 (рис. 2б).
Для полимеризации ММА, протекающей под действием бинарных систем радикальный инициатор/нитроксил эффект накопления стабильных радикалов несколько отличается от рассмотренного выше.
На кинетических кривых изменения концентрации нитроксила (рис. 3) появляется дополнительный короткий начальный участок резкого падения, что обусловлено быстрым распадом инициатора и захватом первичных радикалов нитроксилами. Затем наблюдаются те же участки роста и запределивания концентрации ингибитора, что и при полимеризации ММА, инициированной алкоксиаминами. Роль последних выполняют аддукты первичных радикалов роста с нитроксилами, сформировавшиеся in situ.
Рис. 3.
Зависимости концентрации ТЕМПО от времени при полимеризации ММА (а, б) и стирола (а) при 120°С. а: 1 – ММА, 2 – стирол; [ДАК] = 0.01 моль/л, [ТЕМПО]0 = 0.01 (1) и 0.012 моль/л (2); б: [ДАК] = [ТЕМПО]0 = 0.1 (1), 0.01 (2) и 0.0025 моль/л (3).

И в этих системах концентрация свободного нитроксила оказывается довольно высокой. Так, при полимеризации ММА под действием системы ДАК–ТЕМПО при 120°С она на 1.5 порядка выше, чем для стирола в тех же условиях (рис. 3а).
Понижение исходной концентрации ТЕМПО при сохранении мольного соотношения ДАК–ТЕМПО приводит к уменьшению его доли на стационарном участке. Как и при полимеризации стирола [37], разбавление мономера растворителем вызывает резкое возрастание стационарной концентрации ТЕМПО (табл. 2).
Из сопоставления кривых изменения концентрации нитроксила (рис. 2, 3б) с кинетическими данными [25, 36] следует, что полимеризация ММА как при инициировании алкоксиаминами, так и в бинарных системах радикальный инициатор–нитроксил возможна только в начальный период, когда концентрация свободного нитроксила мала. Это отвечает первому участку на кривых рис. 2 и “яме” на кривых рис. 3б. На стационарном участке полимеризация практически полностью прекращается. В этом и заключается принципиальная разница между полимеризацией стирола и ММА под действием нитроксилов. Если для стирола стационарные условия – основной этап полимеризации, в котором происходит рост “живых” цепей в режиме обратимого ингибирования, то для ММА – это “мертвая” область, в которой из-за высокой концентрации свободного нитроксила равновесие между растущими и спящими цепями сдвигается в сторону последних, и полимеризация затухает вплоть до полного прекращения. Заметим, что при исходной концентрации ДАК и ТЕМПО 0.1 моль/л, когда концентрация свободного ТЕМПО не опускается ниже 10–3 моль/л, происходит полное ингибирование полимеризации.
Таким образом, во всех изученных системах (с участием пиперидиновых и имидазолиновых нитроксилов, при полимеризации в массе и в растворе, при инициировании алкоксиаминами и под действием бинарных систем ДАК–нитроксил) для полимеризации ММА характерен гипер-эффект накопления стабильных радикалов. Причина данного явления будет рассмотрена ниже.
Уменьшить степень его проявления при полимеризации ММА можно, понизив температуру реакции. Как видно на рис. 4, при уменьшении температуры до 80–90°С концентрация свободного ТЕМПО понижается до 10–4–10–5 моль/л. В таких условиях, как уже отмечалось выше, полимеризация ММА протекает до конверсии 50–60%. При этом не только уменьшается вероятность диспропорционирования, но и вырождается эффект накопления стабильных радикалов. Однако даже в данном случае полимеризация ММА с участием ТЕМПО затухает со временем.
Рис. 4.
Зависимость концентрации 4-гидрокси-ТЕМПО от времени при полимеризации ММА в массе при 80 (1), 84 (2), 90 (3) и 96°С (4) в присутствии 0.01 моль/л ДАК и 0.01 моль/л 4-гидрокси-ТЕМПО.

Проведенные эксперименты показывают, что существует еще одна причина, кроме диспропорционирования, из-за которой становится невозможной полимеризация ММА по механизму обратимого ингибирования: равновесие между “спящими” и растущими цепями сдвигается в сторону первых под действием высокой концентрации свободного нитроксила, накапливающегося в системе. Для нитроксилов, не склонных к диспропорционированию, таких как N2, именно она служит основной причиной затухания полимеризации.
Следует отметить, что впервые предположение о затухании полимеризации ММА вследствие накопления нитроксилов в ходе полимеризации, было высказано в 1998 г. на основании результатов кинетического моделирования [13]. Однако расчеты были сделаны с использованием далеких от реальности констант элементарных актов, неизвестных в то время.
ВЫСОКАЯ СКОРОСТЬ ИНИЦИИРОВАНИЯ И АНОМАЛЬНО НИЗКАЯ СКОРОСТЬ ОБРЫВА
Естественно, встает вопрос: в чем причина столь ярко выраженного эффекта накопления стабильных радикалов? На наш взгляд, это результат большой разницы между константами скоростей обратимого и необратимого обрыва и высокого значения скорости реинициирования. Скорость накопления нитроксилов будет зависеть от соотношения скоростей указанных реакций.
То, что степень проявления эффекта Фишера будет расти с увеличением разницы в значениях скоростей необратимого и обратимого обрыва, очевидно. Менее очевидно то, что она будет увеличиваться с повышением скорости реинициирования. Физический смысл данного явления – возрастание вероятности квадратичного обрыва большого количества первичных радикалов в начале полимеризации. На эту причину, как на основную, препятствующую реализации “живого” механизма нитроксильной полимеризации ММА, указывали в работе [33].
Согласно простейшей модели Фишера, концентрация нитроксила Х пропорциональна соотношению [34, 35]
Если для полимеризации стирола и алкилакрилатов с участием нитроксилов значения обеих констант обрыва примерно равны [33], то для ММА величина kX на один–два порядка ниже, чем kt [25]. Кроме того, величина kd для нитроксильных аддуктов ПММА на один–два порядка выше, чем для аддуктов ПС [25]. Подробно такая особенность была рассмотрена в нашей работе на примере имидазолиновых нитроксилов [25], поэтому здесь мы ограничимся лишь сравнением указанных констант для других известных систем (табл. 3).
Таблица 3.
Сравнение констант элементарных актов для реакций нитроксилов с радикалами роста стирола и ММА
| Нитроксил | Радикал роста | kd × 103 с–1/Т, °С | kX × 10–6 М–1 с–1 | $k_{t}^{*}$ × 10–6 М–1 с–1 |
|---|---|---|---|---|
| ТЕМПО | ММА | 3/93 [36] | 1.6 [36] | 60 |
| Стирол | 0.2/100 [37, 38] | 15 [33] | 30 | |
| SG1 | MMA | 2400/120 [39] | 0.014 [1] | – |
| Стирол | 3.4/120 [40] | 0.26 [27] | – | |
| 11/120 [41] | 0.5 [42] | – | ||
| 0.6 [40] | – |
* Усредненные данные [43].
Используя табличные значения, получаем, что параметр (kt$k_{d}^{2}$/$k_{X}^{2}$)1/3 возрастает при переходе от радикала полистирола к радикалу полиметилметакрилата в 30 и 500 раз для нитроксилов ТЕМПО и SG1 соответственно. Все это должно приводить к увеличению концентрации свободного нитроксила в десятки–сотни раз, что и наблюдается в эксперименте.
ЗАВИСИМОСТЬ СКОРОСТИ ПОЛИМЕРИЗАЦИИ ОТ КОНЦЕНТРАЦИИ ИНИЦИАТОРА
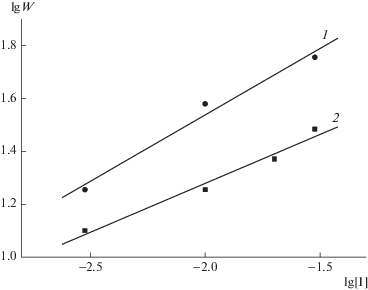
Согласно модели Фишера [34, 35], полимеризация виниловых мономеров на стадии накопления нитроксилов характеризуется порядком скорости реакции n по концентрации алкоксиаминового инициатора 1/3.
Наши данные дают величины n = 0.37 и 0.53 для начальной скорости полимеризации ММА с участием инициаторов А2 и А1 соответственно (рис. 5). В первом случае кинетика полимеризации подчиняется указанному закону. Во втором порядок по инициатору свидетельствует о преимущественном квадратичном обрыве растущих радикалов по сравнению с обратимым. К сожалению, найти в литературе сведения о порядке по инициатору для других случаев полимеризации ММА с участием нитроксилов нам не удалось.
ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА КИНЕТИКУ ПОЛИМЕРИЗАЦИИ ММА С УЧАСТИЕМ НИТРОКСИЛОВ
Из сказанного выше следует, что отсутствие реакции диспропорционирования между радикалами роста ММА и нитроксилами – необходимое, но не достаточное условие протекания процесса в режиме обратимого ингибирования. Кроме этого, свободный нитроксил не должен накапливаться в таких количествах, которые полностью подавляют полимеризацию за счет сдвига равновесия в сторону “спящих” цепей. В настоящее время существует два подхода, чтобы сдвинуть равновесие в сторону диссоциации – сополимеризация с более активным мономером и понижение фоновой концентрации нитроксила.
Первый подход основан на том, что даже небольшое добавление (иногда всего несколько процентов) активного мономера (например, стирола) может направить процесс по механизму обратимого ингибирования за счет того, что преимущественно будут образовываться живые аддукты нитроксила с концевым радикалом активного мономера. Данный способ был впервые описан в работе [44], обоснован в работе [45] и подробно обсуждается в обзоре [11]. Здесь мы остановимся на втором подходе – уменьшение фоновой концентрации нитроксила. Выше упоминалось, что это можно сделать путем понижения температуры полимеризации. Того же результата можно достичь и с использованием специальных добавок. К ним относятся сильные кислоты, полярные соединения и радикальные инициаторы.
Слабые кислоты никакого заметного влияния на полимеризацию с участием нитроксилов не оказывают. Об этом убедительно свидетельствует тот факт, что механизм полимеризации акриловой кислоты не отличается от механизма полимеризации алкилакрилатов [46, 47]. Напротив, сильные органические и минеральные кислоты существенно увеличивают скорость такой полимеризации. Данный подход был впервые применен в 1996 г. на примере использования камфоросульфоновой кислоты в полимеризации ММА под действием ТЕМПО [48]. При добавлении эквивалента кислоты по отношению к нитроксилу выход ПММА повысился с 16 до 40%, значение Mn при этом выросло от 2 × 104 до 4 × 104, но одновременно уширилось ММР (величина Ð изменилась от 1.5 до 2.5). “Ускоряющий” эффект объясняли протонированием нитроксильной группы в полимерном аддукте, что по мнению авторов, облегчало диссоциацию связи С–ОN.
Необычные результаты были получены в работах [49, 50] при полимеризации ММА под действием ТЕМПО в присутствии серной кислоты. При добавлении семикратного избытка кислоты по отношению к алкоксиаминовому инициатору на основе ТЕМПО и 4-оксо-ТЕМПO полимеризация ММА протекала по механизму обратимого ингибирования как при 70, так и при 130°С. Конверсия ММА в обоих случаях достигала 90%, дисперсность ПММА на предельных конверсиях составляла 1.04–1.15. Наконец не только кинетическая зависимость ln([M]0/[M]) = f(t), но и зависимость конверсии от времени полимеризации были линейными вплоть до предельных конверсий. По мнению авторов, роль серной кислоты состоит в том, что она “выводит” свободный ТЕМПО из полимеризата, превращая его в гидроксиламин, тем самым удается избежать диспропорционирования между радикалом роста и нитроксилом:

Такой механизм гидролиза ТЕМПО был установлен экспериментально при кипячении ТЕМПО в серной кислоте [51].
По нашим данным, серная кислота выполняет двоякую функцию: во-первых, она мгновенно “связывает” близкую к эквивалентной часть нитроксила, во-вторых, она продолжает реагировать со свободным ТЕМПО в ходе полимеризации, что способствует существенному уменьшению его концентрации. Добавление серной кислоты в количестве 70 и 100% от исходной концентрации ТЕМПО приводит к понижению его фоновой концентрации при полимеризации ММА на 2 и 2.5 порядка соответственно (рис. 6). Естественно, это вызывает резкое возрастание скорости полимеризации (рис. 7). Однако и в таких случаях полимеризация протекает в затухающем режиме, и предельная конверсия не превышает 60%.
Рис. 6.
Зависимость концентрации ТЕМПО при полимеризации ММА, инициированной ДАК в присутствии H2SO4 при 80°С. 1 – [Т•] = [ДАК] = 0.1, [H2SO4] = = 0.7 моль/л; 2 – [Т•] = [ДАК] = 0.05, [H2SO4] = = 0.035 моль/л; 3 – [Т•] = [ДАК] = 0.05, [H2SO4] = = 0.05 моль/л; 4 – [Т•] = [ДАК] = 0.05 моль/л (без кислоты) ; 5 – [Т•] = [ДАК] = 0.01 моль/л (без кислоты).
Рис. 7.
Интегральные (а) и дифференциальные (б) кинетические кривые полимеризации ММА в системе ТЕМПО–ДАК–H2SO4 при 80°С. [TЕМПО] = [ДАК] = 0.1 (1, 2) и 0.05 моль/л (3), [H2SO4] = 0.7 (1), 0.07 (2) и 0.035 моль/л (3).
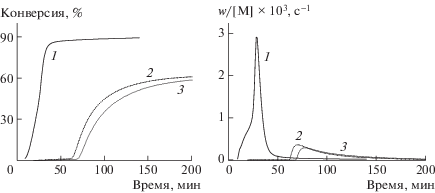
При многократном (семикратном и выше) избытке серной кислоты образуется протонированная форма ТЕМПО в виде красного масла, не совместимого с ММА и не принимающего участия в полимеризации. В данном случае мономер полимеризуется по обычному механизму с типичным ярко-выраженным гель-эффектом (рис. 7, кривая 1).
Следует отметить, что подход с введением небольших добавок серной кислоты позволил нам осуществить контролируемую сополимеризацию ММА с политиленгикольметакрилатом. Как видно на рис. 8а, скорость сополимеризации и выход сополимера увеличивается с ростом содержания кислоты. Причиной этого, как и в гомополимеризации ММА, служит понижение концентрации свободного ингибитора, на фоне которого идет полимеризация (рис. 8б). Несмотря на то, что сополимеризация протекает в затухающем режиме, молекулярная масса сополимеров линейно увеличивается с конверсией при низком значении коэффициента полидисперсности (табл. 4).
Рис. 8.
Кинетические кривые (а) и изменение концентрации ТЕМПО (б) при сополимеризации ММА–ПЭГМА (10 мас. %) под действием системы ДАК–ТЕМПО–H2SO4 при 90°С. [ТЕМПО] = [ДАК] = 0.1 моль/л, [H2SO4] = = 0.07 (1) и 0.035 моль/л (2).

Таблица 4.
Выход и молекулярно-массовые характеристики сополимеров ПММА–ПЭГМА ([ДАК] = [ТЕМПО] = = 0.05 моль/л, [H2SO4] = 0.035 моль/л, Т = 80°С)
| Доля ПЭГМА, мас. % | Время мин | Конверсия, % | Mn ×10–3 | Ð |
|---|---|---|---|---|
| 10 | 105 | 22 | 33.7 | 1.2 |
| 125 | 40 | 44.6 | 1.3 | |
| 150 | 59 | 52.4 | 1.4 | |
| 250 | 88 | 67.7 | 1.4 | |
| 30 | 115 | 11 | 28.5 | 1.2 |
| 122 | 22 | 41.7 | 1.4 | |
| 250 | 53 | 71.4 | 2.5 |
Отметим, что на полимеризацию ММА с участием N2 данная добавка влияет отрицательно. Полимеризация протекает в течение первых нескольких минут, достигая конверсии 8–10%, а далее тормозится.
Среди высокополярных соединений наиболее эффективно в полимеризации ММА с участием нитроксилов применяют малононитрил [52] и трифторуксусный ангидрид (ТФА) [53]. Природа их действия окончательно не ясна. Интересно, что уксусный ангидрид, широко использующийся для повышения скорости полимеризации стирола с участием ТЕМПО [54–56], оказался неэффективным в полимеризации ММА. Он почти не влияет на концентрацию нитроксила при полимеризации ММА с участием А1 (табл. 5). Напротив, ТФА позволил получить узкодисперсный и высокомолекулярный ПММА в присутствии ТЕМПО [53]. При эквивалентном добавлении ТФА выход ПММА повышается до 50%, а дисперность понижается до 1.17; при 2.5-кратном избытке ТФА эти значения составляют 96% и 1.14 соответственно. Однако процесс нельзя назвать контролируемым, так как Mn линейно растет лишь до конверсии 60% и значения ММ на порядок выше теоретических.
Таблица 5.
Выход и молекулярно-массовые характеристики ПММА, полученного с участием 0.01 моль/л аддукта A1 при 80°С
| Добавка | Концентрация добавки, моль/л | Время, мин | Конверсия, % | Mn × 10–3 | Ð |
|---|---|---|---|---|---|
| – | − | 500 | 7 | 5.5 | 1.6 |
| УА | 0.010 | 500 | 6 | 3.5 | 1.9 |
| ТФА | 0.010 | 500 | 20 | 7.4 | 1.6 |
| ДАК | 0.005 | 500 | 36 | 18.7 | 2.6 |
| ДАК | 0.010 | 500 | 54 | 16.2 | 3.0 |
По нашим данным, влияние ТФА на полимеризацию ММА с участием А1 менее выражено. При добавлении от эквивалента до пятикратного избытка ТФА концентрация нитроксила понижается на порядок (рис. 9). Это повышает выход полимера до 20% (рис. 10). ММ полимера увеличивается не столь заметно (табл. 5). ТФА практически “не работает” в случае имидазолиновых нитроксилов: степень конверсии почти не меняется (рис. 11а), и концентрация нитроксила остается на том же уровне (рис. 11б). Значения ММ сопоставимы с результатами, полученными при полимеризации без добавления ТФА (табл. 6). Причину этого объяснить пока не удается.
Рис. 9.
Зависимость концентрации нитроксильных радикалов от времени при полимеризации ММА в массе под действием 0.01 М аддукта A1 при 80°С без добавок (1) и при использовании 0.01 моль/л ТФА (2) и ДАК (3).

Рис. 10.
Зависимости конверсии от времени при полимеризации ММА в массе под действием 0.01 М аддукта A1 при 80°С без добавок (1) и при использовании 0.01 (2), 0.05 моль/л ТФА (3), а также 0.005 (4) и 0.01 моль/л ДАК (5).
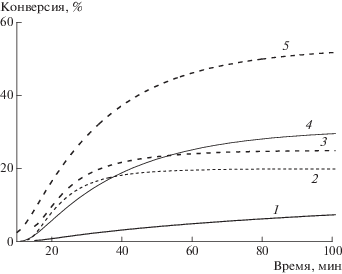
Рис. 11.
Зависимости конверсии (а) и концентрации нитроксила (б) при полимеризации ММА в массе под действием 0.01 М аддукта A2 при 80°С без добавок (1) и при использовании 0.01 моль/л ТФА (2) и ДАК (3).

Таблица 6.
Молекулярно-массовые характеристики ПММА, полученного полимеризацией ММА в массе в присутствии А2 и добавок ТФУ и ДАК (концентрация добавки 0.01 моль/л , Т = 80°С)
| Добавка | Время, мин | Конверсия, % | Mn × 10–3 | Ð |
|---|---|---|---|---|
| ТФА | 5.5 | 5 | 6.8 | 1.5 |
| 12 | 8 | 7.3 | 1.6 | |
| 20 | 10 | 7.7 | 1.8 | |
| 30 | 15 | 12.2 | 1.5 | |
| 500 | 33 | 12.4 | 2.1 | |
| ДАК | 9 | 20 | 8.4 | 2.6 |
| 17 | 27 | 10.5 | 2.3 | |
| 29 | 32 | 20.4 | 1.5 | |
| 64 | 45 | 27.8 | 1.6 | |
| 500 | 86 | 40.8 | 1.8 |
Подробно было исследовано влияние полярных добавок на полимеризацию ММА в присутствии ароматического п-диметоксидифенилнитроксила [57]. В ряду диэтилмалонат < ацетилацетон < малононитрил скорость полимеризации возрастает на порядок. Она увеличивается пропорционально количеству добавки в интервале от эквивалента до трехкратного (по отношению к инициатору) избытка, затем перестает меняться. Во всех системах наблюдается линейный рост Mn полимера до конверсии 70%, а дисперсность составляет 1.3–1.5. Интересно, что действие малононитрила основано не на понижении фоновой концентрации свободного нитроксила при полимеризации, а на повышении вдвое константы скорости реинициирования. Заметим, что камфоросульфокислота на полимеризацию ММА под действием данного нитроксила заметного действия не оказывает.
Для ускорения полимеризации ММА с участием нитроксилов пробовали использовать соли металлов, такие как ZnCl2 или сульфат железа-аммония, но полимеризация оставалась неконтролируемой [50].
Увеличить скорость нитроксильной полимеризации ММА и перевести процесс в режим обратимого ингибирования можно добавлением высокотемпературного инициатора. Последний, поставляя дополнительные активные радикалы в систему, “связывает” свободный нитроксил и тем самым сдвигает равновесие в сторону диссоциации. Термин “высокотемпературный инициатор”, в данном случае – применительно к ДАК – не совсем удачен. Он подразумевает, что скорость дополнительного инициирования с его помощью много меньше скорости реинициирования. Для полимеризации ММА инициированной А1 или А2, таким инициатором может служить ДАК, период полураспада которого на порядок больше, чем у названных алкоксиаминов.
Для получения заметного эффекта на полимеризацию ММА, инициированную алкоксиамином А1, необходимо добавить ДАК в мольном отношении (0.5–1.0) : 1 по отношению к А1. Это позволяет более чем на порядок понизить концентрацию свободного нитроксила, и соответственно увеличить степень конверсии более чем в 10 раз (рис. 9, 10). Чем больше инициатора в системе, тем лучше идет полимеризация. Сравнение с системой без добавки показывает, что Mn полимера увеличивается почти в 2 раза, однако дисперсность также растет (табл. 5). Последнее, очевидно, связано с постоянным поступлением в систему дополнительных радикалов, инициирующих зарождение новых цепей.
Те же закономерности характерны и для полимеризации ММА с участием А2. ДАК существенно понижает концентрацию нитроксила, что позволяет довести конверсию мономера до 86% (рис. 11), т.е. до предельной величины. Молекулярная масса полимера повышается в ходе полимеризации в 5 раз (табл. 6).
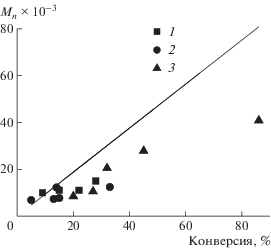
Важно, что зависимость молекулярной массы ПММА от конверсии не меняется при добавлении ТФА и ДАК. Хотя экспериментальные точки и лежат ниже теоретической прямой, но они близки к тем, что наблюдаются в отсутствие добавок при тех же степенях превращения (рис. 12). Это означает что добавки не влияют на механизм роста ММ макромолекул, а лишь продлевают данный процесс до бо́льших конверсий. Таким образом, добавление ДАК и ТФА повышает скорость полимеризации ММА, однако “затухание” до конца не предотвращает.
Рис. 12.
Зависимость Mn полимера от конверсии при полимеризации ММА с участием А2 без добавок (1) и при использовании ТФА (2) и ДАК (3). Прямая – теоретическая зависимость. Условия синтеза приведены в табл. 6.
К разряду процессов “с добавками высокотемпературных инициаторов” можно отнести и полимеризацию ММА под действием DPAIO. Спецификой этого нитроксила является то, что в ходе полимеризации происходит его частичная деструкция с образованием активного О-центрированного радикала [4, 58, 59].
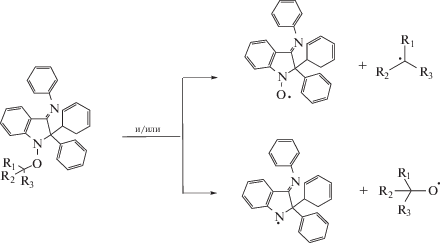
Иными словами, сам нитроксил играет роль высокотемпературного инициатора. В результате реализуются два положительных эффекта: во-первых, понижается концентрация свободного ингибитора, во-вторых, поставляются новые активные радикалы роста, что препятствует затуханию полимеризации ММА. Отрицательным эффектом такой “деструкции”, естественно, становится уширение ММР продукта. В типичных условиях полимеризация ММА с участием DPAIO при 130°С протекает до конверсии 60%. При этом Mn полимера линейно растет, достигая значений (40–70) × 103, а дисперсность ПММА составляет 1.4–2.4.
Отметим, что все описанные выше закономерности и эффекты относятся к полимеризации ММА с участием низкомолекулярных нитроксилов. Кроме них для осуществления контроля процесса предлагали использовать и макронитроксилы, которые получаются in situ из спиновых ловушек (нитронов) [60–63]. В результате удалось понизить температуру полимеризации до 40–50°С, повысить выход ПММА до 80%; наблюдался линейный рост Mn с конверсией [60–62]. Однако ММ полимера была более, чем на порядок выше, чем предсказывает теория, а ширина ММР того же порядка, что и при использовании низкомолекулярных нитроксилов. К сожалению, нам не удалось найти сведений об изменении концентрации нитроксилов, наличии диспропорционирования и значении констант элементарных актов в случае полимеризации ММА с участием макронитроксилов.
ЗАКЛЮЧЕНИЕ
В заключение приведем примеры успешной контролируемой полимеризации ММА с участием нитроксилов за последние 5 лет, т.е. с момента опубликования последнего обзора [11].
Впервые для этих целей стали применять ароматические нитроксилы [57]. Как уже отмечалось выше, полимеризация в их присутствии, заканчивается на начальных конверсиях, как и для большинства нитроксилов. Однако применение различных “ускоряющих” добавок позволило осуществить контролируемую полимеризацию ММА до конверсии 70% и синтезировать ПММА с молекулярной массой до 60 × 103 и дисперсностью ~1.3–1.4.
Продолжается поиск новых нитроксилов, которые бы давали возможность осуществить одновременно контролируемую полимеризацию ММА и других мономеров. Сообщается о получении блок-сополимеров ПС–блок–полибутилметакрилат и ПММА–блок–ПС под действием Dispolreg-007 [32]. Однако значение Ð ~ 2 для сополимеров явно выше, чем в контролируемой полимеризации. Статистические сополимеры ММА с бутилакрилатом, полученные с тем же нитроксилом, характеризуются высоким выходом (более 80%), ММ, совпадающей с рассчитанной по закону живых цепей, и Ð ~ 1.5–1.6 [10].
Одним из эффективных рычагов для улучшения контроля в полимеризации ММА с участием нитроксилов служит понижение температуры реакции, которое позволяет минимизировать вклад побочных реакций. В этой связи наиболее интересны работы по фотополимеризации. Так, синтезирован фотоактивный алкоксиамин на основе хинолин-производного ТЕМПО, который позволяет осуществить полимеризацию ММА при комнатной температуре. Хотя с точки зрения контроля ММ этот нитроксил не оправдал ожиданий, но он оказался эффективным в стерео-контроле макромолекул: в его присутствии доля синдио-триад в ПММА повышается с 46 до 60% [64].
Наконец, продолжает развиваться и “сополимеризационное направление”. Традиционно в роли активного сомономера для улучшения контроля полимеризации ММА используют стирол. Недавно с этой целью успешно применен 4-винилпиридин и целый ряд галогензамещенных стиролов [65, 66]. Небольшого количества (5–11%) названных сомономеров оказывается достаточно, чтобы перевести неконтролируемую полимеризацию ММА с участием SG1 в режим обратимого ингибирования. Данный подход остается основным для дизайна сополимеров различного строения на основе ММА c помощью полимеризации под действием SG1 [67–71].
Суммируя все накопленные данные по полимеризации ММА с участием нитроксилов, можно сделать некоторые выводы, полезные с практической точки зрения. “Метод добавок” на сегодняшний момент представляется основным способом осуществления контролируемого синтеза ПММА и его сополимеров с помощью полимеризации с участием нитроксилов.
Продолжающиеся попытки поиска новых нитроксилов, не способных к диспропорционированию с радикалами роста ММА, могут быть безрезультатными, поскольку отсутствие диспропорционирования – не единственная причина нарушения “живого” механизма полимеризации. На наш взгляд, основные усилия надо направлять на поиск таких агентов контролируемого синтеза, которые позволяли бы осуществлять полимеризацию ММА при низких, близких к комнатной, температурах. Именно в этих условиях не только понижается вклад диспропорционирования, но и минимизируется эффект Фишера.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-03-00707).
Список литературы
Guillaneuf Y., Gigmes D., Marque S.R.A., Tordo P., Bertin D. // Macromol. Chem. Phys. 2006. V. 207. P. 1278
Edeleva M., Marque S.R.A., Kabytaev K., Guillaneuf Y., Gigmes D., Bagryanskaya E. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 1323.
Еделева М.В. // Дис. … д-ра хим. наук. Новосибирск: Новосибирский ин-т органич. химии Сибирского отделения РАН, 2019.
Guillaneuf Y., Gigmes D., Marque S.R.A., Astolfi P., Greci L., Tordo P., Bertin D. // Macromolecules. 2007. V. 40. № 9. P. 3108.
Greene A.C., Grubbs R.B. // Macromolecules. 2010. V. 43. № 24. P. 10320.
Grignard B., Phan T., Bertin D., Gigmes D., Jerome C., Detrembleur C. // Polym. Chem. 2010. № 1. P. 837.
Yoshida E. // Colloid Polym. Sci. 2011. V. 289. P. 1625.
Fukuda T., Terauchi T., Goto A., Ohno K., Tsujii Y., Miyamoto T. // Macromolecules. 1996. V. 29. № 20. P. 6393.
Schmidt-Naake G., Stenzel M. // Angew. Makromol. Chem. 1998. B. 254. № 4414. S. 55.
Simula A., Ruipérez F., Ballard N., Leiza J.R., van Esa S., Asua J.M. // Polym. Chem. 2019. V. 10. P. 106.
Guegain E., Guillaneuf Y., Nicolas J. // Macromol. Rapid Commun. 2015. V. 36. № 13. P. 1227.
Смирнов Б.Р. // Тез. докл. Всесоюз. конф. “Радикальная полимеризация”. Горький, 1989. Р. 230.
Moad G., Anderson A.G., Ercole F., Johnson H.J., Krstina J., Moad C.L., Rizzardo E., Spurling T.H., Thang S.H. // ACS Symp. Ser. 1998. V. 685. P. 332.
Burguiere C., Dourges M.A., Charleux B., Vairon J.P. // Macromolecules. 1999. V. 32. P. 3883.
Souaille M., Fischer H. // Macromolecules. 2001. V. 34. P. 2830.
Parkhomenko D., Bagryanskaya E.G., Marque S.R.A., Siri D. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 13862.
Edeleva M., Marque S.R.A., Bertin D., Gigmes D., Guillaneuf Y., Morozov S. V., Bagryanskaya E.G. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 6828.
Edeleva M.V., Kirilyuk I.A., Zubenko D.P., Zhurko I.F., Marque S.R., Gigmes D., Guillaneuf Y., Bagryanskaya E.G. // J. Polym. Sci., Polym.Chem. 2009. V. 47. P. 6579.
Li I., Howell B.A., Matyjaszewski K., Shigemoto T., Smith P.B., Priddy D.B. // Macromolecules. 1995. V. 28. P. 6692.
Ohno K., Tsujii Y., Fukuda T. // Macromolecules. 1997. V. 30. P. 2503.
Grachev V.P., Golubev V.A., Korolev G.V. // Polymer Science A. 2005. V. 47. № 7. P. 662.
Edeleva M., Audran G., Marque S., Bagryanskaya E. // Materials. 2019. V. 12. P. 688.
Bagryanskaya E.G., Marque S.R.A. // Chem. Rev. 2014. V. 114. P. 5011.
Edeleva M.V., Parkhomenko D.A., Morozov D.A., Dobrynin S A., Trofimov D.G., Kanagatov B., Kirilyuk I.A., Bagryanskaya E.G. // J. Polym. Sci., Polym. Chem. 2014. V. 52. P. 929.
Zaremski M.Yu., Odintsova V.V. // Polymer Science B. 2020. V. 62. № 1. P. 1.
Ananchenko G.S., Fischer H.J. // Polym. Sci., Polym. Chem. 2001. V. 39. P. 3604.
Ananchenko G.S., Souaille M., Fischer H., Le Mercier C., Tordo P. // J. Polym. Sci., Polym. Chem. 2002. V. 40. P. 3264.
Dire C., Belleney J.L., Nicolas J., Bertin D., Magnet S., Charleux B. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 6333.
McHale R., Aldabbagh F., Zetterlund P.B. // J. Polym. Sci., Polym. Chem. 2007. V. 45. P. 2194.
Gryn'ova G., Lin C.Y., Coote M.L. // Polym. Chem. 2013. V. 4. P. 3744.
Ballard N., Aguirre A.M., Simula A., Agirre M., Leiza J.R., Asua J.M., van Es S. // ACS Macro Lett. 2016. V. 5. P. 1019.
Simula A., Aguirre M., Ballard N., Veloso A., Leiza J.R., van Es S., Asua J.M. // Polym. Chem. 2017. V. 8. P. 1728.
Zaremski M.Yu. // Polymer Science C. 2015. V. 57. № 1. P. 65.
Fischer H. // Macromolecules. 1997. V. 30. № 19. P. 5666.
Fischer H. // Chem. Rev. 2001. V. 101. № 12. P. 3581.
Zaremski M.Yu., Zhaksylykov A.B., Orlova A.P., Garina E.S., Badun G.A., Lachinov M.B., Golubev V.B. // Polymer Science A 2005. V. 47. № 6. P. 526.
Zaremski M.Yu., Chen Xin, Orlova A.P., Golubev V.B., Kurochkin S.A., Grachev V.P. // Polymer Science B. 2010. V. 52. № 9–10. P. 528.
Bon S.A.F., Chambard G., German A.L. // Macromolecules. 1999. V. 32. № 25. P. 8269.
Fischer H. // ACS Symp. Ser. 2003. V. 854. P. 10.
Benoit D., Grimaldi S., Robin S., Finet J.-P., Tordo P., Gnanou Y. // J. Am. Chem. Soc. 2000. V. 122. № 25. P. 5929.
Goto A., Tsujii Y., Fukuda T. // Chem. Lett. 2000. P. 788.
Borisova O.V., Zaremskii M.Yu., Golubev V.B., Plutalova A.V., Borisov O.V., Billon L. // Polymer Science B. 2013. V. 55. № 9–10. P. 508.
Brandrup E., Immergut H., Grulke E.A. // Polymer Handbook. New York: Wiley, 1999.
Zaremski M.Yu., Plutalova A.V., Lachinov M.B., Golubev V.B. // Macromolecules. 2000. V. 33. P. 4365.
Charleux B., Nicolas J., Guerret O. // Macromolecules. 2005. V. 38. P. 5485.
Couvreur L., Lefay C., Belleney J., Charleux B., Guerret O., Magnet S. // Macromolecules. 2003. V. 36. № 22. P. 8260.
Borisova O., Billon L., Zaremski M., Grassl B., Bakaeva Z., Lapp A., Stepanek P., Borisov O. // Soft Matter. 2012. V. 8. P. 3649.
Steenbock M., Klapper M., Pinhai N., Hubrich M. // Acta Polymerica. 1996. V. 47. P. 276.
Ansong O.E., Jansen S., Wei Y., Pomrink G., Li S., Patel A. // Polym. Int. 2008. V. 57. P. 863.
Ansong O.E., Jansen S., Wei Y., Pomrink G., Lu H., Patel A., Li S. // Polym. Int. 2009. V. 58. P. 54.
Ma Y., Loyns C., Price P., Chechik V. // Org. Biomol. Chem. 2011. V. 9. № 15. P. 5573.
Jianying H., Jian L., Minghua L., Lizong D., Yousi Z. // J. Polym. Sci., Polym. Chem. 2005. V. 43. P. 5246.
Shuying Z., Jian L., Wanli L., Mingfa Y., Jianying H., Yousi Z. // J. Xiamen University (Natural Science) 2002. V. 41. № 4. P. 468.
Malmstrom E., Miller R.D., Hawker C.J. // Tetrahedron. 1997. V. 53. № 45. P. 15225.
Goto A., Tsuji Y., Fukuda T. // Chem. Lett. 2000. № 8. P. 788.
Butz S., Baethge H., Schmidt-Naake G. // Angew. Makromol. Chem. 1999. B. 270. № 4685. S. 42.
Zhu Z., Shan G., Pan P. // RSC Adv. 2016. V. 6. P. 97995.
Astolfi P., Greci L., Stipa P., Rizzoli C., Ysacco C., Rollet M., Autissier L., Tardy A., Guillaneuf Y., Gigmes D. // Polym. Chem. 2013. V. 4. P. 3694.
Gigmes D., Gaudel-Sirib A., Marque S.R.A., Bertin D., Tordo P., Astolfi P., Greci L., Rizzoli C. // Helv. Chim. Acta. 2006. V. 89. P. 2312.
Гришин Д.Ф., Семенычева Л.Л., Колякина Е.В. // Докл. РАН. 1998. Т. 362. № 5. С. 634.
Kolyakina E.V., Grishin D.F., Semenycheva L.L., Sazonova E.V. // Polymer Science B. 2004. V. 46. № 1–2. P. 10.
Grishin D.F., Semenycheva L.L., Kolyakina E.V. // Rus. J. Appl. Chem. 2001. V. 74. № 3. P. 494.
Zang L., Wong E.H.H., Barner-Kowollik C., Junkers T. // Polymer. 2010. V. 51. № 17. P. 3821.
Su J., Liu X., Li M., Zhang T., Cui Y. // Int. J. Polym. Sci. 2016. Article ID 6482050.
Qiao X.G., Zhoub Z., Pangc X.C., Lansalota M., Bourgeat-Lamia E. // Polymer. 2019. V. 172. P. 330.
Schmidt A.C., Turgut H., Le D., Beloqui A., Delaittre G. // Polym. Chem. 2020. V. 11. P. 593.
Maupu A., Kanawati Y., Métafiot A., Maric M. // Materials. 2019. V. 12. P. 1547.
Guégain E., Michel J.-P., Boissenot T., Nicolas J. // Macromolecules. 2018. V. 51. P. 724.
Cazotti J.C., Fritz A.T., Garcia-Valdez O., Smeets N.M.B., Dubé M.A., Cunningham M.F. // Carbohydr. Polymers. 2020. V. 228. P. 115384.
Wylie K. // MsD Thes. Montréal: McGill Univ., 2016.
Cherifi N., Khoukh A., Benaboura A., Billon L. // Polym. Chem. 2016. V. 7. P. 5249.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)