

Высокомолекулярные соединения (серия С), 2021, T. 63, № 1, стр. 54-94
РЕАКТОПЛАСТЫ НА ОСНОВЕ БИС-ФТАЛОНИТРИЛОВ В КАЧЕСТВЕ ТЕРМОСТОЙКИХ МАТРИЦ ДЛЯ ПОЛИМЕРНЫХ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ
Б. А. Булгаков a, *, О. С. Морозов a, И. А. Тимошкин b, А. В. Бабкин a, А. В. Кепман a, b
a Московский государственный университет им. М В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b Акционерное общество “Институт новых материалов и технологий”
119991 Москва, Ленинские горы, 1, стр. 11, Россия
* E-mail: bbulgakov@gmail.com
Поступила в редакцию 26.08.2020
После доработки 15.10.2020
Принята к публикации 14.01.2021
Аннотация
Обзор посвящен описанию процесса создания термостойких матриц для полимерных композиционных материалов путем полимеризации связующих, состоящих из фталонитрильных мономеров с сомономерами, и особенностям технологий получения полимерных композиционных материалов. Рассмотрены закономерности протекания полимеризации в присутствии инициаторов различного типа и температурные режимы отверждения. Обсуждаются методы получения и механические свойства полимерных композиционных материалов на основе фталонитрильных связующих с различными армирующими наполнителями. Сопоставляются свойства стеклопластиков и углепластиков, полученных отверждением различных олигомеров фталонитрильного типа.
Полимерные композиционные материалы (ПКМ) находят все большее распространение в самых различных областях современной техники. Так, замена металлических частей изделий военной и аэрокосмической промышленности на ПКМ позволяет существенно снизить их вес при высоких удельных показателях прочности. ПКМ широко используется в современном самолетостроении [1]. Например, Boeing 787 Dreamliner на 50% состоит из композитных материалов. Для дальнейшего увеличения доли ПКМ необходимо производить замену металлических деталей в двигателях, что затруднено низкой термостабильностью полимеров. Исходя из этого актуальной является задача по получению новых полимеров, пригодных для высокотемпературных применений.
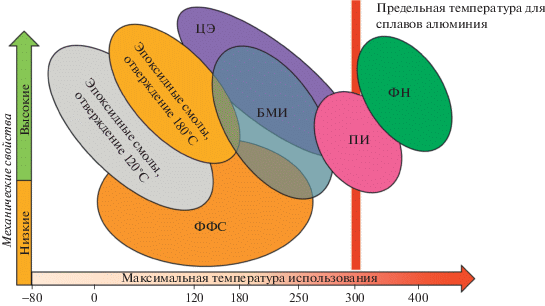
На данный момент наибольшее распространение имеют матрицы для ПКМ на основе фенолформальдегидных [2–4] и эпоксидных [5–7] смол, циановых эфиров [8–10], бисмалеимидов [11–13], полиимидов [14–17], полиуретанов [18, 19]. В качестве полимерных матриц для длительного использования при высоких температурах в большинстве случаев применяются полиимиды, но и они имеют температуру стеклования около 300°С [20]. В настоящее время активно ведется поиск новых полимеров для эксплуатации при температурах выше 350°С. Исследования фталонитрильных связующих показали, что данный материал является перспективным для применения при высокой температуре [21–26]. Обладая исключительно высокой термостойкостью и хорошими механическими свойствами, фталонитрильные связующие уже сейчас могут заменить смолы, используемые в военной и аэрокосмической областях. Примерное соотношение прочностных свойств и максимальной температуры эксплуатации приведены на рис. 1.
Рис. 1.
Сравнение свойств фталонитрильных ПКМ с другими связующими. БМИ – бисмалеимиды, ЦЭ – циановые эфиры, ПИ – полиимиды, ФН– фталонитрилы. Цветные рисунки можно посмотреть в электронной версии.
Термин “фталонитрилы” впервые был введен в 1896 г. Он относился к органическим структурам, содержащим бензольное кольцо, замещенное двумя нитрильными группами в соседних положениях. Началом изучения, а в дальнейшем и применения фталонитрильных смол по праву можно считать 1934 г., когда C.E. Dent и R.P. Linstead [27] обнаружили, что фталоцианины сублимируются без разложения при 560°С. Это открытие послужило толчком к исследованиям в области синтеза термостабильных полимеров, имеющих фрагмент фталоцианина

Фталонитрилы полимеризуются при нагревании с образованием трехмерной сшитой структуры. Полимеры могут быть получены из молекул, содержащих не менее двух фталонитрильных фрагментов. Основоположником тематики бис-фталонитрильных связующих для применения в ПКМ является Тедди Келлер [28]. Первым методом синтеза бис-фталонитрильных мономеров стала прямая реакция между 4-аминофталонитрилом и алифатическими дихлорангидридами:

Таким способом была получена целая серия молекул с алифатическим мостиком от 4 до 18 атомов углерода. Авторы отметили снижение температуры плавления мономеров с ростом длины мостика. Однако после 12–13 гомолога температура плавления выходит на плато T ~ 170°С, после чего дальнейшего падения не наблюдается.
При этом отвержденные бис-фталонитрильные смолы характеризовались значительно меньшей термооксилительной стабильностью, чем фталоцианины. Следовательно, именно мостик, соединяющий фталонитрильные фрагменты, подвержен окислению в первую очередь, а его строение влияет на свойства и характер отверждения бис-фталонитрильных связующих.
Термореактивные полимеры, полученные из бис-фталонитрилов, начали активно разрабатываться в 1980-х годах в лаборатории ВМС США с целью получения термостойких негорючих материалов для использования в конструкции подводных лодок [21, 29]. Первой высокотемпературной смолой на основе фталонитрила являлся 4,4'-бис-(3,4-дицианофенокси)бифенил [30]

Полимеризацию данного мономера осуществляли в присутствии солей металлов. Полученные реактопласты имели достаточно высокую ММ и обладали трехмерной сшитой структурой. Однако, полимеризация мономера была затруднена стерически, а высокая температура переработки (выше 250°C) затрудняла процесс синтеза ПКМ.
Другим методом получения мономеров стал синтез молекул на основе диэфирной структуры, что значительно упростило и удешевило процесс производства смол. В основе метода получения фталонитрильных связующих лежит реакция 4-нитрофталонитрила с фенольными производными по механизму нуклеофильного ароматического замещения. Этот способ был предложен T.M. Keller с сотрудниками в 1980 г. [23]. Синтез, как правило, проводят в полярных апротонных растворителях, таких как ДМАА [31, 32], ДМФА [33, 34], ДМСО [35, 36] или N-метилпирролидон (МП) [37, 38] в присутствии основания, обычно K2CO3. Ниже приведена реакция получения бис-фталонитрильного мономера с диэфирной структурой.

В большинстве случаев реакции протекают с количественным выходом [23, 25].
Кроме мономера 1 в данных условия была получена целая серия бис-фталонитрильных мономеров с различными ароматическими мостиками [39]:

Их отверждение проводили без добавления инициаторов полимеризации при 280°С в течение 6 дней. Термостабильность полимеров, по данным ТГА (рисунок), превосходила все известные на тот момент смолы. Температура потери 5% массы (T5%) для различных соединений составила 400–500°С. Отличительной особенностью фталонитрильных матриц также стала высокая температура стеклования (Tс > 400°С).
Рис. 2.
Термогравиметрический анализ первых фталонитрильных мономеров. Номера кривых соответствуют номерам мономеров.

Позже было показано, что применение ароматических диаминов позволяет значительно сократить время полимеризации [23]. Для полученных смол были проведены эксперименты по старению в течение 100 ч при разной температуре. Сравнение результатов проводили с коммерческой смолой PMR-15 на основе полиимидов, в настоящее время используемой в аэрокосмических композиционных материалах (табл. 1). Как видно из данных таблицы, потеря массы фталонитрильных связующих оказалась меньше, чем у коммерчески доступного полиимидного связующего, особенно при Т = 315°С.
Таблица 1.
Потери массы для различных фталонитрильных смол после 100 ч нагревания
| Мономер | Потеря массы, г/м2 | |
|---|---|---|
| 315°С | 343°С | |
| 1 | 0.002 | 0.019 |
| 3 | 0.003 | 0.016 |
| 5 | 0.002 | 0.007 |
| PMR-15 | 0.014 | 0.027 |
Рис. 3.
Зависимость вязкости расплавов фталонитрильных форполимеров от типа используемых диаминов в качестве отвердителей при температуре 260°С на примере 4,4'-бис-(3,4-дицианофенокси)бифенила [47]. 1 – п-АФБ, 2 – м-АФБ, 3 – п-БАФС, 4 – м-БАФС.

Основным недостатком указанных бис-фталонитрилов является высокая температура плавления мономеров (около 200°С), в то время как самопроизвольная полимеризация протекает крайне медленно, а добавление инициатора приводит к быстрому отверждению смол уже при ~220°С, что оставляет крайне узкое технологическое окно для формования ПКМ.
ПОЛИМЕРИЗАЦИЯ ФТАЛОНИТРИЛЬНЫХ СМОЛ
Самопроизвольная полимеризация фталонитрилов в большинстве случаев не происходит, либо протекает крайне медленно: заметное изменение вязкости наблюдается лишь спустя несколько дней перемешивания расплавленных мономеров при 260°С [40]. В связи с этим для отверждения фталонитрильных смол используют различные инициаторы полимеризации.
В ранних опытах по полимеризации фталонитрильных мономеров отвердителями служили соли металлов (магния, меди, цинка, железа, кобальта, никеля, палладия и платины [21–23]), наибольшую активность проявляет хлорид олова [41]. Полимер, полученный в его присутствии, содержал также меньше пор по сравнению со смолами, отвержденными с помощью других солей металлов. В таких случаях образовывалась сшитая полимерная сетка, состоящая из фталоцианиновых циклических структур [42]. Из-за низкой диспергируемости металла или его соли в системе, в которой происходит полимеризация, нельзя достичь полной конверсии фталонитрильных связующих во фталоцианиновый полимер. Данную проблему пытались решить, используя в качестве отвердителей совместимые с мономером органические восстановители, такие как гидрохинон [43]. Оказалось, что основная часть гидрохинона остается не включенной в структуру полимера.
В работе [44] в качестве отверждающего агента применяли пентакарбонил железа (Fe(CO)5). Форполимер представлял собой комплекс фталоцианина с железом, который при дальнейшем нагревании превращался в полимер с высокой термической стабильностью и магнитными свойствами.
Помимо относительно малой скорости полимеризации использование солей металлов требовало строгих стехиометрических соотношений. В случае недостатка ионов металла полимеризация протекала не полностью, а в случае избытка металл мог атаковать бензольное кольцо при температурах выше 280°С [26]. Также существовала проблема неоднородного диспергирования катализатора, поскольку соли металлов образуют с расплавом мономера гетерогенную систему, требующую длительного перемешивания.
В дальнейшем спирты и фенолы рассматривались как отвердители для фталонитрильных смол. Гидроксильные группы этих соединений могут атаковать нитрильные группы мономера с образованием изоиндолиновой структуры, которая в дальнейшем реагирует с другими циано-группами с образованием полизиоиндолиновой структуры:

Фенолы оказались предпочтительнее алифатических спиртов ввиду более высокой активности гидроксильных групп. Для получения полимера без пор температура кипения отвердителя должна быть хотя бы на 50–70°C выше температуры первого шага отверждения. В работе [28] было установлено, что добавка 10 мол. % бисфенола А (Ткип = 360°С) к фталонитрильному мономеру на его основе значительно уменьшает время и температуру полимеризации. Данные ИК-спектроскопии показали, что, нагревание смолы без отвердителя при T = 300°С приводит к снижению интенсивности сигнала цианогрупп на 50% за 27 ч. С добавкой бисфенола А полимеризация половины цианогрупп была достигнута за 7 ч при T = = 210°С. Был предложен механизм полимеризации фталонитрилов, включающий триазиновые и фталоцианиновые структуры:

Высокой скорости полимеризации бис-фталонитрилов удается добиться, применяя новолачную фенолформальдегидную смолу (НС) в качестве сомономера [45]. Огнестойкий материал, не содержащий пор, был получен из смеси, содержащей от 5 до 25 мас. % бис-фталонитрила, после ее выдержки при 200°C в течение 1 ч, затем нагревания до 220°C и выдержке при этой температуре 3 ч. Однако указанные материалы обладают довольно низкой для фталонитрильных связующих температурой стеклования 120–200°C.
Следующим шагом стало исследование возможности инициации полимеризации ароматическими аминами. Механизм полимеризации в данном случае очень схож с механизмом полимеризации с помощью фенолов: аминогруппа атакует цианогруппы мономера, образуя при этом изоиндолиновый цикл, который далее может атаковать цианогруппы других мономеров [29]. Кроме значительного снижения времени и температуры полимеризации, в полученных реактопластах практически не наблюдалось пор, и они обладали более высокими механическими свойствами [30].
T.M. Keller в 1980 г. впервые использовал ароматические диамины для инициирования реакции полимеризации фталонитрилов [46]. В их присутствии повышается скорость реакции, а также уменьшается температура отверждения [47]. Используемые диамины должны обладать высокой термической стабильностью и низкой летучестью при высоких температурах, так как увеличение их летучести приводит к уменьшению скорости реакции и способствует образованию пор в полимерной матрице. Данным условиям удовлетворяют высокомолекулярные ароматические диамины [47].
В качестве инициаторов полимеризации фталонитрилов наибольшее распространение получили коммерчески доступные высококипящие ароматические диамины (табл. 2). Особенное место среди ароматических диаминов занял 1,3-бис-(4-аминофенокси)бензол (м-АФБ), термически стабильный до 250°С [31]. Позже в качестве отвердителей были предложены амины бис-[4-(3-аминофенокси) фенил] сульфон (м-БАФС) и бис-[4-(4-аминофенокси) фенил] сульфон (п-БАФС) [48] на основе ароматических сульфонов, термостойкость которых оказалась еще выше, чем у м-АФБ. Динамическая вязкость расплавов фталонитрильных форполимеров зависит как от реакционной способности и концентрации амина, так и от температуры расплава. Ароматические диамины, содержащие сульфоновую группу, имеют низкую летучесть при высоких температурах по сравнению с другими ароматическими диаминами. Также из-за присутствия электроноакцепторных групп п-БАФС, м-БАФС, 4,4'-диаминодифенилсульфон (ДДС) являются менее реакционноспособными по сравнению с ароматическими диаминами, содержащими электронодонорные группы (п-АФБ, м-АФБ). Использование в качестве инициаторов полимеризации ароматических диаминов, содержащих сульфоновую группу, позволяет увеличить интервал перерабатываемости благодаря их малой реакционной способности.
Таблица 2.
Ароматические диамины, используемые для отверждения фталонитрильных мономеров [47–50]
| Диамин | Формула диамина | Тпл, °С |
|---|---|---|
| Бис-[4-(4-аминофенокси) фенил]сульфон (п-БАФС) | 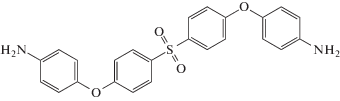 |
195 |
| Бис-[4-(3-аминофенокси) фенил]сульфон (м-БАФС) | 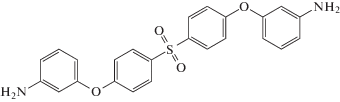 |
132–135 |
| 4, 4'-диаминодифенилсульфон (ДДС) |  |
155 |
| 1,4-бис-(4-аминофенокси)бензол (п-АФБ) | 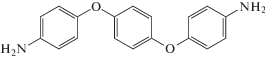 |
173 |
| 1,3-бис-(3-аминофенокси)бензол (м-АФБ) |  |
114–116 |
| 1,4-бис-(4-перфтораминофенокси) бензол(п-АФБF) | 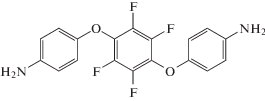 |
185 |
| 4,4'-оксидианилин (ОДА) |  |
188–192 |
Совсем недавно описан синтез фторсодержащего диамина п-АФБF и его применение в качестве отвердителя бисфталонитрилов (табл. 2) [48]. Полимер, полученный на основе мономера 2 с новым отвердителем, демонстрирует повышенную термоокислительную стабильность.
Из эмпирических данных было найдено, что оптимальная концентрация п-АФБ и м-АФБ для отвержения фталонитрильных мономеров составляет 1.5–2.3 мас. %. Из-за меньшей реакционной способности и более высокой молекулярной массы ММ оптимальная концентрация п-БАФС и м-БАФС для получения форполимера составляет 2–4 мас. %. Вязкость фталонитрильных форполимеров также меняется в зависимости от концентрации [51] и химической природы [47] отверждающего агента. Вязкость расплавов фталонитрильных форполимеров зависит от типа используемых диаминов в качестве отвердителей (рис. 3) при температуре 260°С.
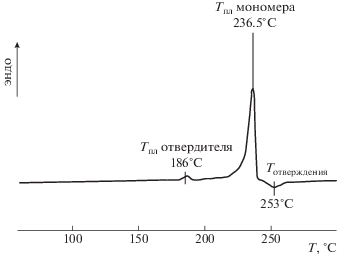
Полимеризация фталонитрильных мономеров, как правило, происходит в две стадии. На первой стадии при температуре 250–260°С [52] при нагревании смеси фталонитрильного связующего с ароматическим диамином на термограмме ДСК проявляются эндотермические пики и один экзотермический пик. Эндотермические пики относятся к процессу плавления исходных компонентов смеси, а появление экзотермического пика связано с протеканием быстрой реакции (10–15 мин) между диамином и мономером. На типичной кривой ДСК полимеризации 4,4'-бис-(3,4-дицианофенокси) бифенила (мономер) и 4 мас. % п-БАФС (отвердитель) наблюдается два пика, относящихся к плавлению отвердителя и мономера, и пик полимеризации (рис. 4) [47].
Рис. 4.
Типичная ДСК-кривая отверждения фталонитрильных мономеров в присутствии ароматических диаминов на примере 4,4’-бис-(3,4-дицианофенокси)бифенил (мономер)/(4 мас. %) п-БАФС (отвердитель).
Известно, что в результате реакции нуклеофильного присоединения амина к фталонитрилу образуется изоиндолин [53]. На основании этого факта считается, что при термической полимеризации фталонитрильного мономера в присутствии ароматических диаминов образуется полиизоиндолин [54]. Механизм образования полиизоиндолиновой структуры при отверждении фталонитрильных связующих ароматическими диаминами при 250°С приведен ниже.

где 
На второй стадии форполимер нагревают в течение длительного времени при повышенных температурах (325–375°С) (постотверждение). При этом происходит перегруппировка с образованием трехмерной полимерной сетки, которая состоит из триазиновых структур [23]:
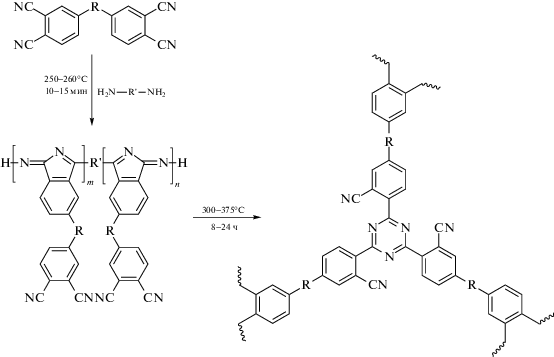
Полученный термостойкий, нерастворимый термореактивный полимер имеет высокую температуру стеклования, а также обладает высокой термической и термоокислительной стабильностью [55]. Условия проведения постотверждения оказывают влияние на механические свойства полимера (табл. 3). При выдерживании полимера при 315°C в течение 100 ч наблюдается снижение предела прочности из-за окисления на воздухе.
Таблица 3.
Влияние условий постотверждения на механические свойства полимера [55]
| T, °C | Условия отверждения/ постотверждения | Предел прочности при растяжении, МПа | |
|---|---|---|---|
| время, ч | атмосфера | ||
| 315 | 24 | воздух | 94 ± 17 |
| 350 | 12 | аргон | 94 ± 21 |
| 375 | 12 | аргон | 80 ± 7 |
| 315 | 100 | воздух | 72 ± 5 |
Аморфный форполимер, получаемый на первой стадии в результате реакции фталонитрильного мономера с диамином, называется B-стадией смолы и обычно характеризуется низким значением температуры стеклования. Например, форполимер, образованный при отверждении мономера 1 имеет температуру стеклования в температурном интервале 70–90°С. Форполимеры растворяются во многих органических растворителях и могут в течение долгого времени храниться при комнатной температуре.
Исходя из преимуществ ароматических аминов как отвердителей, авторы большинства современных исследований используют м-АФБ, п-БАФС и другие ароматические амины.
САМООТВЕРЖДАЮЩИЕСЯ МОНОМЕРЫ
Как было отмечено ранее, полимеризация фталонитрильных мономеров без специальных отвердителей, таких как соли металлов, фенолы, амины, протекает крайне медленно. Смола, получаемая в случае использования отвердителя, является двухкомпонентной системой, свойства которой сильно зависят от равномерности распределения компонентов, особенно в случае малых концентраций отвердителя. Основная идея разработки самоотверждающихся мономеров – введение инициирующих полимеризацию фталонитрилов амино- и гидроксигрупп в структуру самого мономера. В таком случае необходимость в дополнительном отвердителе отпадет, поскольку полимеризация будет протекать самопроизвольно.
Первым описанным самоотверждающимся бис-фталонитрилом был 3,5-бис-(3,4-дицианофенокси)анилин [56].

Как видно из структуры мономера, он содержит аминогруппу, способную к инициированию полимеризации по описанному ранее механизму. Анализ ДСК соединения показал, что температура его плавления оказалась равной 214°C. Исследователи наблюдали пик тепловыделения около 260°С, подтвердивший самоотверждение мономера.
В дальнейшем по реакции замещения 4-нитрофталонитрила аминофенолами был получен ряд самоотверждающихся мономеров и исследованы смолы на их основе [57].
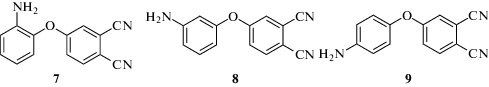
Все изомеры были получены с выходом более 90%. Интересно, что, согласно правилу Карнелли, симметричные молекулы обладают более высокой температурой плавления [58], в данном случае наоборот, температура плавления соединений составляет 122, 173, 131°С для орто-, мета- и пара-изомеров соответственно. Все три изомера активно полимеризуются уже при 170°С; смолы, полученные при ступенчатом нагреве до максимальной температуры 375°С, обладают рекордными для фталонитрильных смол температурами стеклования выше 500°С и T5% больше 520°С. Интересно, что по приведенным в работе кривым ТГА коксовый остаток превышает содержание углерода в исходных мономерах. При этом при карбонизации фталонитрильных смол потеря массы превышает теоретические значения из-за того, что часть углерода покидает смесь в виде СО и СО2 [60]. Позже было показано, что по данным синхронного термического анализа соединения 2 максимум потери массы совпадает с максимумом пика полимеризации кривой ДСК. Хотя авторы объясняют потерю массы только испарением мономера без выделения других летучих компонентов, в работе приводится механизм, включающий выделение аммиака. Известно, что изоиндолины, образующиеся на первом этапе полимеризации фталонитрилов, действительно способны реагировать с ароматическими аминами с выделением аммиака [60, 61]. В работе [57] также приводятся данные динамического механического анализа для композиционных материалов с матрицами на основе всех трех изомеров аминофеноксифталонитрила 7–9, армированных стеклотканью. Величина модули упругости для трех композитов находится в диапозоне 0.8–1.1 ГПа, что является крайне низкими значениями для фталонитрильных смол и тем более неправдоподобно низкими для стеклопластиков. Приведенные данные косвенно указывает на то, что в процессе полимеризации при высокой температуре происходит частичная карбонизация смолы с образованием пористого материала с низкими механическими свойствами.
В более поздней работе [62] данные изомеры исследовали в качестве отверждающих добавок к бис-фталонитрилу 2. Подробное исследование влияния содержания отвердителя показало, что наибольшая термическая стабильность достигается при применении 10 мас. % соединения 9 (табл. 4).
Таблица 4.
Свойства отвержденных при 320°С смол на основе бис-фталонитрила 2
| Отвердитель (мас. %) | T5%, °С | Коксовый остаток, % (при 900°С) | Tс, °С | E, МПа | |
|---|---|---|---|---|---|
| в азоте | на воздухе | ||||
| 9 (2) | 455 | 465 | 64.16 | 291 | 3040 |
| 9 (4) | 466 | 498 | 62.98 | 252 | 3154 |
| 9 (6) | 490 | 503 | 66.83 | 278 | 3618 |
| 9 (8) | 504 | 505 | 67.94 | 286 | 3860 |
| 9 (10) | 523 | 529 | 70.26 | 337 | 3825 |
| 9 (20) | 515 | 522 | 69.80 | 349 | 3267 |
| 9 (30) | 509 | 515 | 70.43 | 364 | 3199 |
| 9 (40) | 504 | 509 | 70.61 | 353 | 3863 |
| 9 (50) | 497 | 498 | 70.74 | 354 | 3406 |
| 7 (10) | 502 | 558 | 69.40 | 332 | 3820 |
| 8 (10) | 502 | 554 | 67.40 | 345 | 3352 |
В литературе также описан самоотверждающийся мономер с нафтильным фрагментом [63].
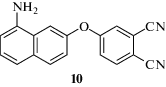
В работе исследуются свойства двух полимеров, полученных при конечной температуре 260 и 280°С. Как и в случае с соединениями 7–9 авторы не приводят потерю массы при полимеризации, а описывают только свойства конечного полимера. При этом по данным ТГА видно, что полученный при меньшей температуре полимер начинает терять массу уже при 250°С, что может косвенно свидетельствовать о выделении аммиака при полимеризации.
Самоотверждающиеся мономеры, содержащие гидроксильные группы, получают частичным замещением бисфенолов или гидрохинона фталонитрильной группой [64, 65].

Если реагенты берутся в стехиометрическом соотношении, то выход не превышает 25–35%, и продукты реакции выделяют с помощью колоночной хроматографии. В аналогичной реакции при применении шестикратного избытка резорцина выход продукта составляет 54% [66]. При этом чистый продукт удается выделить также только с помощью хроматографии, что сильно ограничивает возможность практического применения данных соединений как индивидуальных компонентов смол. Реакция 4-нитрофталонитрила с резорцином в мольном соотношении 2: 3 приводит к образованию смеси моно- и дизамещенного резорцина в соотношении 28 : 72 с общим выходом 87% [67]. Отверждение данной смеси без выделения отдельных компонентов по ступенчатой программе от 180 до 375°С способствует образованию полимера с температурой стеклования выше 400°С. По данным ТГА и ДМА, такая смола незначительно уступает аналогам, отвержденным в присутствии ароматических диаминов.
Похожий подход описан для смеси моно-, ди- и тризамещенного фталонитрильной группой α,α,α'-трис-(4-гидроксифенил)-1-этил-4-изопропилбензола [68].
 ,
,
где 
Варьируя соотношения реагентов, были получены четыре смеси мономеров с соотношением OH : CN = 0.03, 0.25, 0.35 и 0.55. Показано, что с увеличением этого соотношения в смеси мономеров снижается термическая стабильность и коксовый остаток полученных из них полимеров. Смесь с наибольшим содержанием гидроксильных групп также применяли в качестве инициатора полимеризации бис-фталонитрила 2 и сравнивали с ДДС. По результатам ТГА полимеры с новым отвердителем немного превосходят реактопласты с ДДС по термической стабильности, хотя, как следует из данных ДМА,они уступают им по модулю упругости.
Фталонитрильные соединения на основе НС способны полимеризоваться без использования дополнительных инициаторов. Химическая модификация новолака фталонитрильными группами впервые описана в работе [69]. Смола, полученная по реакции азосочетания фенолформальдегидной смолы и диазониевой соли фталонитрила, самопроизвольно полимеризуется при температуре 350°C за 2 ч, при этом никаких данных о механических свойствах не приводится. Другой подход к химической модификации НС фталонитрильными группами описан в работе [70], в которой приводится способ получения частично замещенной по гидроксильным группам смолы.
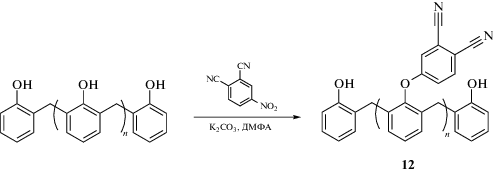
Полимеризация полученной смолы начинается уже при 150°C и максимальная степень сшивки достигается при 350°C, однако образующийся полимер обладает весьма низкими по сравнению с бис-фталонитрилами термическими (T5% = 449°C) и механическими (модуль упругости составляет всего 700 МПа) свойствами.
В работе [71] было показано, что аллильные группы также способны инициировать полимеризацию фталонитрилов. Термостойкий полимер образуется при ступенчатом нагреве от 260 до 360°С мономера, полученного взаимодействием 2,2'-диаллилбисфенола А и 4-нитрофталонитрила.
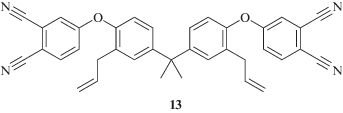
По данным ДСК, фталонитрильные группы вовлекаются в реакцию только после полимеризации аллильных групп, что требует значительно большего нагрева по сравнению с инициаторами на основе аминов или фенолов. К тому же образующийся полимер уступает по термической и термоокислительной стабильности классическим бис-фталонитрилам.
Позже были обнаружены и другие функциональные группы, которые могут инициировать полимеризацию фталонитрилов. Недавние исследования показали, что реакция может активироваться также и соединениями на основе пропаргиловых эфиров [72]. Был предложен механизм сопополимеризации пропаргиловых и фталонитрильных групп, основанный на перегруппировке Кляйзена с последующим образованием хромена:

Введение пропаргиловой группы открыло дополнительные возможности влияния на полимеризацию. Как известно, соли Ni2+ катализируют процессы гомополимеризации и тримеризации тройных связей [73, 74], Co2+ – циклотримеризацию [75], а соли Cu2+ – реакции кросс-сочетания [76]. Влияние указанных катализаторов на процесс полимеризации мономера 14, содержащего пропаргиловую и фталонитрильную группы был исследован с помощью метода ДСК [32]:

На представленных кривых ДСК (рис. 5) экзотермическим пикам соответствуют процесс полимеризации пропаргиловых и фталонитрильных групп. В случае полимеризации без катализатора (рис. 5а) наблюдалось два пика, первый из которых относят к полимеризации пропаргильных групп, второй – к полимеризации фталонитрильных. В случае добавки 4% ароматического диамина м-АФБ пики смещаются по температуре и сливаются в один, больший по интенсивности (рис. 5б). Также исследователи отмечают увеличение общего выделения тепла в ходе процесса с 800 до 1121 Дж/г. Добавки солей металлов, главным образом воздействуя на механизм полимеризации именно пропаргильных групп, влияют на температурные интервалы полимеризации, а также на ее интенсивность. Это отчетливо видно из положения и формы экзо-пиков на рис. 5в–5е.
Рис. 5.
Кривые ДСК полимеризации пропаргилсодержащего фталонитрильного мономера с различными катализаторами.

Недавно принципиально иной механизм инициации фталонитрильных групп был описан в серии работ группой исследователей под руководством G. Yang [77–80]. В работе [77] было показано, что алициклические фрагменты способны вступать в сополимеризацию с фталонитрилами. Позже было обнаружено, что наличие кратной связи в алицикле не является необходимым для инициации полимеризации и в качестве отвердителей могут выступать производные тетра- и гексагидрофталимида разного строения [78].

На основе этого открытия был синтезирован самоотверждающийся мономер 15, исследована кинетика его полимеризации и оценены термические и механические свойства полимера. Поскольку мономер обладает крайне высокой температурой плавления (279°С), первый этап полимеризации с образованием форполимера проводят при 280°С, который затем постотверждают при 375°С. Интересно, что по данным инфракрасной спектроскопии полимерная структура состоит из фталоцианиновых и полииндолиновых фрагментов и не содержит триазинов, которые образуются при высокой температуре при отверждении ароматическими аминами и фенолами. Несмотря на высокие термические свойства (T5% = 523°С, Tс > > 470°С), по данным ДМА полимер обладает чрезвычайно низкими механическими свойствами.
Отдельная работа [78] посвящена исследованию механизма инициации фталонитрилов алициклами. Ее авторы вводят новый термин для этого процесса – термическая синергетическая полимеризация [77], хотя суть термина не раскрывается, а другие упоминания в литературе отсутствуют. Изучение механизма реакции проводилось на модельной системе, состоящей из взятых в эквимольном соотношении алициклического и фталонитрильного мономера:

Приведенный в работе механизм предполагает радикальную атаку алицикла по нитрильной группе с образованием радикала изоиндолина, который инициирует дальнейшую полимеризацию. Для подтверждения возможности радикальной атаки по нитрильной группе авторы приводят ссылку на работу [81], в которой представлена схема, изображающая присоединение фенильного и циклогексильного радикала к нитрильной группе бензонитрила. Однако данная работа носит расчетно-теоретический характер, а приведенная схема, вероятно, содержит ошибку поскольку опирается на работу [82], в которой показано, что присоединение радикалов к бензонитрилу происходит исключительно в ароматическое кольцо. Для экспериментального доказательства радикального механизма активации фталонитрилов также приводится описание эксперимента с добавлением дифенилпикрилгидразила (ДФПГ) в качестве ловушки радикалов. При этом известно, что ДФПГ разлагается при плавлении при 129°С [83], а полимеризацию проводят при 300°С. По приведенным в работе данным масс-спектрометрии с матрично-активированной лазерной десорбцией/ионизаций разница между составом с ДФПГ и без него не очевидна. В итоге стоит отметить, что кроме указанных работ никаких упоминаний о возможности протекания радикальной полимеризации фталонитрильных групп нет.
ЛЕГКОПЛАВКИЕ ФТАЛОНИТРИЛЫ
Для расширения технологического “окна” (т.е. разницы между температурой плавления и температурой начала полимеризации) фталонитрильных смол было предложено несколько подходов: синтез бис-фталонитрильных олигомеров, введение гибких мостиков, содержащих гетероатом, и приготовление смесей мономеров.
Впервые синтез олигомерного бис-фталонитрила описан в работе [84]. Ниже приведен пример синтеза данного соединения.
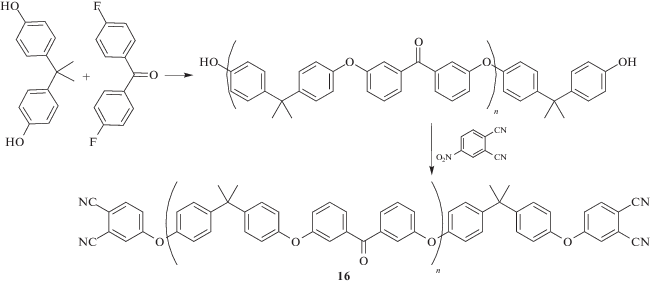
Целью работы было показать возможность получения мономеров с различной молекулярной массой, при этом не приводятся данные по механическим свойствам полимеров.
Позже было предложено несколько новых подходов к синтезу мономеров c мостиками из полиэфиркетона и полиэфира. Так, были получены фталонитрильные олигомеры на основе бис-фенолов [85–87] и резорцина [88–90].
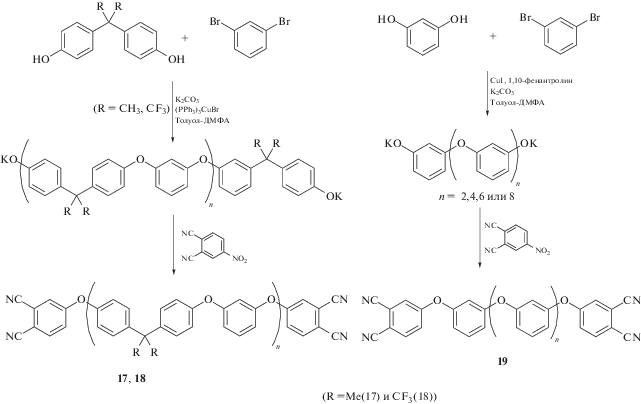
Новые олигомерные соединения обладают аморфной структурой, поэтому они характеризуются более низкой температурой стеклования по сравнению с температурой плавления низкомолекулярных аналогов [91]. Несмотря на низкие температуры стеклования (50–90°С), активная полимеризация этих олигомеров в присутствии ароматических диаминов начинается только при 250–270°С.
Показано, что при увеличении гибкости структуры линкера между концевыми фталонитрилами наблюдается снижение температуры стеклования. Однако с понижением Тс олигомеров уменьшается и устойчивость полимеров на их основе к повышенным температурам. Термические свойства отвержденных полимеров были несколько ниже, чем у классических бис-фталонитрилов (Tс = 350–400°С, T5% < 500°С). По приведенным результатам ДМА модуль упругости полимеров при комнатной температуре составляет всего 1.1–1.3 ГПа. В итоге при модификации фталонитрилов олигомерными фрагментами возникает ряд трудностей. Во-первых, синтез олигомеров значительно сложнее по сравнению с меньшими молекулами, также становится необходимым контроль олигомерного состава. В ряде случаев выделение чистого продукта осложняется трудноотделимыми примесями. Во-вторых, внедрение олигомера в ядро фталонитрильного связующего может привести к повышению пластификации мономера, что в результате вызывает снижение теплостойкости и ухудшение механических свойств отвержденной матрицы.
Отдельно стоит отметить олигомерные соединения, содержащие эфир триарилфосфиноксида [92]. Олигомеры разного строения были получены с резорцином или бисфенолом А по стандартной методике:

Как и в случае других олигомеров, фосфорсодержащие мономеры характеризуются низкими температурами стеклования (75–90°С), при этом их активная полимеризация не происходит при температуре ниже 230°С. Полимеры на основе указанных соединений обладают лучшими механическими свойствами по сравнению с ранее полученными олигомерами, а также проявляют повышенную термоокислительную стабильность.
Другим интересным примером низкоплавкого олигомера, содержащим фосфор, является гексафталонитрил 21 на основе фосфазенового цикла [93].

Основным его преимуществом является высокий зольный остаток, однако ввиду высокой стоимости и сложности синтеза, а также сравнительно высокой температуры стеклования (Tс = 87°С), данные фталонитрильные мономеры не представляют интереса для практического применения.
Дальнейшая эволюция структур олигомеров, несущих фталонитрильные группы, развивалась с целью повышения термической и термоокислительной стабильности полимеров на их основе. Наибольший прирост в устойчивости оказался у мономеров с азотосодержащими гетероциклами. Так, была получена серия мономеров с 1,3,5-триазиновыми [94–96], пиразиновым [97] циклами и мостиком, содержащим фталозиноновый фрагмент [98, 99].
Расширить технологическое “окно” мономеров удалось путем введения в их структуру дивинилпиридинового фрагмента [100]. Новый мономер был получен, исходя из 2,6-диметилпиридина, с выходом в 32%.
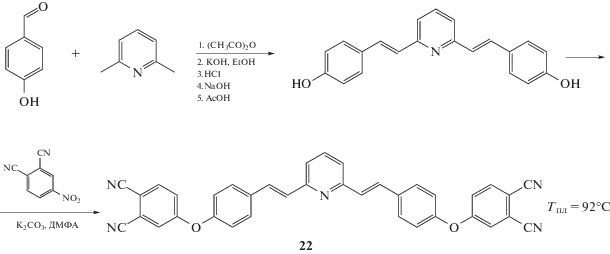
Несмотря на относительно низкую температуру плавления, расплав мономера в диапазоне температур 130–210°С характеризуется весьма высокой вязкостью (более 1.5 Па с). Полимер, полученный с 9 в качестве инициатора, демонстрирует механические и термические свойства на уровне классических бисфталонитрилов.
Другим подходом к снижению температуры размягчения мономеров стало введение в их структуру гетероатомов, таких как кремний и фосфор. Впервые введение кремниевого фрагмента в структуру фталонитрильного мономера осуществили взаимодействием 4-(4-аминофенокси)фталонитрила и дихлорсиланов [101]. Прямым взаимодействием реагентов в присутствии триэтиламина в качестве основания удалось с высоким выходом получить ряд кремнийсодержащих мономеров, демонстрирующих рекордно низкие на тот момент температуры плавления 40–59°С:

Полученные мономеры содержат активную группу NH с донорным заместителем и поэтому способны к самопроизвольной полимеризации уже при 170°С. Использование силазанового линкера позволило значительно расширить технологический интервал с сохранением термических свойств отвержденной матрицы на уровне классических фталонитрилов (Tс > 450°С, T5% > 550°С).
В отличие от аминосодержащих соединений силоксановые эфиры фенолов, несущих фталонитрильные группы, оказались неустойчивы: гидролиз происходит даже в присутствии влаги воздуха [31]. Значительного расширения технологического интервала удалось достичь благодаря получению бензиловых эфиров силоксанов [13, 102, 103]. В ходе работы был получен ряд новых кремнийсодержащих мономеров с Tс = –1…+27°С с сохранением термических свойств отвержденной матрицы на характерном для фталонитрилов уровне.

Основным недостатком полученных мономеров является сложность синтеза, связанная с необходимостью восстановления альдегидной группы, что приводит к низкой технологичности процесса при масштабировании.
Позже было предложено использование олигомеров, содержащих, в том числе, кремниевый фрагмент, обладающих температурой стеклования Tс = 35–60°С

Однако термические свойства отвержденной матрицы оказались существенно ниже опубликованных ранее данных для классических фталонитрилов (табл. 5) [104].
Таблица 5.
Общие сведения о фталонитрильных мономерах и отвержденных матрицах
| Образец, № | Мономер | Отвержденная матрица | Литература | ||
|---|---|---|---|---|---|
| Tпл(с), °С | Tс, °С | T5%, °С (воздух/N2) | Коксовый остаток, % | ||
| 3 | 194–200 | >450 | 450/490 | 72 | [23] |
| 5 | 235–238 | >450 | 460/500 | 68 | [23] |
| 1 | 235 | >450 | 500 | 71 | [23] |
| 2 | 185–190 | >450 | 527 | 70 | [50] |
| 17 | 60 | >550 | – | 78 | [89] |
| 19 | 42 | 270 | 486/493 | 62 | [90] |
| 22 | 92 | >400 | 505/524 | 48 | [100] |
| 24 | 59 | >450 | 558/539 | 80 | [101] |
| 25 | 40 | >450 | 543/541 | 82 | [101] |
| 23 | 42 | >450 | 562/570 | 83 | [101] |
| 29 | 35 | 210 | 420 | 58 | [104] |
| 20 | 75 | >450 | 490/490 | 65 | [92] |
| 21 | 87 | >380 | 510/530 | 83 | [93] |
| 32 | 21 | 455 | 524/523 | 80 | [31] |
| 26 | –1 | 413 | 527/545 | 81 | [102] |
Значительного снижения температуры стеклования вплоть до 4°С удалось достичь введением в структуру мономера дисилоксанового фрагмента [105].

Полимер на основе этого соединения демонстрирует высокие термические свойства на уровне классических бисфталонитрилов, сохраняя при этом высокую устойчивость к гидролизу.
Помимо кремниевого фрагмента было также предложено введение в мономер фосфорсодержащих групп [92]. В ходе работы был получен фосфорсодержащий мономер с Tс = 75°С. Основным наблюдением была меньшая потеря массы при нагревании на воздухе по сравнению с классическими фталонитрильными полимерами, что свидетельствует о большей термоокислительной стабильности полимерной матрицы.
Дальнейшее изучение фосфорсодержащих фталонитрилов привело к получению мономеров с Тс близкой к мономерам, модифицированных кремнием. Ниже приведена схема синтеза легкоплавких фосформодифицированных фталонитрилов

где R = Ph, OPh. OMe, OBu, OiPr.
Была выявлена закономерность: с увеличением длины углеродной цепи в ядре происходило уменьшение Тс. Были изучены и термические свойства мономеров: потерь в свойствах не обнаружили [31]. Ввиду присутствия фосфатной группы было важно изучить гидролитическую устойчивость полученных мономеров. В работе [106] было выяснено, что гидролиз фосфатов происходит активно только в щелочной среде и практически прекращается с понижением pH до 4, что дает возможность масштабировать синтез фосфатсодержащих мономеров. Существенным плюсом также стала повышенная устойчивость реактопластов к окислению. Авторы исследования связывают это явление с антипиреновыми свойствами фоcфатной группы, установленными при исследовании горючести композитов [107, 108].
Еще один подход к повышению технологичности фталонитрильных связующих заключается в использовании сразу нескольких мономеров, которые способны к образованию эвтектической смеси с низкой температурой плавления. Для практического применения в изготовлении композитов смесь должна обладать также низкой вязкостью в широком диапазоне температур. Поэтому обычно одним из компонентов таких смесей являются низкоплавкие мономеры, которые выступают в роли активного разбавителя связующего. Связующие на основе кремний- и фосфорсодержащих мономеров

имеют низкую вязкость расплава (<250 мПа с) при температуре выше 120°С [31, 108, 109]. Применение именно этих смесей позволило впервые осуществить пропитку армирующего наполнителя энергоэффективным методом вакуумной инфузии.
В качестве активных разбавителей также использовали соединения, не содержащие фталонитрильные группы, но способные к сополимеризации с ними. Применение цианофенольного фосфата

позволило получить связующие с температурой стеклования –2…+10°С в зависимости от содержания разбавителя. Интересно, что даже при содержании 50 мас. % соединения 33 в смеси полимер на основе этого связующего демонстрирует механические и термические свойства на уровне классических бисфталонитрилов [110]. Как и фосфорсодержащие фталонитрилы, фосфат 33 оказался устойчив к гидролизу в кислой и нейтральной среде [111].
Монофталонитрильные соединения также нашли применение в качестве активных разбавителей:

Смеси на их основе с классическими бисфталонитрилами плавятся при 120–150°С, однако вязкость расплава превышает 1 Па с, что ограничивает спектр технологий их переработки [37, 112]. Кроме того, полимеры на их основе значительно превосходят по механическим свойствам аналоги из легкоплавких олигомерных соединений.
Таким образом, в литературе имеется достаточно большое количество работ, посвященных синтезу и исследованию свойств фталонитрилов различного строения (табл. 5).
МЕХАНИЧЕСКИЕ И ТЕРМОМЕХАНИЧЕСКИЕ СВОЙСТВА ФТАЛОНИТРИЛЬНЫХ СМОЛ
Механические и термомеханические свойства фталонитрильных смол зависят от множества факторов, таких как структура мономера, температурный режим отверждения, тип используемого отвердителя и т.д. Поскольку существует множество вариаций всех указанных параметров, провести сравнение между всей литературной информацией становится сложно, зачастую работы разных авторов отличаются более чем варьированием одного параметра. В связи с этим мы рассмотрим общие закономерности, однозначно прослеживаемые между всеми данными.
В работе [39] исследованы механические свойства полимеров на основе классических бисфталонитрилов. Полимеризацию проводили без использования каких-либо отвердителей в течение 6 дней непрерывного нагревания при 280°С. Механические свойства исследовали при комнатной температуре и сравнивали полученные параметры с коммерческой эпоксидной смолой Narmco 5208 (табл. 6). Значения механических характеристик довольно сильно разнятся от мономера к мономеру, но в среднем находятся выше или на том же уровне по сравнению с эпоксидной смолой.
Таблица 6.
Механические характеристики смол на основе фталонитрилов “второго поколения”
| Смола | Трещиностойкость, G1C, Дж/м2 | Модуль упругости, ГПа | Модуль сдвига, ГПа | Тс, °С |
|---|---|---|---|---|
| 3 | 99 | 3.92 | 1.20 | 320 |
| 4 | 202 | 2.25 | 1.32 | 300 |
| 1 | 140 | 4.35 | 1.22 | 300 |
| 5 | 53 | 4.02 | 1.25 | 225 |
| Эпоксидная смола 5208 | 76 | 3.89 | 1.25 | 260 |
Авторы другой работы [25] изучали влияние режима отверждения на свойства смолы. Исследователи отверждали мономер 1 в присутствии 1 мас. % 4,4'-метилендианилина (МДА) в четырех разных режимах. Образец А был получен в результате нагревания при 260°С в течение 16 ч с последующей выдержкой на 300°С в течение 15 ч, образец B – нагреванием образца А при 350°С в течение 4 ч, а образцы C и D – нагреванием образца B при 375°С в течение 4 и 24 ч соответственно. Результаты динамо-механического анализа показали, что образец А обладал наибольшим модулем упругости (~3.3 ГПа), но терял 90% от этого значения при нагревании до 375°С, претерпевая расстекловывание в районе 290°С. Остальные образцы имели меньший модуль упругости (2.0–2.5 ГПа). Наиболее интересным результатом стало то, что модуль упругости образцов C и D в значительной степени сохранялся вплоть до 375°С, свидетельствуя о том, что их температура стеклования лежит за этим пределом. Это означает, что повышение температуры постотверждения смолы увеличивает ее жесткость при повышенных температурах и снижает подвижность звеньев полимера. Авторы логично связывают эти явления с ростом степени сшивки смолы.
В другой работе [47] было исследовано влияние различных ароматических аминов на температуру стеклования и модуль упругости смол из мономера 1. Результаты показали, что использование м-АФБ в случае отверждения при 280°С обеспечивает немного более высокий модуль упругости смолы, чем в случае других отвердителей. Также смола, полученная с использованием м-АФБ обладала температурой стеклования на ~50°С выше, чем остальные. При постотверждении смол при температуре 350°С указанные выше различия сглаживались: свойства смол становились почти идентичными для отвердителей м-АФБ и п-БАФС. Исследователи связывают это с более высокой активностью м-АФБ, в результате чего больше функциональных групп вступает в полимеризацию при более низкой температуре отверждения. При повышении температуры степень превращения функциональных групп достигает определенного предела, величина которого при использовании отвердителей разного строения примерно одинакова.
Механические свойства смол на основе олигомерных фталонитрилов сильно зависят от количества и типа мостиковых групп [89, 92, 113]. Авторы работы [113] изучили полимеризацию смол на основе олигомеров с разной длиной алифатического мостика (от 6 до 36 звеньев). Отверждение проводили при 220°С в течение 24 ч без использования отвердителей. С ростом длины мостика между фталонитрильными фрагментами наблюдалось падение модуля сдвига отвержденной смолы и понижение ее температуры стеклования (табл. 7).
Таблица 7.
Изменение модуля сдвига смолы с ростом количества звеньев в алифатическом мостике мономера
| Количество звеньев олигомера | Модуль сдвига, ГПа | Тс, °С |
|---|---|---|
| 6 | 1.6 | 350 |
| 10 | 1.2 | 286 |
| 22 | 0.7 | 110 |
| 36 | 0.3 | 90 |
Механические свойства смол на основе этих олигомеров оказались не такими высокими, как у смол на основе классических бисфталонитрилов, но сохранялись до более высоких температур. В работе [89] был выполнен ДМА смол на основе олигомеров 38 и 39:

Отверждение проводили в присутствии 3 мас. % п-БАФС в инертной атмосфере по следующей программе: 12 ч при 270°С, 3 ч при 300°С, 6 ч при 350°С, 8 ч при 375°С и 8 ч при 425°С. По данным ДМА [89] модуль упругости оказался меньше 2 ГПа в обоих случаях, но его значение сохранялось вплоть до 450°С. Это свидетельствует о том, что значение температуры стеклования смолы выше 450°С.
В литературе представлены примеры, где за счет строения мономера авторам удавалось добиться более высокого модуля упругости, но эти свойства не сохранялись при высоких температурах. В работе [92] исследованы свойства смол на основе фосфорсодержащего олигомера
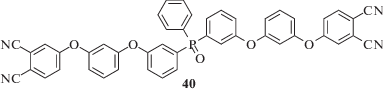
Отверждение проводили в присутствии 3 мас. % п-БАФС следующим образом: 12 ч при 270°С, 4 ч при 300°С, 4 ч при 350°С и 8 ч при 375°С. Модуль упругости полученной смолы при комнатной температуре оказался выше, чем у смол на основе других олигомеров (2780 МПа), но падал в ⁓5 раз при нагревании до 400°С.
Механические и термомеханические свойства смол на основе легкоплавких мономеров с гетероатомами исследованы в работах [13, 103, 109]. Авторами был получен большой массив данных по свойствам смол на основе мономеров с гетероатомами, при этом условия их отверждения были идентичными: 4 мол. % м-АФБ, отверждение при 200°С и постотверждение при 375°С.
Механические свойства смол на основе силоксановых мономеров оказались относительно невысокими для класса фталонитрилов (E = 2.6 ГПа). Механическое испытание смол на основе фосфатных мономеров было затруднено высокой хрупкостью смолы, в связи с этим их тестировали в смеси с другими фталонитрилами. Добавки фосфорсодержащих мономеров положительно сказывались на механических свойствах отвержденных матриц и позволили достичь высоких значений модуля упругости (4.4–5.7 ГПа). Смолы из фосфорсодержащих мономеров, продемонстрировали комплексное превосходство над классическими бисфталонитрилами и коммерческим полиимидным связующим.
Механические свойства реактопластов зависят от степени сшивки макромолекулярных цепей. В олигомерных соединениях расстояние между функциональными группами значительно больше, чем в классических бисфталонитрилах, что приводит к меньшей плотности сшивки и, как следствие, пониженным механическим свойствам. В работе [114] исследовали смеси фталонитрильного олигомера и трифункционального мономера 41 в разных соотношениях.

В результате было показано, что при увеличении концентрации олигомера в смеси, модуль упругости отвержденного образца уменьшается. Интересно, что смола, отвержденная при 375°С, с 75 мас. % содержанием трифункционального мономера демонстрирует довольно низкий модуль упругости (1.4 ГПа против 1.2 ГПа у чистого олигомера). В другой работе этот же трифункциональный мономер, отвержденный до максимальной температуры в 400°С, имел модуль упругости 3.7 ГПа [33].
Наибольшего прироста механических свойств удалось добиться, применяя трифункциональный мономер на основе фосфата. Хотя сам мономер обладает относительно высокой температурой плавления в 172°С, его смеси с дифункциональным аналогом представляют собой аморфные смеси с температурами стеклования 28–43°С в зависимости от соотношения компонентов. Лучшие результаты по механическим свойствам достигаются для смеси мономеров 42 и 32 в соотношении 4 : 1.
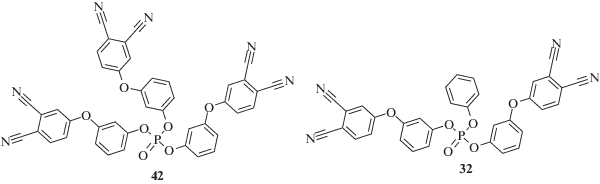
Полимер, полученный после отверждения этой смеси до максимальной температуры 375°С, продемонстрировал рекордно высокий модуль упругости в 7.2 ГПа, что является максимальным описанным значением для реактопластов [115]. По данным ДМА температура стеклования полимера превышает 450°С, при этом модуль упругости при 400°С составляет ⁓4 ГПа.
КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ С ФТАЛОНИТРИЛЬНЫМИ СВЯЗУЮЩИМИ
Механические свойства композиционных материалов зависят от различных факторов, включая параметры переработки (время, температура и давление), ориентацию армирующего волокна, качество препрега, условия постотверждения, состав композита, его пористость и т.д. Несмотря на то, что поиск новых мономеров ведется в основном с целью улучшения технологичности фталонитрильных смол при формовании ПКМ, в подавляющем большинстве работ композиты формовали из препрегов, полученных пропиткой наполнителя раствором связующего [112, 116, 117, 120, 122, 126–146]. Чтобы можно было сравнивать результаты исследований между собой, ниже приводится обзор композитов с фталонитрильными матрицами, структурированный в зависимости от типа наполнителя и направлений армирования.
Однонаправленные углепластики с фталонитрильными матрицами
Как и в случае с разработкой связующих, первые опыты по получению ПКМ с фталонитрильными матрицами были выполнены группой Т. Келлера. В работе [52] описан углепластик на основе мономера 1 и однонаправленной углеродной ленты из волокна марки Hexcell IM7 12K. Из-за высокой температуры плавления мономера (234°C) для получения связующего проводили форполимеризацию: в расплав мономера при 250°С был добавлен инициатор полимеризации м-АФБ в количестве 2.1 мас. %. После интенсивного перемешивания при 250°С в течение 15 мин получали связующее с Тс ~ 80°C, растворимое в органических растворителях. Содержание инициаторa было выбрано на основании времени желирования препрега, при концентрациях диамина 4–5% полимер образовывался за 5 мин, что не позволяло полностью пропитать армирующий наполнитель, в то же время концентрация около 2% обеспечивала время желирования в районе 1 ч. Раствор форполимера наносили на углеродную ленту, и из полученного препрега формовали композит горячим прессованием (250°C) при давлении 13 бар и постотверждали по ступенчатой программе с максимальной температурой 325°C или 375°C. При этом значительной разницы в механических свойствах композитов, постотвержденных при разной температуре, не наблюдалось, однако разница в температуре стеклования была существенной – 350°C и >450°C соответственно. В работе [51] изучали влияние природы отвердителя на свойства углепластиков с матрицей на основе 1. Углепластики получали в автоклаве при давлении 14 бар с конечной ступенью отверждения 8 ч при 375°C. Результаты исследований показали, что м-АФБ и п-БАФС в качестве инициаторов полимеризации оказывают влияние только на реологию смолы и оптимизацию цикла отверждения, тогда как механические свойства материалов изменяются не сильно. При этом в работе впервые проведены исследования механических свойств углепластиков с фталонитрильными матрицами при повышенных температурах. Величина прочности межслоевого сдвига при 316 и 371°C составляла около 65 МПа, что равно примерно 70% от значения при комнатной температуре (табл. 8).
Таблица 8.
Механические свойства однонаправленных углепластиков с фталонитрильными матрицами
| Связующее/ наполнитель | Максимальная температура пост-отверждения | Прочность при растяжении | Прочность при изгибе | Прочность при межслоевом сдвиге τ13, МПа | Литера- тура | ||
|---|---|---|---|---|---|---|---|
| $\sigma _{{11}}^{ + }$, МПа | Е, ГПа | σf, МПа | Е, ГПа | ||||
| 1 + АФБ/углеродная лента “IM7”, 0° | 375°C, 4 ч | 2000 | 183 | 2350 | 174 | 85 | [52] |
| 1 + АФБ/углеродная лента “IM7”, 90° | 375°C, 4 ч | 41 | 10 | 80 | 11 | – | [52] |
| 1 + п-БАФС/углеродная лента “IM7” | 375°C, 8 ч | 1975 | 159 | 2287 | 144 | 93 | [51] |
| 43/Углеродная лента из волокна Т700 | 375°C, 8 ч | – | – | 1339–1855 (25°C)/ 255 (450°С) | 113–164 | 71 (25°С)/ 36 (450°С) | [94] |
| 2 + 31 + 32 + АФБ/ углеродная лента из волокна IMS65 24k | 375°C, 8 ч | – | – | – | – | 72 (25°С)/ 61 (300°С) | [108] |
Несмотря на то, что механические свойства однонаправленных углепластиков с матрицей на основе 1 до сих пор являются наилучшими из всех, представленных в литературе для однонаправленных композитов, процесс их получения наглядно продемонстрировал все недостатки фталонитрильных связующих первого поколения: необходимость форполимеризации для снижения Тс связующего и растворный метод пропитки препрегов.
В работах [94, 95] было предложено использовать матрицы на основе олигомеров, содержащих триазиновый фрагмент в ядре.

Из-за высоких температур плавления олигомеров (>200°C) процесс получения композитов требовал форполимеризации тугоплавких олигомеров в присутствии 5% п-БАФС при 250°C. Раствор форполимера в ДМФА использовали для пропитки углеродного волокна Т700, которое впоследствии наматывали на прямоугольную раму для создания однонаправленного препрега. Композиты отверждали в оснастке при 280°C и давлении 25 бар и постотверждали в свободном виде с максимальной температурой 375°C.
Прочность и модуль упругости композитов при изгибе немного уступали значениям, полученным ранее группой Т. Келлера, но, тем не менее, оказались на достаточно высоком уровне (табл. 9). В то же время из-за высокой подвижности олигомерных цепей при 450°C композит сохранял только 15 и 51% от исходной прочности при изгибе и при межслоевом сдвиге соответственно.
Таблица 9.
Механические свойства ПКМ с фталонитрильными матрицами, полученных из препрегов на основе стеклянных тканей (E-стекло)
| Связующее/наполнитель | Пост- отверждение | Прочность при изгибе | τ13, МПа | Литера-тура | |
|---|---|---|---|---|---|
| σf, МПа | Е, ГПа | ||||
| 1+АФБ | 325°C, 8 ч | 834 | 31 | – | [126] |
| 1+АФБ | 375°C, 4 ч | 819 | 32.9 | 54 | [126] |
| 3+БФА (2:2) | 300°C, 4 ч | 571 | – | – | [127] |
| 3+ДДС+ДГБФ | 320°C, 2 ч | 601 | 21.7 | – | [128] |
| 3+ДДС+ДГБФ/оксид кремния | 320°C, 2 ч | ≤650 | ≤24.9 | – | [128] |
| 1+49 (1:2)+ZnCl2+ДДС | 370°C, 2 ч | 670 (25°C), 310 (400°C) | 27 (25°C) 16 (400°C) | 85 | [129] |
| ФНС-4П | 330°C | – | – | 53 (25°C), 28 (350°C) | [123] |
Однонаправленный углепластик получали в работе [108] методом пропитки в форме под давлением. Использовали связующее, состоящее из смеси мономеров 2, 31 и 32. Полученный углепластик схожей величиной прочности при межслоевом сдвиге, что и описанный в работах [94, 95], но провести сравнение по другим показателям не представляется возможным из-за расхождения в типе испытаний. В работе [108] приводятся также величины прочности на сжатие (767 МПа при 25°C и 515 МПа при 300°C) и при сдвиге в плоскости. Однако стоит отметить низкую воспламеняемость композита: кислородный индекс составляет более 80%, что существенно выше опубликованных значений кислородного индекса для других углепластиков. Для композитов с матрицами на основе фенолформальдегидных смол он равен ⁓ 55%.
Углепластики из препрегов на основе углеродной ткани по растворной технологии
Углепластики с использованием ткани в большинстве описанных случаев также получали через раствор связующего. В работе [112] описаны углепластики на основе углеродной ткани MT300 (атлас из 5 жгутов), пропитанной раствором многокомпонентного связующего ФНС-1:

Введение нескольких мономеров в состав связующего позволяет получать легкоплавкие смеси для улучшения технологичности [109, 118], но даже несмотря на использование в качестве реактивного разбавителя 4-феноксифталонитрила, композиты формовали в автоклаве (8 бар) из препрега, полученного по растворной технологии.
Было изучено влияние максимальной температуры постотверждения (315, 330, 350 и 375°C) на механические свойства композитов. Показано, что при увеличении температуры постотверждения до 350°C и выше в матрице начинают появляться трещины в поперечном направлении к плоскости армирования. Эта картина характерна для композитов с матрицами с высокой степенью сшивки, и объясняется высокой химической усадкой реактопласта. Таким образом, повышение температуры постотверждения приводит к ухудшению механических свойств композита (табл. 10). Такое же влияние температуры пост-отверждения на свойства углепластиков наблюдалось и в других работах [107, 119].
Таблица 10.
Механические свойства углепластиков, полученных на углеродной ткани из препрегов
| Связующее/наполнитель | Максимальная температура постотверждения | Прочность при изгибе σf, МПа | Прочность при сжатии $\sigma _{{11}}^{ - }$, МПа | Прочность при межслоевом сдвиге τ13, МПа | Ударная прочность по Изоду, кДж/м2 | Литература |
|---|---|---|---|---|---|---|
| ФНС-1/углеродная ткань MT300 | 315°С, 2 ч | 633 (25°C) 206 (400°C) |
455 (25°C) 89 (400°C) |
30 (25°C) 15 (400°C) |
– | [112] |
| ФНС-1/углеродная ткань MT300 | 330°С, 2 ч | 675 (25°C) 301 (400°C) |
350 (25°C) 140 (400°C) |
26 (25°C) 13 (400°C) |
– | [112] |
| ФНС-1/углеродная ткань MT300 | 350°С, 2 ч | 575 (25°C) 356 (400°C) |
390 (25°C) 213 (400°C) |
21 (25°C) 14 (400°C) |
– | [112] |
| ФНС-1/углеродная ткань MT300 | 375°С, 2 ч | 517 (25°C) 414 (400°C) |
329 (25°C) 239 (400°C) |
26 (25°C) 16 (400°C) |
– | [112] |
| 44/углеродная ткань Т-300 | 350°С, 3 ч | 370 | 65 | 20 | – | [120] |
| 46/углеродная ткань Т-300 | 350°С, 3 ч | 250 | 32 | 17 | – | [120] |
| 46/углеродная ткань Т-300 | 330°С, 3 ч | 147 | – | 15 | 75 | [121] |
| 46+10% 47/углеродная ткань Т-300 | 330°С, 3 ч | 324 | – | 23 | 128 | [121] |
| 46+20% 47/углеродная ткань Т-300 | 330°С, 3 ч | 483 | – | 40 | 186 | [121] |
| 46+30% 47/углеродная ткань Т-300 | 330°С, 3 ч | 257 | – | 22 | 100 | [121] |
| 46+30% 48/углеродная ткань Т-300 | 330°С, 3 ч | 284 | – | 19 | 119 | [121] |
| ФНС-4П/углеродная ткань 22502 | 330°C, 8 ч | – | 548 | 41 (25°C) 26 (400°C) |
– | [123] |
В цикле работ группы Р. Наира были изучены новолачные фталонитрильные смолы, замещенные фталонитрилом по гидроксильным группам в качестве матриц для получения углепластиков [120].

Используя раствор олигомеров с различной степенью замещения гидрокси-групп в метилэтилкетоне, получали препреги на основе углеродной ткани марки Т-300 (70 мас. %). Листы препрега сушили под вакуумом при температуре 60°С и отверждали в оснастке с максимальной температурой 350°C. Была изучена зависимость свойств ПКМ от содержания фталонитрильных групп в связующем и было показано возрастание термических и падение механических свойств композитов. Прочность на сжатие ПКМ с матрицей 44 (65 МПа) оказалась почти в 2 раза выше, чем у композита с матрицей 46 (30 МПа). Авторы работы связывают это с улучшением адгезии матрицы к волокну с увеличением концентрации групп ОН и с тем, что степень сшивки реактопласта с более высоким содержанием фталонитрильных групп намного выше, что приводит к увеличению хрупкости матрицы. Стоит отметить, что уровень прочности описанных в работе композитов при сжатии и межслоевом сдвиге по сравнению с другими описанными ПКМ находится на значительно более низком уровне (табл. 9), и исходя из представленных в работе кривых ТГА можно предположить частичную карбонизацию матрицы в процессе постотверждения при 350°C. Известно, что прочность на сжатие углерод-углеродного материала, полученного карбонизацией углепластика с фталонтрильной матрицей, составляет 72.2 МПа [59], что ближе к описанным результатам, чем характерные для углепластиков на ткани значения в 350–700 МПа (табл. 10, 11). Представленные фотографии растровой электронной микроскопии образцов композитов с матрицами 45 и 46 демонстрируют низкую степень пропитки волокна матрицей, что свидетельствует в пользу гипотезы авторов об адгезионной прочности границы волокно–матрица в зависимости от содержания групп OH и может быть индикатором разрушения границы в процессе пост-отверждения при частичной карбонизации смолы. Гидроксильные группы являются инициаторами полимеризации фталонитрильних групп, и при их низкой концентрации процесс проходит медленнее, в результате чего образование термостойкого реактопласта происходит параллельно с разложением новолачной основы олигомера.
Таблица 11.
Механические свойства углепластиков, полученных методом вакуумной инфузии, углеродная ткань 22502
| Углепластик | Условия отверждения | $\sigma _{{11}}^{ + }$, МПa | E, ГПa | $\sigma _{{11}}^{ - }$, МПa | E, ГПa | τ13, МПa | τ12, MПa | E, ГПa |
|---|---|---|---|---|---|---|---|---|
| растяжение | сжатие | межслоевой сдвиг | сдвиг в плоскости | |||||
| ФНС-2 [109] | 8 ч, 375°C | 669 (25°C) 691 (300°C) | – | 429 (25°C) 382 (300°C) | – | 37 (25°C) 35 (300°C) |
78 (25°C) 72 (300°C) |
– |
| 14+АФБ [124] | 8 ч, 375°C | 548 | 67 | 325 | 63 | 32 | 57 | – |
| ФНС-3 [125] | 8 ч, 375°C | 657 | 65 | 513 | 57 | 39 | 77 | 5.8 |
| 715 | 69 | 623 | 64 | 36 | 85 | 5.7 | ||
| ФНС-4 [107] | 8 ч, 375°C | 685 | 61 | 538 | 60 | 44 | 75 | 5.7 |
| 8 ч, 330°C | 834 | 60 | 852 | 67 | 53 | 106 | 6.0 | |
В работе [121] для улучшения механических характеристик углепластиков, в частности снижения хрупкости материала, было предложено использовать олигомеры полиэфирэфиркетона (ПЭЭК) с Mn ⁓ 4 × 103 в комбинации с новолак-фталонитрильным олигомером 46 и диамином 4,4'-ДДС в качестве инициатора полимеризации.

Внесение высокомолекулярного термопласта ПЭЭК для улучшения ударных характеристик бис-фталонитрильных связующих невозможно из-за его нерастворимости, поэтому был предложен подход с использованием олигомеров 47 и 48, способных образовывать с 46 единую трехмерную сетку за счет наличия реакционноспособных концевых групп. Углепластики готовили пропиткой углеродной ткани T-300 (50 мас. %) раствором связующего в ТГФ с последующим отверждением при 330°C в оснастке.
Результаты механических испытаний показали улучшение свойств композита с увеличением содержания 47 до 20%, при этом при повышении его содержания в связующем до 30% наблюдалось падение прочности на изгиб, прочности при межслоевом сдвиге и ударной прочности по Изоду. Объяснение данному феномену в статье не приводится. Тем не менее, ожидаемый эффект увеличения ударопрочных характеристик композита за счет добавления термопластичных молекул был достигнут, причем за счет включения термопластичных фрагментов в трехмерную структуру реактопласта удалось сохранить высокую термостойкость матриц.
Исследование межфазной границы и механизма адгезии углеродного волокна и фталонитрильной матрицы проводили в работе [122]. Для этого получали микропластики из углеродного волокна (аналог Т700, произведенный в Китае) и фталонитрильного связующего на основе 4-(4-аминофенокси)фталонитрила 9 и поли(арилен эфир нитрила), точный состав которого не раскрывается в статье. Связующее для исследования постотверждали при максимальной температуре 320, 350 или 380°C в течение 15 мин, для сравнения использовали микропластик с эпоксидной смолой. Результаты изучения межфазной прочности при сдвиге показали, что адгезия фталонитрильного связующего к волокну возрастает с увеличением температуры постотвержения за счет образования большего числа ароматических структур при сшивке: с 56 до 78 МПа при повышении температуры постотверждения с 320 до 380°C. Поверхность углеродного волокна состоит из сопряженных графеновых фрагментов, и увеличение адгезионной прочности объясняется π-стэкингом. В силу того, что образующиеся гетероциклические структуры изонидолина, триазина и фталоцианина имеют электронно-дефицитные сопряженные системы, возрастает энергия взаимодействия с относительно донорной поверхностью углеродного волокна. Этим объясняется более сильная адгезия фталонитрильной матрицы к волокну, чем эпоксидной. Кроме того, авторы отмечают, что причиной более высокой адгезии фталонитрила к волокну может служить возможность взаимодействия фенольных гидроксилов и карбоксилов поверхности волокна (образовавшихся в результате его окисления в процессе производства) с компонентами связующего.
Возможный механизм взаимодействия фталонитрильного связующего с поверхностью углеродного волокна за счет образования ковалентных связей [122] приведен ниже.

Углепластики, полученные по безрастворным технологиям
Возможность применения фталонитрильных связующих для формования ПКМ эффективными инжекционными методами определяется прежде всего их вязкостью при температуре пропитки [7]. Для получения связующего, обладающего достаточно низкой вязкостью расплава при температуре переработки (не выше 500 мПа с), необходимо получить мономеры с низкой температурой плавления. Приемлемыми для пропитки можно считать температуры в интервале 120–150°, предельной – 180°C, поскольку это предельная температура для стандартных вспомогательных материалов, применяемых при формовании, например, эпоксидных смол.
В цикле работ [107, 109, 119, 123, 125], выполненных группой российских ученых, впервые продемонстрирована возможность получения углепластиков с фталонитрильными матрицами методом вакуумной инфузии и пропитки в форме под давлением, а также описывается безрастворный способ получения препрегов и композитов из них. Вакуумная инфузия имеет целый ряд преимуществ по сравнению с препреговыми технологиями: не требует автоклава, практически не ограничивает размер и форму деталей, не ограничивает срок годности исходных материалов. Но в то же время накладывает жесткие требования к связующим, такие как низкая вязкость и высокая жизнестойкость при температуре пропитки. Именно последнее и являлось основной причиной невозможности применения фталонитрилов в инжекционных методах формования композитов. Благодаря использованию бисфталонитрилов с фосфатным мостиком [109] удалось получить связующие со значениями вязкости 300–600 мПа с при 150°C, подходящие для вакуумной инфузии. Связующее ФНС-2, состоящее из смеси 2 и 32 с м-АФБ в качестве отвердителя обладало вязкостью расплава около 600 мПа с, и полученный из него углепластик демонстрировал низкие механические свойства из-за плохой смачиваемости волокна матрицей (прочность на растяжение 487 МПа, прочность на сжатие 341 МПа). Для снижения вязкости применили реактивный разбавитель 14, что позволило улучшить механические свойства композита (табл. 11). Особо стоит отметить сохранение механических свойств при температуре 300°C, причем прочность на растяжение композитов оказалась даже больше при повышенной температуре за счет снятия части внутренних напряжений с матрицы, возникших при охлаждении после постотверждения, и увеличения максимального удлинения при разрушении, что выразилось в росте доли прочности углеродного волокна в формировании прочности ПКМ. В работе [124] мономер 14 использовали в качестве основного компонента связующего для получения композита. Несмотря на то, что этот мономер способен к термической гопомолимеризации за счет инициирования полимеризации продуктами перегруппировки Кляйзена [73], для увеличения скорости гелеобразования в состав связующего был введен диамин м-АФБ. Благодаря низкой вязкости (45 мПа с при 130°C) удалось снизить температуру пропитки углеродной ткани до 130°C, однако полученные углепластики обладали плохими механическими свойствами из-за сильной усадки связующего, обусловленной механизмом полимеризации. Кроме того, высокое тепловыделение при полимеризации не позволяло рассматривать это связующее как перспективное для получения крупноразмерных деталей.
В работе [125] изучали связующее ФНС-3 на основе тех же мономеров 2 и 32, однако использовали пластификатор, позволивший снизить вязкость связующего до 50 мПа с при 150°C. Также была снижена температура отверждения до 180°C, после чего углепластики постотверждали при 375°C. Изучали влияние эпоксидного аппрета, наносимого на коммерческое углеродное волокно, на механические свойства ПКМ. В качестве образца сравнения использовали углепластик, полученный из исходного углеродного волокна. Был сделан вывод о том, что при использовании аппретированного волокна механические свойства углепластика оказываются несколько ниже, что вызвано разложением аппрета на границе волокно–матрица при постотверждении (табл. 11). При этом, судя по всему, часть аппрета смывается потоком связующего во время инфузии, что объясняет относительно невысокий рост механических свойств после предварительного снятия аппрета.
После открытия нового класса реактивных разбавителей [110], а именно бисбензонитрилов, удалось разработать состав связующего ФНС-4, которое позволило снизить температуру вакуумной инфузии до 120°C [107, 123]. Кроме того, в работе исследовали влияние температуры постотверждения на механические свойства ПКМ. Как и в работе [112], была выявлена тенденция, показывающая образование микротрещин поперек направлению армирования, вызванное химической усадкой материала при 375°C (рис. 6).

Общий уровень механических свойств углепластиков, полученных на ткани методом вакуумной инфузии, оказывается выше (табл. 11), чем полученных по растворной препреговой технологии (табл. 10). Это может быть связано с недостаточно качественной пропиткой наполнителя и присутствием остаточного растворителя в препрегах. В работе [123] были получены препреги путем нанесения тонких пленок связующего на углеродную ткань при помощи горячего утюга. Такая техника максимально приближена к технике промышленных пропиточных линий. Из препрега были приготовлены образцы методом горячего прессования по следующей программе отверждения: 180°C, 8,75 бар, постотверждение 330°C (рис. 7). Механические свойства таких углепластиков уступали аналогичным образцам, полученным вакуумной инфузией (табл. 11), что свидетельствует о лучшей пропитке наполнителя при использовании инжекционных методов. При этом прочность увеличилась по сравнению с растворной технологией получения препрегов.

Температурную стойкость углепластиков можно оценивать по сохранению величины прочности при межслоевом сдвиге, так как данная величина определяется матрицей и силой межфазного взаимодействия, а не волокном. Композиты, полученные по безрастворным технологиям продемонстрировали лучшее сохранение свойств при температурах вплоть до 400°С (табл. 12) по сравнению c растворными препрегами (табл. 10), что может быть связано с худшим качеством (низкая степень пропитки и пористость) композитов, полученных через раствор связующего.
Таблица 12.
Прочность при межслоевом сдвиге композитов с матрицей на основе ФНС-4, полученных по безрастворным технологиям [123]
| T, °C | Вакуумная инфузия | Углеродный препрег | Стеклянный препрег, горячее прессование | ||
|---|---|---|---|---|---|
| вакуумное формование | горячее прессование | ||||
| 8 ч, 330°C | 8 ч, 375°C | 8 ч, 330°C | 8 ч, 330°C | 8 ч, 330°C | |
| 25 | 53 | 44 | 41 | 41 | 47 |
| 300 | 50 | 43 | 35 | 41 | 38 |
| 350 | 46 | 41 | 31 | 33 | 28 |
| 400 | – | 38 | 21 | 26 | – |
Стеклопластики с фталонитрильными матрицами
Большое количество работ посвящено получению стеклопластиков с фталонитрильными матрицами. Несмотря на то, что стеклопластики в отличие от углепластиков не могут обеспечить сохранение прочности при экстремальных температурах (выше 350°С), они обладают радиопрозрачностью и высокой диэлектрической проницаемостью. В работе [126] впервые были описаны стеклопластики с фталонитрильными матрицами на основе мономера 1. Помимо растворного метода получения стеклянного препрега продемонстрирована возможность применения порошковой вакуумной инфузии для получения композита. Стеклопластик получали, распределяя порошок связующего между слоями стеклянной ткани в вакуумном пакете. Связующее было изготовлено из мономера 1 и инициатора м-АФБ, и демонстрировало необходимые для пропитки значения вязкости менее 0.6 Па с только при 260°C, несмотря на использование форполимера. Полимеризация фталонитрильных групп при этой температуре происходит с существенной скоростью, в связи с чем количество инициатора м-АФБ было снижено до 1.68% для увеличения времени набора вязкости. Стоит отметить высокую чувствительность связующего к содержанию инициатора: при использовании 1.8% м-АФБ не удавалось полностью пропитать композитную панель, а при 1.71% м-АФБ получали панель неравной толщины. Сравнение постотвержденных композитных панелей, полученных порошковой инфузией и формованием препрега, показало близость величин прочности при межслоевом сдвиге. Сами авторы делают вывод о сложности формования ПКМ пропиткой сухого наполнителя при 260°C, и в подавляющем большинстве последующих работ приводятся только технологии получения композитов через растворные препреги.
В работе [127] изучали стеклопластики на основе бисфталонитрила 3, только в качестве инициатора полимеризации использовали бисфенол А (БФА) при мольном отношении к мономеру 2 : : 0.5, 2 : 1, 2 : 1.5 и 2 : 2. Матрицы с низким соотношением мономер: БФА демонстрировали более высокие механические и термические свойства. Так, при снижении соотношения с 2 : 0.5 до 2 : 1.5 возрастали прочность и модуль упругости при изгибе с 525 МПа и 29 ГПа до 633 МПа и 33 ГПа соответственно. При дальнейшем увеличении соотношения 3 : БФА наблюдали снижение механических свойств в пределах погрешности измерений. Тем не менее, авторы объясняют это присутствием непрореагировавшего БФА в трехмерной полимерной матрице, ввиду чего по их предположению была нарушена целостность границы волокно–матрица. Композиты также подвергались старению при 300°C в течение 6 ч, в результате которого прочность при изгибе не изменилась, а прочность при растяжении упала с 410 до 355 МПа. При сравнении свойств стеклопластиков с матрицей на основе 1 и 3, можно сделать вывод, что использование 1 позволяет получать ПКМ с лучшими механическими характеристиками (табл. 9), однако с точки зрения технологии использование смесей 3/БФА при низких соотношениях с мономером удешевляет и упрощает производство связующего.
Добавление мелкодисперсного (0.5–1.0 мкм) оксида кремния для усиления межфазной границы в композитах со стеклотканью и матрицей на основе 3 изучали в работе [128]. Оксид кремния в количестве 4–16% вводили в связующее на стадии форполимеризации (в качестве инициаторов использовали ДДС и 4,4'-дигидроксибифенил (ДГБФ)) в растворе в N-метилпирролидоне при 200°C и перемешивании со скоростью 600 об/мин. После этого полимер осаждали и заново растворяли в МЭК для получения препрегов на стеклянной ткани, которые формовали в прессе при давлении 10 МПа. Введение оксида кремния в состав связующего привело к увеличению прочности и модуля при изгибе (табл. 9), а также к лучшему сохранению механических свойств после старения композита на воздухе при 400°C. После 24 ч прочность при изгибе композита без оксида кремния упала ниже 40% от изначальной, а наполненного композита (16% SiO2) сохранилась чуть менее, чем на 60%. Авторы объясняют этот феномен более высокой термоокислительной стабильностью композитов с добавкой оксида кремния, основываясь на данных о потере массы по ТГА. Увеличение температуры потери массы авторы связывают с тем, что оксид кремния изолирует подвод тепла к связующему и также затрудняет колебания макромолекул, ведущие к разложению. Кроме того, снижение доли полимерной матрицы в композите при введении дополнительного наполнителя естественным образом увеличивает температуру потери массы всего композита, так как только матрица теряет массу в этих условиях.
Существенного увеличения прочности при межслоевом сдвиге стеклопластиков удалось достичь в результате использования связующего, состоящего из смеси 1 и мономера 49, вошедшего в состав связующего в работе [129], со смешанным отвердителем ZnCl2–ДДС.
В ряде работ получали композиты из связующих ФНС-5, состоящих из смесей фталонитрилов с бензоксазинами и циановыми эфирами [130–136].

Во всех случаях стеклопластики формовали из препрегов, полученных через раствор связующих. В работе [135] использовали полиариловый эфир, содержащий боковые кислотные и цианогруппы, за счет чего было обеспечено усиление межфазной границы стекловолокно–фталонитрильная матрица.
Композит, полученный из безрастворного препрега описывается в работе [123]. Так как для формования препрега не важна текучесть связующего, для снижения внутренних напряжений, возникающих в реактопласте после отверждения, в состав легкоплавкого связующего ФНС-4 была введена кварцевая мука в соотношении 10 : 4. Светло-зеленую пасту ФНС-4П использовали для изготовления препрега [123]. Композиты, полученные горячим прессованием этого препрега, продемонстрировали схожий уровень прочности при межслоевом сдвиге с материалами, описанными ранее [126, 134], но по величине прочности при растяжении (σ+ = 296 МПа, E = 31) уступали стеклопластику с матрицей на основе мономера 3 (σ+ = 410 МПа, табл. 9). Это можно объяснить снижением максимального удлинения при растяжении из-за введения в матрицу кварцевой муки. При этом прочность при сжатии материала находится на уровне немногим ниже 500 МПа с модулем упругости 29 ГПа.
Единственный пример получения композитов с рубленным стекловолокном опубликован в работе [137]. Связующее на основе олигомера 50
смешивали в двухшнековом экструдере с рубленным стеклянным волокном, после чего отверждали в форме с максимальной температурой 320°C и давлении 1.1 МПа. Прочность на изгиб композитов ожидаемо возрастала с увеличением доли волокна и достигла максимального значения 280 МПа для образца с содержанием наполнителя 40%. Также с повышением содержания волокна увеличивалась прочность на растяжение и ударная прочность.
Фталонитрилы, армированные кварцевой тканью
В последние годы наметилась тенденция к получению фталонитрильных композитов, армированных кварцевым волокном. Кварц, также как и стекло, обладает радиопрозрачностью, но при этом кварцевые волокна прочнее и сохраняют механические свойства при более высокой температуре. В работах [138–140] изучали композиты со связующими на основе флуоренсодержащих олигомеров.
Из-за того, что падение вязкости происходило выше 230°C до уровня 4–14 Па с, для получения препрегов использовали растворы связующих в ацетоне. Композит формовали методом горячего прессования под давлением 5–6 МПа, после чего постотверждали в свободном виде.
В работе [138] исследовали влияние соотношения гидрохиноновых звеньев к бисфенол-флуореновым в составе олигомера 51 на механические свойства композита. Было показано, что композиты на основе связующего, полученного в синтезе с исходным соотношением гидрохинона и бисфенолфлуорена 1 : 1 и соотношением фенолов к дихлорбензонитрилу 3 : 4 (Mn = 1309, Ð = 1.98) продемонстрировали наибольшую прочность при изгибе и межслоевом сдвиге. Этот факт авторы связывают с химическим составом олигомера, а именно с оптимальной комбинацией жесткости линейных участков, обеспечиваемой флуореновыми звеньями, и длиной линейных участков в трехмерной сетке (т.е. с Mn исходных олигомеров), обеспечивающей нужную степень сшивки. Было показано, что увеличение степени сшивки полимерной матрицы (при повышении температуры постотверждения) приводит к снижению механической прочности материала за счет возникновения в матрице внутренних напряжений. В статье было подробно изучено влияние химического состава олигомера 51 на свойства композита и показаны возможности для “настройки” свойств матрицы для получения нужных свойств композита.
Чтобы улучшить технологичность олигомеров, в работе [140] в структуру флуоренсодержащего олигомера было решено ввести силоксановые мостики, ранее приводившие к существенному снижению вязкости фталонитрилов [13, 101, 103–105, 141]. Олигомер 52

был получен с Тс = 147°C, Мn = 1104, Ð = 1.42 и содержал 2 мас. % кремния. Несмотря на присутствие силоксановых мостиков вязкость мономера опускалась до минимальных значений 4–10 Па с при температурах выше 230°C, при этом в присутствии 3% п-БАФС рост вязкости, связанный с полимеризацией фталонитрильных групп, начинался при указанной температуре. По этой причине для получения композита был выбран растворный метод пропитки кварцевой ткани.
Полученные композиты уступали по механическим свойствам предшественникам (табл. 13), но продемонстрировали низкую массовую скорость абляции, равную 0.0528 г/с, и устойчивость к возгоранию выше, чем у традиционных композитов с фенолформальдегидной смолой. В работе [140] для получения связующего использовали смеси классического мономера 2 с флуоренсодержащим олигомером 53 (5, 10 и 15 мас. %) и 9 в качестве отвердителя.
Таблица 13.
Механические свойства ПКМ с фталонитрильными матрицами, полученными из препрегов на основе кварцевых тканей
| Связующее/ наполнитель |
Постотверждение | Прочность при изгибе | τ13, МПа | Литература | |
|---|---|---|---|---|---|
| σf, МПа | Е, ГПа | ||||
| 51+ БАФС | 350°C, 5 ч | 1000 | 33 | 55 | [138] |
| 51 + БАФС | 375°C, 5 ч | 942 | 35 | 40 | [138] |
| 51 + БАФС | 400°C, 5 ч | 550 | 24 | 28 | [138] |
| 52 + БАФС | 375, 5 ч | 550 (25°C), 357 (400°C) | 24.5 (25°C), 27.4 (400°C) | 34 (25°C), 18 (400°C) | [139] |
| 53 + 2 + 9 | 375, 5 ч | 535–855 | 22–37 | 30–42 | [140] |
| 56 + ДДС | 375, 8 ч | 300–340 | 18–24 | 20–28 | [142] |

Таким образом удалось получить смеси, вязкость которых выходила на минимальные значения уже при 185°C и при этом сами значения вязкости (80–120 мПа ∙ с при 200°C) были немногим выше, чем для смеси 2 и 9, что согласуется с температурой плавления 2, составляющей 180–185°C [50]. Из-за такого реологического поведения связующего препреги получали растворным методом, после чего формовали образцы ПКМ методом горячего прессования. Наибольшие прочность и модуль при изгибе (855 МПа и 37 ГПа соответственно), прочность при межслоевом сдвиге и ударную прочность продемонстрировал ПКМ с содержанием олигомера в матрице 5%. Авторы связывают это с падением плотности сшивки матрицы (за счет роста длины линейных участков молекул) и с увеличенной вязкостью расплава по мере роста доли олигомера в связующем. В то же время прочность при межслоевом сдвиге, определяемая в большей степени связующим, падает равномерно при повышении доли олигомера, а модуль упругости матрицы при этом растет. Можно предположить, что с увеличением модуля упругости снижается максимальное удлинение при разрыве, за счет чего и наблюдается резкое падение прочности при изгибе (комбинация растяжения и сжатия) и плавное падение прочности при межслоевом сдвиге. При значениях растяжения при разрыве у связующего ниже, чем у волокна, при растягивающих нагрузках вклад матрицы в прочность материала возрастает по отношению ко вкладу волокна. Тем не менее, подход, описанный в статье, позволил получить ПКМ на кварцевой ткани с наилучшими механическими характеристиками.
Трифункциональные фталонитрильные олигомеры были впервые применены для получения композитов на кварцевой ткани в работе [142]. Композиты получали прессованием под давлением 3–5 МПа при 250°C из препрега, полученного пропиткой кварцевой ткани раствором олигомеров 54–56 с 5% ДДС.
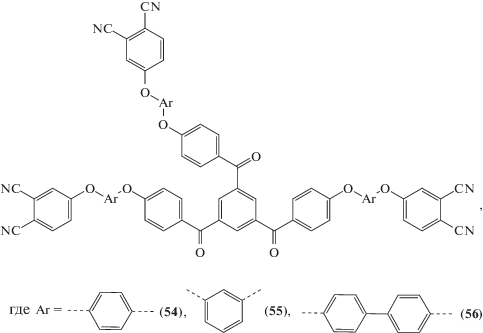
Механические свойства композитов (табл. 13) оказались ниже, чем для составов, исследованных ранее, по всей видимости, из-за увеличения степени сшивки матрицы за счет использования трехфункциональных фталонитрилов. Различия в механических свойствах ПКМ с разными олигомерами лежали в пределах погрешности, хотя стоит отметить, что олигомер с бифенильным линкером 56 более жесткий, чем два других композита. Диэлектрические свойства материалов в гигагерцовом диапазоне оказались на том же уровне (табл. 14), что и для композитов, описанных в работе [139]. Интересно отметить, что диэлектрические свойства практически не меняются в зависимости от температуры в диапазоне 25–500°C. Этот факт согласуется с высокой жесткостью трехмерной сетки матрицы и отсутствием расстекловывания до 500°C.
Композиты с фталонитрильными матрицами для радиационной защиты
Следует отметить цикл работ, посвященных получению композитов с фталонитрильными матрицами на основе мономера 3 с отвердителем 9 для радиационной защиты и баллистических применений [143–146]. Поглощение самой матрицы и композита с базальтовым волокном исследовали в работе [143]. На образец направляли γ-излучение интенсивностью 1.17 МэВ, и измеряли интенсивность сигнала, прошедшего через пластину. Линейный коэффициент затухания μ находили по формуле –ln(I/I0) = μx, где I/I0 – отношение интенсивности сигнала, прошедшего через образец, к начальной интенсивности, а х – толщина образца. За счет высокого содержания ароматических фрагментов фталонитрильная матрица имела наибольший коэффициент затухания, чем другие термореактивные смолы, при этом композиты оказались худшей защитой от гамма-излучения, по всей видимости, за счет прозрачности базальта для гамма-лучей. При этом наполнение фталонитрильной матрицы наночастицами вольфрама позволило на порядок увеличить линейный коэффициент затухания [144]. В связи с этим в работе [145] для одновременного увеличения механических свойств матрицы и создания защиты от излучения исследовали сандвич-конструкции, состоящие из фталонитрильной матрицы с добавлением наночастиц карбида вольфрама (WC), армированной углеродным волокном, размещенной между двух пластин алюминий-литиевого сплава. Линейный коэффициент затухания увеличивался с повышением содержания наночастиц карбида вольфрама как за счет увеличения плотности материала, так и за счет поглощения γ-излучения самим карбидом вольфрама. При этом были достигнуты достаточно высокие значения μ для различных композиционных материалов (табл. 15).
Таблица 15.
Экранирующие свойства материалов по отношению к γ-излучению мощностью 1.17 МэВ, источник Co-60
| Материал | Линейный коэффициент затухания, μ, см–1 | Литература |
|---|---|---|
| 3 + 9 | 0.071 | [143] |
| 3 + 9/Базальтовая ткань | 0.020 | [143] |
| 3 + 9/40% W | 0.141 | [144] |
| 3 + 9/Al–Li сплав2 | 0.270 | [145] |
| 3 + 9/Al–Li сплав + 10% WC | 0.330 | [145] |
| 3 + 9/Al–Li сплав + 20% WC | 0.370 | [145] |
| Эпоксидная смола + Pb2O3 | 0.103 | [147] |
| Полиэфир + 50% Fe2O3 | 0.160 | [148] |
| Свинец | 0.632 | [148] |
В заключение отметим, что в подавляющем большинстве работ композиты получают по растворной технологии. С одной стороны, это дает возможность сравнивать влияние матриц на свойства композитов, но с другой, из-за часто невысокого уровня описания эксперимента, когда авторы упускают важные детали методик, бывает невозможно объективно оценить полученные данные. Кроме того, большинство работ так или иначе направлено именно на создание новых связующих, но не во всех случаях изучается прочность при сжатии и межслоевом сдвиге, которая в большей степени определяется связующим и прочностью межфазной границы, чем волокном [149] . Таким образом, методология работы с фталонитрильными связующими до сих пор находится в стадии развития, и с ростом числа публикаций и появлением первых коммерческих продуктов стоит ожидать улучшения глубины и разнообразия новых исследований.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 19-13-50529).
Список литературы
Кива Д.С. // Авиационно-космическая техника и технология. 2014. Т. 6. № 113. С. 5.
Asim M., Saba N., Jawaid M., Nasir M., Pervaiz M., Alothman O.Y. // Curr. Anal. Chem. 2018. V. 14. № 3. P. 185.
Pizzi A., Ibeh C.C. Phenol–Formaldehydes // Handbook of Thermoset Plastics / Ed. by H. Dodiuk. New York: William Andrew Publ., 2014. P. 13.
Bulgakov B., Kalugin D., Babkin A., Makarenko I., Malakho A., Kepman A., Avdeev V., Garadja N., Rogozin A. // Can. J. Chem. Eng. 2016. V. 94. № 1. P. 46.
Gibson G. // Brydson’s Plastics Materials. Oxford: Butterworth Heinemann, 2017. P. 773.
Cicala G., Mannino S., Latteri A., Ognibene G., Saccullo G. // Adv. Polym. Technol. 2018. V. 37. № 6. P. 1868.
ASM Handbook / Ed. by D.B. Miracle, S.L. Donaldson. ASM International: Novelty, OH, 2001. V. 21.
Bauer M., Wurzel R., Uhlig C., Bauer J. Pat. 20080119630 USA. 2008.
Chemistry and Technology of Cyanate Ester Resins / Ed. by I. Hamerton. Heidelberg: Springer Netherlands, 2012.
Uhlig C., Bauer M., Bauer J., Kahle O., Taylor A.C., Kinloch A.J. // React. Funct. Polym. 2018. V. 129. P. 2.
Бабкин А.В., Эрдни-Горяев Э.М., Солопченко А.В. К.А.В. // Изв. вузов. Химия и хим. технология. 2015. Т. 58. № 1. С. 54.
Evsyukov S.E., Pohlmann T., Stenzenberger H.D. // Polym. Adv. Technol. 2015. V. 26. № 6. P. 574.
Babkin A.V., Zodbinov E.B., Bulgakov B.A., Kepman A.V., Avdeev V.V. // Eur. Polym. J. 2015. V. 66. P. 452.
Ji M. // Advanced Polyimide Materials / Ed. by S.-Y. Yang. Amsterdam: Elsevier, 2018. P. 93.
Lubowitz H.R., Sheppard C.H. Pat. 5116935 USA. The Boeing Company, 1992.
Mittal K.L. // Polyimides and Other High Temperature Polymers: Synthesis, Characterization and Applications. Boca Raton: CRC Press, 2009.
Stenzenberger H.D. // High Performance Polymers. Heidelberg: Springer-Verlag, 2018. P. 165.
Zia K.M., Bhatti H.N., Ahmad Bhatti I. // React. Funct. Polym. 2007. V. 67. № 8. P. 675.
Atiqah A., Mastura M., Ali B., Jawaid M., Sapuan S. // Curr. Org. Synth. 2017. V. 14. № 2. P. 233.
Pater R.H. // Polym. Eng. Sci. 1991. V. 31. № 1. P. 20.
Keller T.M., Griffith J.R. Pat. 4234712A USA. 1980.
Keller T.M., Griffith J.R. Pat. 4259471A USA. 1979.
Sastri S.B., Keller T.M. // J. Polym. Sci., Polym. Chem. 1999. V. 37. № 13. P. 2105.
Keller T.M. Pat. 5003039A USA. 1991.
Keller T.M. // J. Polym. Sci., Polym. Chem. 1988. V. 26. № 12. P. 3199.
Phthalonitrile Resins and Composites: Properties and Applications / Ed. by M. Derradji, J. Wang, W. Liu. Amsterdam: Elsivier, 2018.
Dent C.E., Linstead R.P. // J. Chem. Soc. 1934. P. 1027.
Griffith J.R., O’Rear J.G., Walton T.R. // Advanced Chemistry / Ed. by N.A.J. Platzer. New York: Am. Chem. Soc., 1975. P. 458.
Keller T.M., Griffith J.R. // Am. Chem. Soc. 1980. P. 25–34.
Keller T.M. // J. Polym. Sci., Polym. Chem. 1988. V. 26. № 12. P. 3199.
Bulgakov B.A., Babkin A.V., Dzhevakov P.B., Bogolyubov A.A., Sulimov A.V., Kepman A.V., Kolyagin Y.G., Guseva D.V., Rudyak V.Y., Chertovich A.V. // Eur. Polym. J. 2016. V. 84. P. 205.
Bulgakov B.A., Sulimov A.V., Babkin A.V., Kepman A.V., Malakho A.P., Avdeev V. V. // J. Appl. Polym. Sci. 2017. V. 134. № 18.
Sheng H., Peng X., Guo H., Yu X., Tang C., Qu X., Zhang Q. // Mater. Chem. Phys. 2013. V. 142. № 2–3. P. 740.
Zhao F., Liu R., Kang C., Yu X., Naito K., Qu X., Zhang Q. // RSC Adv. 2014. V. 4. № 16. P. 8383.
Peng X., Sheng H., Guo H., Naito K., Yu X., Ding H., Qu X., Zhang Q. // High Perform. Polym. 2014. V. 26. № 7. P. 837.
Yu X.-Y., Naito K., Kang C., Qu X.-W., Zhang Q.-X. // Macromol. Chem. Phys. 2013. V. 214. № 3. P. 361.
Han Y., Tang D., Wang G., Guo Y., Zhou H., Qiu W., Zhao T. // Eur. Polym. J. 2019. V. 111. P. 104.
Han Y., Wang G., Qiu W., Guo Y., Sun Y., Zhang Y., Zhou H., Zhao T. // Asian J. Org. Chem. 2018.
Ting R.Y., Keller T.M., Price T.R., Poranski C.F. // Cyclopolymerization and Polymers with Chain-Ring Structures / Ed. by G.B. Butler, J.E. Kresta. Washington: Am. Chem. Soc., 1982. P. 337.
Keller T.M., Griffith J.R. Pat. 4223123 USA. 1980.
Keller T.M., Griffith J.R. // Chem. Informationsd. 1979. V. 10. № 26.
McKeown N.B. // J. Mater. Chem. 2000. V. 10. № 9. P. 1979.
Snow A.W., Griffith J.R., Marullo N.P. // Macromolecules. 1984. V. 17. № 8. P. 1614.
Jia K., Zhao R., Zhong J., Liu X. // J. Mater. Sci. Mater. Electron. 2010. V. 21. № 7. P. 708.
Sumner M.J., Sankarapandian M., McGrath J.E., Riffle J.S., Sorathia U. // Polymer (Guildf). 2002. V. 43. № 19. P. 5069.
Keller T.M., Price T.R., Griffith J.R. // Am. Chem. Soc. Div. Org. Coat. Plast. Chem. Prepr. 1980. V. 43. P. 804.
Sastri S.B., Keller T.M. // J. Polym. Sci. Part A Polym. Chem. 1998. V. 36. № 11. P. 1885.
Terekhov V.E., Morozov O.S., Afanaseva E.S., Bulgakov B.A., Babkin A.V., Kepman A.V., Avdeev V.V. // Mendeleev Commun. 2020. V. 30. № 5. P. 671.
Hinkley J.A. // J. Appl. Polym. Sci. 1984. V. 29. № 11. P. 3339.
Keller T.M., Dominguez D.D. // Polymer (Guildf). 2005. V. 46. № 13. P. 4614.
Dominguez D.D., Jones H.N., Keller T.M. // Polym. Compos. 2004. V. 25. № 5. P. 554.
Sastri S.B., Armistead J.P., Keller T.M. // Polym. Compos. 1996. V. 17. № 6. P. 816.
Siegl W.O. // J. Org. Chem. 1977. V. 42. № 11. P. 1872.
Yu G., Liu C., Li X., Wang J., Jian X., Pan C. // Polym. Chem. 2012. V. 3. № 4. P. 1024.
Warzel M., Keller T. // Polymer (Guildf). 1993. V. 34. № 3. P. 663.
Zeng K., Zhou K., Tang W.R., Tang Y., Zhou H.F., Liu T., Wang Y.P., Zhou H.B., Yang G. // Chinese Chem. Lett. 2007. V. 18. № 5. P. 523.
Zhou H., Badashah A., Luo Z., Liu F., Zhao T. // Polym. Adv. Technol. 2011. V. 22. № 10. P. 1459.
Carnelley T. // Philos. Mag. Ser. 5. 1882. V. 13. № 79. P. 112.
Aleshkevich V.V., Babkin A.V., Avdeev V.V. // IOP Conf. Ser. Mater. Sci. Eng. 2019. V. 683. P. 12023.
Zhang Z.-Q., Uth S., Sandman D.J., Foxman B.M. // J. Phys. Org. Chem. 2004. V. 17. № 9. P. 769.
Iling S.A., Ponomarev I.I., Kuznetsov V.V., Lokshin B.V., Korshak V.V., Vinogradova S.V. // Bull. Acad. Sci. USSR Div. Chem. Sci. 1983. V. 32. № 8. P. 1588.
Chen X., Liu J., Xi Z., Shan S., Ding H., Qu X., Zhang Q. // High Perform. Polym. Sage Publications. 2017. V. 29. № 10. P. 1209.
Wang J., Chen C., Chen X., Wang H., Yu X., Ma Y., Naito K., Zhang Q. // High Perform. Polym. 2018. V. 30. № 9. P. 1114.
Zeng K., Zhou K., Zhou S., Hong H., Zhou H., Wang Y., Miao P., Yang G. // Eur. Polym. J. 2009. V. 45. № 4. P. 1328.
Ma J.Z., Cheng K., Lv J.B., Chen C., Hu J.H., Zeng K., Yang G. // Chinese J. Polym. Sci. 2018. V. 36. № 4. P. 497.
Lyubimtsev A., Vagin S., Syrbu S., Hanack M. // Eur. J. Org. Chem. 2007. № 12. P. 2000.
Wang J., Hu J., Zeng K., Yang G. // RSC Adv. 2015. V. 5. № 127. P. 105038.
Hu Y., Weng Z., Qi Y., Wang J., Zhang S., Liu C., Zong L., Jian X. // RSC Adv. 2018. V. 8. № 57. P. 32899.
Yang Y., Min Z., Yi L. // Polym. Bull. 2007. V. 59. P. 185.
Zhang B., Luo Z., Zhou H., Liu F., Yu R., Pan Y., Wang Y., Zhao T. // High Perform. Polym. 2012. V. 24. № 5. P. 398.
Zou X., Xu M., Jia K., Liu X. // J. Appl. Polym. Sci. 2014. V. 131. № 23.
Augustine D., Mathew D., Reghunadhan Nair C.P. // Polymer (Guildf). 2015. V. 60. P. 308.
Wang M., Wei L., Zhao T. // Eur. Polym. J. 2005. V. 41. № 5. P. 903.
Douglas W.E., Overend A.S. // Eur. Polym. J. 1993. V. 29. № 11. P. 1513.
Douglas W.E., Overend A.S. // Polymer (Guildf). 1993. V. 34. № 7. P. 1544.
Balaraman K., Kesavan V. // Synthesis (Stuttg). 2010. № 20. P. 3461.
Ji S., Yuan P., Hu J., Sun R., Zeng K., Yang G. // Polymer (Guildf). 2016. V. 84. P. 365.
Yuan P., Ji S., Hu J., Hu X., Zeng K., Yang G. // Polymer (Guildf). 2016. V. 102. P. 266.
Liu Z., Liu Y., Peng W., Lu Z., Liang B., Liu Y., Li C., Hu J., Zeng K., Yang G. // J. Appl. Polym. Sci. 2019. V. 136. № 42. P. 48134.
Lv J., Liu Z., Li R., Liu Y., Hu J., Zeng K., Yang G. // Thermochim. Acta. 2020. V. 683. P. 178446.
De Lijser H.J.P., Arnold D.R. // J. Phys. Chem. A. 1998. V. 102. № 28. P. 5592.
Shelton J.R., Uzelmeier C.W. // J. Am. Chem. Soc. 1966. V. 88. № 22. P. 5222.
Poirier R.H., Kahler E.J., Benington P. // J. Org. Chem. 1952. V. 17. № 11. P. 1437.
Keller T.M. // Chem. Mater. 1994. V. 6. № 3. P. 302.
Laskoski M., Dominguez D.D., Keller T.M. // J. Polym. Sci., Polym. Chem. 2005. V. 43. № 18. P. 4136.
Laskoski M., Schear M.B., Neal A., Dominguez D.D., Ricks-Laskoski H.L., Hervey J., Keller T.M. // Polymer (Guildf). 2015. V. 67. P. 185.
Laskoski M., Neal A., Keller T.M., Dominguez D., Klug C.A., Saab A.P. // J. Polym. Sci., Polym. Chem. 2014. V. 52. № 12. P. 1662.
Dominguez D.D., Keller T.M. // Polymer (Guildf). 2007. V. 48. № 1. P. 91.
Laskoski M., Dominguez D.D., Keller T.M. // Fire and Polymers IV / Ed. by C.A. Wilkie, G.L. Nelson. 2005. P. 378.
Dominguez D.D., Keller T.M. // High Perform. Polym. 2006. V. 18. № 3. P. 283.
Zhang H., Liu T., Yan W., Su Y., Yu H., Yang Y., Jiang Z. // High Perform. Polym. 2014. V. 26. № 8. P. 1007.
Laskoski M., Dominguez D.D., Keller T.M. // Polymer (Guildf). 2007. V. 48. № 21. P. 6234.
Zhao F., Liu R., Yu X., Naito K., Qu X., Zhang Q. // J. Appl. Polym. Sci. 2015. V. 132. № 39. P. 42606.
Zong L., Liu C., Zhang S., Wang J., Jian X. // Polymer (Guildf). 2015. V. 77. P. 177.
Zong L.S., Liu C., Guo Y., Wang J.Y., Jian X.G. // RSC Adv. 2015. V. 5. № 94. P. 77027.
Zu Y., Zong L., Wang J., Jian X. // Polymer (Guildf). 2019. V. 172. P. 372.
Liu Y., Ji P., Zhang Z., Yu X., Naito K., Zhang Q. // High Perform. Polym. 2019. V. 31. № 9–10. P. 1075.
Liu C., Wang J., Lin E., Zong L., Jian X. // Polym. Degrad. Stab. 2012. V. 97. № 3. P. 460.
Liu C., Lin E., Zong L., Liu C., Yu G., Wang J., Hu F., Weng Z., Jian X. // Polym. Bull. 2018. V. 75. № 3. P. 1037.
Wang H., Wang J., Guo H., Chen X., Yu X., Ma Y., Ji P., Naito K., Zhang Z., Zhang Q. // Polym. Chem. 2018. V. 9. № 8. P. 976.
Zhang Z., Li Z., Zhou H., Lin X., Zhao T., Zhang M., Xu C. // J. Appl. Polym. Sci. 2014. V. 131. № 20.
Babkin A.V., Zodbinov E.B., Bulgakov B.A., Kepman A.V., Avdeev V.V. // Polymer Science B. 2016. V. 58. № 3. P. 298.
Bulgakov B.A., Babkin A.V., Bogolyubov A.A., Afanas’eva E.S., Kepman A.V. // Russ. Chem. Bull. 2016. V. 65. № 1.
Laskoski M., Neal A., Schear M.B., Keller T.M., Ricks-Laskoski H.L., Saab A.P. // J. Polym. Sci., Polym. Chem. 2015. V. 53. № 18. P. 2186.
Dzhevakov P.B., Korotkov R.F., Bulgakov B.A., Babkin A.V., Kepman A.V., Avdeev V.V. // Mendeleev Commun. 2016. V. 26. № 6. P. 527.
Belsky K.S., Sulimov A.V., Bulgakov B.A., Babkin A.V., Kepman A.V. // Data Br. 2017. V. 13. P. 10.
Bulgakov B.A., Babkin A.V., Kepman A.V., Avdeev V.V. // Abstrs VII International Baekeland Symposium. Tarragona: Spain, 2019.
Bulgakov B.A., Sulimov A.V., Babkin A.V., Afanasiev D.V., Solopchenko A.V., Afanaseva E.S., Kepman A.V., Avdeev V.V. // Mendeleev Commun. 2017. V. 3. № 3. P. 257.
Bulgakov B., Sulimov A., Babkin A., Timoshkin I., Solopchenko A., Kepman A., Avdeev V. // J. Compos. Mater. 2017. V. 51. № 30. P. 4157.
Terekhov V.E., Aleshkevich V.V., Afanaseva E.S., Nechausov S.S., Babkin A.V., Bulgakov B.A., Kepman A.V., Avdeev V.V. // React. Funct. Polym. 2019. V. 139. P. 34.
Terekhov V.E., Aleshkevich V.V., Afanaseva E.S., Nechausov S.S., Babkin A.V., Bulgakov B.A., Kepman A.V., Avdeev V.V. // Data Br. 2020. V. 28. P. 104858.
Sun B.-G., Lei Q., Guo Y., Shi H.-Q., Sun J.-B., Yang K.-X., Zhou H., Li Y.-Q., Hu N., Wang H., Fu S.-Y. // Compos. Part B Eng. 2019. V. 166. P. 681.
Ting R.Y., Nash H.C. // Polym. Eng. Sci. 1981. V. 21. № 8. P. 441.
Laskoski M., Clarke J.S., Neal A., Keller T.M. 2017. V. 1700207. P. 1.
Yakovlev M.V., Morozov O.S., Afanaseva E.S., Bulgakov B.A., Babkin A.V., Kepman A.V. // React. Funct. Polym. 2020. V. 146. P. 104409.
Laskoski M., Shepherd A.R., Mahzabeen W., Clarke J.S., Keller T.M., Sorathia U. // J. Polym. Sci., Polym. Chem. 2018. V. 56. № 11. P. 1128.
Medjahed A., Derradji M., Zegaoui A., Wu R., Li B. // Mater. Sci. Technol. 2019. V. 35. № 6. P. 661.
Булгаков Б.А., Бабкин А.В., Кепман А.В., Авдеев В.В. Пат. 2695606C1. АО “ИНУМИТ” Россия, 2019.
Bulgakov B.A., Babkin A.V., Afanasieva E.S., Kep-man A.V. // Abstrs Conference “Proceedings of the Thermosetting Resins.” Berlin, Germany, 2018. P. 127.
Augustine D., Mathew D., Nair C.P.R. // Polym. Int. 2012. V. 62. № 7. P. 1068.
Augustine D., Mathew D., Reghunadhan Nair C. // Polym. Int. 2015. V. 64. № 1. P. 146.
Yin C., Sheng L., Yang Y., Liang G., Xing S., Zeng J., Xiao J. // RSC Adv. 2018. V. 8. № 67. P. 38210.
Timoshkin I.A., Aleshkevich V.V., Afanas’eva E.S., Bulgakov B.A., Babkin A.V., Kepman A.V., Avdeev V.V. // Polymer Science C. 2020. V. 62. № 2. P. 172.
Bulgakov B.A., Belsky K.S., Nechausov S.S., Afanaseva E.S., Babkin A.V., Kepman A.V., Avdeev V.V. // Mendeleev Commun. 2018. V. 28. № 1. P. 44.
Babkin A.V., Sulimov A.V., Bulgakov B.A., Kepman A.V. // SAMPE Conf. Procs. Seattle, USA, 2017. P. 713.
Sastri S.B., Armistead J.P., Keller T.M., Sorathia U. // Polym. Compos. 1997. V. 18. № 1. P. 48.
Chen Z., Guo H., Tang H., Yang X., Xu M., Liu X. // J. Appl. Polym. Sci. 2013. V. 129. № 5. P. 2621.
Yang X., Li K., Xu M., Liu X. // Compos. Part B Eng. 2018. V. 155. P. 425.
Wu Z., Wang S., Zong L., Li N., Wang J., Jian X. // High Perform. Polym. 2018. V. 30. № 6. P. 720.
Li X., Zhou F., Zheng T., Wang Z., Zhou H., Chen H., Xiao L., Zhang D., Wang G. // Polymers (Basel). 2019. V. 11. № 1.
Yang X.L., Li K., Xu M.Z., Liu X.B. // Chinese J. Polym. Sci. 2018. V. 36. № 1. P. 106.
Chen L., Ren D.X., Chen S.J., Pan H., Xu M.Z., Liu X.B. // Express Polym. Lett. 2019. V. 13. № 5. P. 456–468.
Luo Y., Xu M., Pan H., Jia K., Liu X. // Polym. Eng. Sci. 2016. V. 56. № 2. P. 150.
Ren D., Li K., Chen L., Chen S., Han M., Xu M., Liu X. // Compos. Part B Eng. 2019. V. 177. P. 107419.
Ren D., Lei Y., Pan H., Yan L., Xu M., Liu X. // J. Appl. Polym. Sci. 2018. V. 135. № 7. P. 45881.
Ren D., Chen L., Yuan Y., Li K., Xu M., Liu X. // Polymers (Basel). 2018. V. 10. № 10. P. 1128.
Zou Y., Liu X. // J. Appl. Polym. Sci. 2013. V. 129. № 1. P. 130.
Wang G., Han Y., Guo Y., Sun J., Wang S., Zhou H., Zhao T. // Eur. Polym. J. 2019. V. 113. P. 1.
Wang G., Han Y., Guo Y., Wang S., Sun J., Zhou H., Zhao T. // Ind. Eng. Chem. Res. 2019. V. 58. № 23. P. 9921.
Wang G., Guo Y., Han Y., Li Z., Ding J., Jiang H., Zhou H., Zhao T. // High Perform. Polym. 2020. V. 32. № 1. P. 3.
Wang M., Ning Y. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 14. P. 11933.
Zu Y., Zhang F., Chen D., Zong L., Wang J., Jian X. // Polymer (Guildf). 2020. V. 198. P. 122490.
Derradji M., Zegaoui A., Medjahed A., Dayo A.Q., Wang J., Arse Y.B., Liu W., Liu Y.-G. // Polym. Compos. 2019. V. 40. № S1. P. E912.
Derradji M., Zegaoui A., Xu Y.-L., Wang A., Dayo A.Q., Wang J., Liu W., Liu Y.-G., Khiari K. // Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. with Mater. Atoms. 2018. V. 421. P. 13.
Medjahed A., Derradji M., Zegaoui A., Wu R., Li B., Wang Y., Hou L., Zhang J., Zhang M. // Adv. Eng. Mater. 2019. V. 21. № 2. P. 1800779.
Derradji M., Mouloud A., Trache D., Zegaoui A., Medjahed A., Tarchoun A.F., Bessa W. // High Perform. Polym. 2020. V. 32. № 5. P. 550.
Eid G.A., Kany A.I., El-Toony M.M., Madbouly A.M., Bashter I.I., Gaber F.A. // Arab J. Nucl. Sci. Appl. 2013. V. 46. № 2. P. 226.
Eren Belgin E., Aycik G.A., Kalemtas A., Pelit A., Dilek D.A., Kavak M.T. // Radiat. Phys. Chem. 2015. V. 115. P. 43.
Gibson R.F. Principles of Composite Material Mechanics. CRC Press, 2016.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)