

Высокомолекулярные соединения (серия С), 2022, T. 64, № 2, стр. 218-228
СОПОЛИМЕРЫ НА ОСНОВЕ ПОЛИВИНИЛИДЕНФТОРИДА С ПРИВИТЫМИ ЦЕПЯМИ ПОЛИЭТИЛМЕТАКРИЛАТА: СИНТЕЗ, ТЕРМИЧЕСКИЕ И ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
Е. А. Клеймюк a, А. И. Косякова a, А. И. Бузин a, В. Г. Шевченко a, Ю. Н. Лупоносов a, *, С. А. Пономаренко a
a Институт синтетических полимерных материалов им. Н.С. Ениколопова Российской академии наук
117393 Москва, ул. Профсоюзная, 70, Россия
* E-mail: luponosov@ispm.ru
Поступила в редакцию 06.04.2022
После доработки 23.05.2022
Принята к публикации 01.07.2022
- EDN: WVQLEC
- DOI: 10.31857/S2308114722700133
Аннотация
Сополимеры на основе поливинилиденфторида c привитыми цепями полиэтилметакрилата впервые синтезированы методом фотоиндуцированной обратимо-дезактивационной радикальной полимеризации. В качестве исходных полимерных цепей для модификации были использованы двойные и тройные сополимеры винилиденфторида с хлортрифторэтиленом и трифторэтиленом. Исследовано влияние содержания привитых цепей на термические и диэлектрические свойства сополимеров. Показано, что увеличение содержания привитых цепей приводит к снижению температуры деструкции сополимеров, уменьшению диэлектрической проницаемости и диэлектрических потерь, но значительно улучшает их пленкообразующие свойства за счет уменьшения степени кристалличности.
В современном мире постоянно растет спрос на функциональные материалы и устройства, такие как электронный текстиль, дисплеи, наногенераторы, искусственная кожа и т.д., важными параметрами которых являются портативность, миниатюрный размер и экологичность. Отдельные полимерные материалы обладают уникальным набором свойств, среди которых возможность обратимого растяжения, биосовместимость, проводящие свойства, способность реагировать на внешние раздражители. Все это способствует развитию исследований в соответствующих направлениях полимерной науки и технологии [1, 2]. Важный класс функциональных материалов – сегнетоэлектрические полимеры, которые имеют спонтанные, но термодинамически стабильные состояния поляризации, переключаемые приложением достаточно сильного внешнего электрического поля. Исследования сегнетоэлектрических полимеров, обладающих уникальными электрическими свойствами, открывают большие возможности их применения в качестве активных материалов в энергонезависимой памяти, пьезоэлектрических или пироэлектрических сенсорах, электромеханических приводах или искусственных мышцах, наногенераторах электроэнергии, а также электронике с автономным питанием и т.д. [3–6].
Сегнетоэлектрические полимеры, такие как ПВДФ или его сополимер с трифторэтиленом (ТФЭ) (П(ВДФ–co–ТФЭ)) имеют большие пьезоэлектрические коэффициенты и высокий электрический выход, поэтому могут быть перспективными материалами для гибких наногенераторов [7]. Пьезоэлектрические и пироэлектрические эффекты делают сегнетоэлектрические полимеры подходящими функциональными материалами для создания на их основе гибких сенсоров давления и температуры, которые могут применяться для изготовления искусственной и портативной электроники. Такие полимеры способны давать достаточно быстрый отклик, изменяя свое состояние поляризации при незначительном внешнем воздействии [8].
Благодаря уникальным сегнетоэлектрическим и диэлектрическим свойствам, поливинилиденфторид является очень перспективным материалом для применения в гибкой и носимой электронике [9, 10]. Однако для проявления необходимых свойств нужно, чтобы ПВДФ находился в кристаллической β-фазе, перевод в которую является весьма энергозатратным процессом, поскольку требует проведения высокотемпературного отжига, поляризации под действием сильного электрического поля или высоких механических деформаций [11, 12]. Все это несовместимо со многими технологическими процессами производства тонкопленочных устройств.
Для уменьшения степени кристалличности и снижения температуры Кюри часто получают различные сополимеры на основе поливинилиденфторида [13]. Среди недостатков такого приема можно отметить невысокую диэлектрическую проницаемость, большие диэлектрические потери и необходимость снижения рабочих напряжений, которые значительно ограничивают возможности их использования, особенно в части носимой электроники.
Одним из вариантов решения некоторых проблем сополимеров на основе ПВДФ, а также наделением их новыми ценными функциями может стать прививка к ним полярных полимерных цепей. Данный подход позволит тонко настраивать свойства сополимеров, так как функциональные группы могут повышать полярность макромолекулы, уменьшать значения рабочего напряжения и остаточной поляризации, сохраняя при этом проявление сегнетоэлектрических свойств, что, несомненно, расширит их области применения. На сегодняшний день известны работы, проводимые в этой области, где в качестве привитых цепей используют ПАН, ПММА или ПС [14–17]. Однако привитые сополимеры с полиэтилметакрилатом (ПЭМА) остаются малоизученными [18]. Актуальным направлением исследования полимеров на основе ПВДФ является получение их композитов с различными наночастицами, например титанатом бария, с целью улучшения диэлектрических и сегнетоэлектрических свойств [19–21]. Важными вопросами в этом направлении остаются возможность получения высоконаполненных композитов и отсутствие агрегации наночастиц. В данном случае использование ПЭМА, содержащего более длинные по сравнению с ПММА алкильные фрагменты, может оказать положительный эффект и способствовать лучшему изолированию наночастиц друг от друга в полимерной матрице. Также введение ПЭМА в полимеры с ПВДФ может более эффективно снижать степень кристалличности по сравнению с ПММА, тем самым ослабляя взаимодействие и изменяя направление полярных доменов ПВДФ, что дает возможность сегнетоэлектрику уменьшить диэлектрические потери, повысить прочность на пробой и снизить высокие потери энергии в сильном электрическом поле [22].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы анализа
Спектры ЯМР 1H регистрировали на спектрометре ”Bruker WP-250 SY“ на частоте 250.13 МГц с использованием сигнала ДМСО-d6 (2.49 м.д.) и ацетон-d6 (2.04 м.д.) в качестве внутреннего стандарта. Для спектроскопии ЯМР 1Н готовили 2%-ные растворы анализируемых веществ, результаты обрабатывали на компьютере с использованием специального программного обеспечения “ACDLabs”.
Термогравиметрический анализ образца проводили в динамическом режиме в диапазоне от 50 до 700°C с использованием системы “TG50 Mettler Toledo” с точностью определения веса образца до 1 мкг. Скорость нагревания составляла 10 град/мин в атмосфере воздуха и азота.
Методом ДСК образец исследовали на дифференциальном сканирующем калориметре “Mettler Toledo DSC30” со скоростью нагревания или охлаждения 20 град/мин в атмосфере аргона.
Оптическую поляризационную микроскопию выполняли на приборе “Axioscop 40 A Pol”(“Carl Zeiss”, Германия) с нагревательным модулем.
ИК-спектроскопические исследования с преобразованием Фурье (FTIR) проводили на приборе “Varian Scimitar 2000 FT-IR” в спектральном диапазоне от 2800 до 11000 см–1 с максимальным разрешением 0.5 см–1.
Диэлектрические свойства изучали на пленках толщиной 100 мкм, изготовленных из растворов сополимеров в ДМФА методом полива на тефлоновые чаши. Пленки оставляли до полного высыхания при температуре 70–80°C. Диэлектрическую проницаемость, диэлектрические потери и проводимость измеряли при комнатной температуре на анализаторе импеданса “Novocontrol Alpha-A” с активной ячейкой для образцов ZGS Alpha и позолоченными дисковыми электродами диаметром 20 мм. Частота измерений изменялась в диапазоне 0.1–106 Гц, напряжение, подаваемое на электроды, составляло 1 В.
Материалы
Для синтеза привитых сополимеров использовали коммерчески доступные исходные реагенты П(ВДФ–co–ХТФЭ) (9 мол. % хлортрифторэтилена –ХТФЭ) и П(ВДФ–co–ТФЭ–co–ХТФЭ) (7 мол. % ХТФЭ) (“PolyK Technologies, State College”, USA), хлорид меди (II) (CuCl2, 99%) и 2,2'-бипиридин (Bpy, 99%) (“Acros Organics B.V.B.A.”), трис-(2-диметиламиноэтил)амин (Me6-TREN, 99%) (“Abcr GmbH”), этилметакрилат (ЭМА, (99%) (“Acros Organics B.V.B.A.”). В качестве растворителя для проведения реакции применяли N-метилпирролидон (МП). Все реакции проводили в атмосфере аргона.
Синтез П(ВДФ–co–ХТФЭ)–прив–ПЭМА
В трехгорлую колбу на 100 мл в атмосфере аргона помещали 2.5 г П(ВДФ–co–ХТФЭ)–прив–ПЭМА (3.2 ммоля) и 0.017 г CuCl2 (0.1 ммоля). К смеси добавили 50 мл МП и нагревали до полного растворения полимера. Затем в колбу загружали 0.118 мл Me6-TREN (0.4 ммоля), и добавляли 8.9 мл ЭМА (70.8 ммоля). Реакционную массу перемешивали при облучении УФ-светом (λ = 365 нм, мощность лампы 26 Вт), поддерживая температурный режим в диапазоне 18–25°С. Пробы объемом 10 мл отбирали в определенное время (табл. 1), осаждали в смеси H2O/CH3OH (1 : 1 об. %). Образец каждой фракции в виде волокон кремового цвета перемешивали 15 мин при интенсивном кипении в 50 мл хлороформа, отфильтровывали и сушили в вакуумном шкафу при температуре 60°С в течение 8 ч. Высушенные образцы составляли от 360 до 720 мг в зависимости от продолжительности полимеризации и содержания ПЭМА.
Таблица 1.
Мольное содержание ПЭМА в полученных привитых сополимерах П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА
| Фракция, № | Время реакции, мин | ПЭМА, мол. % |
|---|---|---|
| П(ВДФ–co–ХТФЭ)–прив–ПЭМА | ||
| 1 | 30 | 1.3 |
| 2 | 60 | 2.1 |
| 3 | 360 | 4.7 |
| 4 | 540 | 6.0 |
| П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА | ||
| 1 | 30 | 4.1 |
| 2 | 60 | 10.8 |
| 3 | 180 | 27.3 |
| 4 | 360 | 50.5 |
| 5 | 540 | 65.4 |
Синтез П(ВДФ–со–ТФЭ–co–ХТФЭ)–прив–ПЭМА
Аналогичным способом, используя 2.5 г П(ВДФ–ТФЭ–ХТФЭ) (2.4 ммоля), 0.013 г CuCl2 (0.1 ммоля), 0.118 мл Me6-TREN (0.4 ммоля), 8.9 мл этилметакрилата (70.8 ммоля) получали привитые сополимеры П(ВДФ–со–ТФЭ–co–ХТФЭ)–прив–ПЭМА. Высушенные образцы составляли от 430 до 670 мг в зависимости от продолжительности полимеризации и содержания ПЭМА.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез и определение содержания ПЭМА в полученных образцах
Для синтеза новых привитых сополимеров на основе ПВДФ в качестве исходных были выбраны двойной и тройной сополимеры винидиленфторида –П(ВДФ–co–ХТФЭ) и П(ВДФ–co–ТФЭ–co–ХТФЭ) соответственно, а в качестве прививки – ПЭМА. Наличие атома хлора у исходных сополимеров позволяет провести по этому фрагменту функционализацию методом радикальной полимеризации с переносом атома (ATRP) [23]. Существует множество различных вариантов проведения ATRP. Один из наиболее перспективных – фотоиндуцированная обратимо-дезактивационная радикальная полимеризация (RDRP) [24, 25], механизм которой детально изучен и описан в работах [26–29]. Этот метод зарекомендовал себя, как эффективный для получения сополимеров П(ВДФ–co–ХТФЭ) и П(ВДФ–co–ТФЭ–co–ХТФЭ) с привитыми цепями ПАН, а также ММА [30, 31]. Преимуществами RDRP являются возможность регулировать молекулярно-массовые характеристики привитых сополимеров путем изменения количества лиганда и времени реакции, а также возможность проведения синтеза при комнатной температуре. Низкое мольное содержание каталитической системы (0.02 ммоля Cu(II) и 0.12 ммоля Me6-TREN для П(ВДФ–co–ХТФЭ)–прив–ПЭМА, а также 0.02 ммоля Cu(II) и 0.12 ммоля Me6-TREN в случае П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА дает возможность уменьшить загрязнение полимера остаточными ионами металла, наличие которых может отрицательно сказываться на диэлектрических и сегнетоэлектрических свойствах материала [31]. Ранее сообщалось, что сополимер П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА [18] был синтезирован методом ATRP с переносом электрона (ARGET-ATRP), используя медь в качестве восстановителя [32]. Однако метод RDRP для получения привитых полимеров c ПЭМА не описан. В данной работе впервые с использованием Cu(II)-катализируемой фотоиндуцируемой RDRP были получены сополимеры П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА

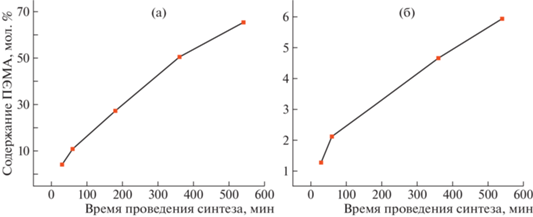
В результате проведенных исследований было найдено, что содержание ПЭМА в полимере возрастает с увеличением продолжительности реакции (рис. 1), что позволяет в определенной степени контролировать процесс. Следует отметить, что осуществлять контроль за ходом полимеризации методом ГПХ оказалось невозможным ввиду известной проблемы детектирования фторированных полимеров – небольшой разницы между показателем преломления полимера и элюента. В связи с этим контроль мольного содержания ПЭМА в образцах проводили методом спектроскопии ЯМР 1H.
Рис. 1.
Зависимость содержания ПЭМА, привитого на боковые цепи П(ВДФ–co–ХТФЭ) (а) и П(ВДФ–co–ТФЭ–co–ХТФЭ) (б) от времени реакции. Цветные рисунки можно посмотреть в электронной версии.
Рассмотрим спектры ЯМР 1H исходного сополимера П(ВДФ–co–ХТФЭ) и привитого П(ВДФ–co–ХТФЭ)–прив–ПЭМА (рис. 2) и рассчитаем содержание ПЭМА в полученных образцах, основываясь на расчетах близких аналогов [24, 31, 33–35].
Рис. 2.
Спектр ЯМР 1H исходного П(ВДФ–co–ХТФЭ) (a) в сравнении с привитым П(ВДФ–co–ХТФЭ)–прив–ПЭМА (б).
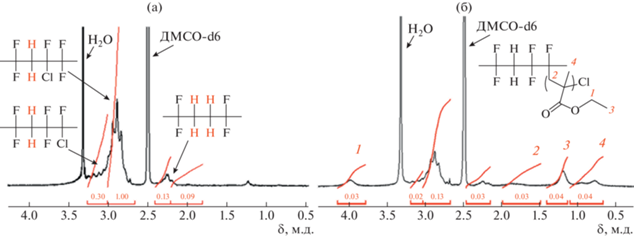
В спектре ЯМР 1H исходного двойного сополимера П(ВДФ–co–ХТФЭ) (рис. 2а), можно выделить три группы пиков: в области 2.2–2.4 м.д. (I1), 2.65–3.0 м.д. (I2) и 3.0–3.25 м.д. (I3). Согласно литературным источникам [34], пик, соответствующий области 3.0–3.25 м.д. (I3) относится к соединению звеньев ВДФ и ХТФЭ по типу “хвост к хвосту”. Две группы множественных пиков в области 2.65–3.0 м.д. (I2) и 2.2–2.4 м.д. (I1) соответствуют соединениям “голова к хвосту” в случае I2 и “хвост к хвосту” для I1 нескольких единиц ВДФ.
На спектре привитого П(ВДФ–co–ХТФЭ)–прив–ПЭМА по сравнению с исходным П(ВДФ–co–ХТФЭ) наблюдается новый пик в области 3.8–4.2 м.д. (м, 2H, –O–CH2–CH3) (рис. 2б, сигнал 1), который можно использовать для расчета мольного содержания привитого ПЭМА. Также на спектре появляются сопутствующие пики в области 1.5–2.0 (м, 2H, –CH2–C(CH3)–) (рис. 2б, сигнал 2), 0.6–1.1 (м, 3H, –CH2–C(CH3)–) (рис. 2б, сигнал 4), 1.1–1.4 (м, 3H, –O–CH2–CH3) (рис. 2б, сигнал 3), интегральная интенсивность которых возрастает с увеличением доли ПЭМА в образце. Согласно литературным данным [18, 34], новый широкий пик, появляющийся в спектре в области 3.8–4.2 м.д. привитого П(ВДФ–co–ХТФЭ)–прив–ПЭМА, идентифицируется как протон этильной группы цепи ПЭМА (I4).
Таким образом, соотнесение литературных данных и результатов анализа ЯМР 1H позволяет применить формулу для расчета мольного содержания привитого ПЭМА [18, 34, 37, 36 ]:
(1)
${\text{ПЭМА}} = \frac{2}{3} \cdot \frac{{{{I}_{4}} \cdot {\text{\;}}91}}{{{{I}_{1}} + {{I}_{2}} + {{I}_{3}}}},$На рис. 3 показаны спектры ЯМР 1H исходного тройного сополимера П(ВДФ–co–ТФЭ–co–ХТФЭ) и привитого сополимера на его основе П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА. В спектре привитого сополимера наблюдается новый пик в области 4.0–4.2 м.д. (м, 2H, –O–CH2–CH3) (рис. 3б, сигнал 5), который можно использовать для расчета мольного содержания привитого ПЭМА [18, 34, 36, 37], а также наличие сопутствующих пиков ПЭМА в областях 0.6–1.1 м.д. (м, 3H, –CH2–C(CH3)–), 1.1–1.4 м.д. (м, 3H, –O–CH2–CH3) и 1.5–2.0 м.д. (м, 2H, –CH2–C(CH3)–), интегральная интенсивность которых возрастает с увеличением доли ПЭМА.
Рис. 3.
Спектр ЯМР 1H исходного П(ВДФ–co–ТФЭ–co–ХТФЭ) (a) в сравнении с привитым П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА (б).

Литературные данные подтверждают, что пик при 3.3–3.4 м.д. (I4) (рис. 3, сигнал 2) соответствует соединению “хвост к хвосту” звеньев ВДФ и ХТФЭ. Группы множественных пиков при 3.3–2.9 м.д. (I2) (рис. 3, сигнал 3) относится к соединению типа “голова к хвосту” и 2.5–2.3 м.д. (I1) (рис. 3, сигнал 4) отвечают соединениям прямой последовательности ВДФ и нескольких единиц ВДФ “хвост к хвосту”. Протон ТФЭ (м, 1H, –CF2–CFH–) соответствует сигналу при 5.9–5.1 м.д. (I3) (рис. 3, сигнал 1).
Таким образом, при помощи анализа спектров ЯМР было определено мольное содержание привитого ПЭМА в полученных полимерах П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА (табл. 1). Существенно меньшее содержание ПЭМА в случае П(ВДФ–co–ХТФЭ)–прив–ПЭМА по сравнению с П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА предположительно можно объяснить влиянием стерических факторов на каталитический процесс.
Структура полученных привитых полимеров дополнительно была подтверждена ИК-спектроскопией. Было найдено, что в ИК-спектрах сополимеров П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА присутствует интенсивная полоса поглощения при 1720 см–1, соответствующая валентным колебаниям карбонильной группы этилметакрилатного фрагмента (рис. 4).

Термические свойства
Термическую и термоокислительную стабильность полученных сополимеров изучали методом термогравиметрического анализа (рис. 5). Температуры деструкции Tд, соответствующие 5%-ной потере массы образцов, суммированы в табл. 2. Сополимеры П(ВДФ–co–ХТФЭ) и П(ВДФ–со–ТФЭ–co–ХТФЭ) демонстрируют высокую термостабильность как в инертной атмосфере, так и на воздухе. Прививка ПЭМА приводит к понижению термостабильности. При высоких содержаниях ПЭМА резко уменьшается Tд, причем деструкция становится двухступенчатым процессом.
Рис. 5.
Термическая и термоокислительная стабильность привитых сополимеров на воздухе (а, в) и в инертной атмосфере (б, г), полученные методом ТГА для образцов П(ВДФ–co–ХТФЭ)–прив–ПЭМА (а, б) и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА (в, г). Содержание ПЭМА 0 (1), 1.3 (2), 2.1 (3), 4.7 (4) и 6.0 мол. % (5) (а, б); 0 (1), 4.1 (2), 10.8 (3), 27.3 (4), 50.5 (5) и 65.4 мол. % (6) (в, г).

Таблица 2.
Теплофизические характеристики привитых сополимеров П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА
| Мольное соотношение* | $T_{{\text{д}}}^{*}$ О2/N2 | $\omega _{{{\text{ВДФ}}}}^{*}$, % | $\omega _{{{\text{ПЭМА}}}}^{*}$, % | Тпл, °С | Тс, °С | ΔHпл, Дж/г | ΔСp, Дж/г К | χ, % | ${{\omega }}_{{{\text{ПЭМА}}}}^{{{\text{ДСК}}}}$, % |
|---|---|---|---|---|---|---|---|---|---|
| П(ВДФ–co–ХТФЭ)–прив–ПЭМА | |||||||||
| 91 : 9 : 0 | 395/392 | 84.7 | – | 167 | – | 16.3 | – | 18.4 | – |
| 91 : 9 : 1.3 | 394/398 | 82.9 | – | 160,167 | – | 16.9 | – | 19.4 | – |
| 91 : 9 : 2.1 | 375/379 | 81.7 | – | 160,167 | – | 16.2 | – | 18.9 | – |
| 91 : 9 : 4.7 | 296/268 | 79.2 | – | 160,167 | – | 11.1 | – | 13.4 | – |
| 91 : 9 : 6.0 | 294/265 | 77.1 | – | 158 | – | 3.4 | – | 4.2 | – |
| П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА | |||||||||
| 80 : 13 : 7 : 0 | 378/425 | 73.1 | – | 122 | – | 16.2 | – | 21.1 | – |
| 80 : 13 : 7 : 4.1 | 379/415 | 68.5 | – | 124 | – | 14.0 | – | 19.5 | – |
| 80 : 13 : 7 : 10.8 | 310/318 | 62.2 | – | 121 | – | 9.4 | – | 14.4 | – |
| 80 : 13 : 7 : 27.3 | 278/305 | 50.6 | – | 107 | – | 3.2 | – | 6.0 | – |
| 80 : 13 : 7 : 50.5 | 263/294 | – | 45.1 | – | 38 | – | 0.12 | – | 43 |
| 80 : 13 : 7 : 65.4 | 267/285 | – | 51.6 | – | 44 | – | 0.14 | – | 50 |
Примечание. Tд – температура 5%-ной потери массы в атмосфере воздуха и азота – по данным ТГА; Тпл, энтальпия плавления ΔHпл, степень кристалличности χ фазы ВДФ – по данным ДСК; температура стеклования Тс, скачок теплоемкости ΔСp и содержание фазы ПЭМА ${{\omega }}_{{{\text{ПЭМА}}}}^{{{\text{ДСК}}}}$ – по данным ДСК.
Обращает на себя внимание тот факт, что в случае П(ВДФ–co–ХТФЭ)–прив–ПЭМА потеря массы на первой стадии деструкции (рис. 5а, 5б) существенно превышает содержание ПЭМА в сополимере, т.е. присутствие ПЭМА облегчает частичную деструкцию основной цепи сополимера. В случае П(ВДФ–со–ТФЭ–co–ХТФЭ)–прив–ПЭМА, потеря массы на первой стадии близка к содержанию ПЭМА в сополимере.
На рис. 6 приведены сканы ДСК второго нагревания привитых сополимеров П(ВДФ–co–ХТФЭ)–прив–ПЭМА и П(ВДФ–со–ТФЭ–co–ХТФЭ)–прив–ПЭМА. Температура плавления Тпл, энтальпия плавления ΔHпл, степень кристалличности фазы ВДФ χ, а также температура стеклования Тс, скачок теплоемкости ΔСp и содержание фазы ПЭМА по данным ДСК ($\omega _{{{\text{ПЭМА}}}}^{{{\text{ДСК}}}}$) сополимеров суммированы в табл. 2. Степень кристалличности фазы ПВДФ в привитых сополимерах была рассчитана следующим образом:
(2)
${\mathbf{\chi }} = \Delta {{H}_{{{\text{пл}}}}}\omega _{{{\text{ВДФ}}}}^{{ - 1}}\Delta H_{{{\text{пл}}}}^{{* - 1}},$Рис. 6.
Сканы ДСК второго нагревания изученных привитых сополимеров П(ВДФ–co–ХТФЭ)–прив–ПЭМА (а) и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА (б). Содержание ПЭМА 0 (1), 1.3 (2), 2.1 (3), 4.7 (4) и 6.0 мол. % (5) (а); 0 (1), 4.1 (2), 10.8 (3), 27.3 (4), 50.5 (5) и 65.4 мол. % (6).

Содержание фазы ПЭМА найдено по формуле
(3)
$\omega _{{{\text{ПЭМА}}}}^{{{\text{ДСК}}}} = \Delta {{С}_{p}}\Delta С_{p}^{{* - 1}} \times 100\% $Здесь $\Delta С_{p}^{*}$ – скачок теплоемкости при расстекловывании 100% аморфного ПЭМА ($\Delta С_{p}^{*}$ = = 0.28 Дж/г К [39]).
На сканах ДСК образцов П(ВДФ–co–ХТФЭ)–прив–ПЭМА наблюдается эндотермический пик в области 150–170°С, соответствующий плавлению фазы ПВДФ. Пик плавления исходного образца П(ВДФ–co–ХТФЭ), проявляющийся при 167°С, имеет небольшое низкотемпературное плечо. Прививка ПЭМА приводит к возникновению ярко выраженной бимодальности пика плавления с максимумами при 160 и 167°С, связанной с переходом из одной кристаллической модификации в другую. Плавление при 167°С характерно как для α, так и для β кристаллических фаз чистого ПВДФ [40]. С повышением содержания ПЭМА степень кристалличности, рассчитанная по формуле (2), уменьшается и при содержании 6.0 мол. % бимодальность исчезает, а температура плавления фазы ВДФ понижается до 158°С.
На сканах ДСК образцов П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА присутствует эндотермический пик в области 110–130°С, соответствующий плавлению фазы ВДФ. С ростом содержания ПЭМА энтальпия плавления, а следовательно, и степень кристалличности фазы ВДФ падает. Кроме того, на сканах ДСК образцов с небольшим содержанием ПЭМА виден низкотемпературный эндотермический пик при 26 (2.1 Дж/г), 28 (2.4 Дж/г) и 33°С (2.8 Дж/г) для образцов с содержанием 4.1, 10.8 и 27.3 мол. % соответственно. В работе [41] у сополимеров П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА с мольным соотношением компонентов основной цепи 80 : 18 : 2 наблюдался похожий низкотемпературный эндотермический пик, хотя и при более высокой температуре (около 90°С), который соответствовал температуре Кюри. При более высоком содержании ПЭМА исчезают как низкотемпературный пик, так и пик плавления кристаллической фазы ВДФ. Вместо этого на сканах ДСК образцов с содержанием 50.5 и 65.4 мол. % возникает скачок теплоемкости, характерный для области стеклования полимеров. Содержание ПЭМА $\omega _{{{\text{ПЭМА}}}}^{{{\text{ДСК}}}}$, рассчитанное из соотношения скачка теплоемкости при расстекловывании этих образцов к табличному значению для чистого ПЭМА по формуле (3) [39], хорошо коррелирует с массовой долей ПЭМА в сополимерах по данным ЯМР 1Н (табл. 1). Полученные результаты подтверждают, что такой скачок теплоемкости относится к расстекловыванию фазы ПЭМА. Для чистого ПЭМА характерно стеклование при 65°С [39], для ПВДФ – при –61°С [38]. Температуры стеклования привитых сополимеров лежат между этими значениями. Подобное смещение температуры стеклования указывает на то, что привитые цепи ПЭМА связаны с основной цепью сополимеров ковалентной связью.
Приведенные данные ДСК подтверждаются снимками поверхности пленок, полученными с помощью оптического микроскопа (рис. 7). Даже малое количество аморфного ПЭМА (1.3%) в сополимере влияет на его кристаллическую структуру и свойства, благодаря чему привитой полимер имеет хорошую пленкообразующую способность в отличие от исходного сополимера П(ВДФ–co–ХТФЭ), который проявляет кристаллические свойства.

Диэлектрические свойства
Диэлектрические свойства исходных двойного и тройного сополимера, а также синтезированных привитых сополимеров изучали в частотном диапазоне 0.1–106 Гц при комнатной температуре. Тонкие пленки 100 мкм были изготовлены методом полива раствора полимера в ДМФА на тефлоновые чаши.
Для привитых полимеров на основе П(ВДФ–co–ХТФЭ) диэлектрическая проницаемость снижается практически в два раза во всем исследованном частотном диапазоне при мольном содержании ПЭМА 1.3% (рис. 8а). С дальнейшим увеличением содержания ПЭМА в сополимерах значения диэлектрической постоянной продолжают уменьшаться, что вызвано, по-видимому, ограничением дипольно-сегментальной подвижности из-за роста массы полимерной цепи. Величина проводимости уменьшается с падением диэлектрической проницаемости при повышении содержания ПЭМА в образцах (рис. 8б).
Рис. 8.
Диэлектрическая проницаемость (а, г), проводимость (б, д) и диэлектрические потери (в, е) для серии привитых сополимеров П(ВДФ–co–ХТФЭ)–прив–ПЭМА (а, б, в) и П(ВДФ–co–ТФЭ–co–ХТФЭ)–прив–ПЭМА (г, д, е). Содержание ПЭМА 0 (1), 1.3 (2), 2.1 (3), 4.7 (4) и 6.0 мол. % (5) (а–в); 4.1 (1), 10.8 (2), 27.3 (3), 50.5 (4), 65.4 (5) и 0 мол. % (6).

Диэлектрическая проницаемость привитых полимеров на основе П(ВДФ–co–ТФЭ–co–ХТФЭ) практически не меняется вплоть до мольного содержания ПЭМА 10.8%, а затем резко уменьшается (рис. 8в), по той же причине, что и полимеров на основе П(ВДФ–co–ХТФЭ) – снижение дипольно-сегментальной подвижности из-за роста массы полимерной цепи с увеличением содержания ПЭМА. В пользу этого свидетельствует также поведение диэлектрических потерь, которые не меняются до мольного содержания ПЭМА 10.8%, а затем резко уменьшаются (рис. 8д). В области 106 Гц наблюдается начало релаксационного процесса, максимум которого лежит на более высоких частотах. При частотах ниже 102 Гц диэлектрическая проницаемость и потери (рис. 8а, 8в, 8д, 8е) возрастают с уменьшением частоты, что вязано с электродной поляризацией из-за наличия примесной проводимости.
ЗАКЛЮЧЕНИЕ
Использование метода RDRP позволяет получать сополимеры на основе П(ВДФ–co–ХТФЭ) и П(ВДФ–co–ТФЭ–co–ХТФЭ) c привитыми цепями ПЭМА. Преимуществами использованного метода является малое количество катализатора, а также проведение реакции при комнатной температуре. Термическая и термоокислительная стабильность полимеров уменьшается с увеличением содержания ПЭМА. Повышение мольного содержания ПЭМА приводит к понижению температуры и теплоты плавления, что косвенно указывает на уменьшение кристалличности обоих серий полимеров. Исследование диэлектрических свойств показало, что возрастание содержания ПЭМА в сополимерах способствует снижению значения диэлектрической постоянной и диэлектрических потерь, что может быть вызвано снижением дипольно-сегментальной подвижности ввиду увеличения массы полимерной цепи с повышением содержания ПЭМА.
Исследования методом ЯМР 1Н проводились в Центре коллективного пользования “Центр исследования полимеров” в рамках госзадания от Министерства науки и высшего образования России (тема FFSM-2021-0005).
Работа выполнена при финансовой поддержке Российского научного фонда (проект 19-73-30028).
Список литературы
Li Q., Wang Q. // Macromol. Chem. Phys. 2016. V. 217. P. 1228.
Chen X., Han X., Shen Q.-D. // Adv. Electron. Mater. 2017. V. 1600460. P. 1.
Fan R.F., Tang W., Wang Z.L. // Adv. Mater. 2016. V. 28. P. 4283.
Prateek, Thakur V.K., Gupta R.K. // Chem. Rev. 2016. V. 116. P. 4260.
Liu Z., Zhang S., Jin Y.M., Ouyang H., Zou Y., Wang X.X., Xie L.X., Li Z. // Sci. Technol. 2017. V. 32. P. 064004.
Wang X. // Nano Energy. 2012. V. 1. P. 13.
Pi Z., Zhang J., Wen Ch., Zhang Z., Wu D. // Nano Energy. 2014. V. 7. P. 33.
Han X., Chen X., Tang X., Chen Y.L. Liu J.H., Shen Q.D. // Adv. Funct. Mater. 2016. V. 26. P. 3640.
Wan Ch., Bowen Ch.R. // J. Mater. Chem. A. 2017. V. 5. P. 3091.
Liu Y., Wang Q. // Adv. Sci. 2020. V. 7. № 1902468.
Correia H.M.G., Ramos M.M.D. // Comput. Mater. Sci. 2005. V. 33. № 1. P. 224.
Li M., Wondergem H.J., Spijkman M.J., Asadi K., Katsouras I., Blom P.W.M., Leeuw D.M. // Nature Mater. 2013. V. 12. № 5. P. 433.
Wang Sh., Li Q. // IET Nanodielectr. 2018. V. 1. № 2. P. 80.
Tan S., Xiong J., Zhao Y., Liu J., Zhang Z. // J. Mater. Chem. C. 2018. V. 6. № 15. P. 4131.
Hu X., Cui G., Zhu N., Zhai J., Guo K. // Polym. Chem. 2018. V. 10. № 68. P. 10.
Guan F.J., Wang Yang L., Tseng J.K., Han K., Wang Q., Zhu L. // Macromolecules. 2011. V. 44. № 7. P. 2190.
Gong H., Miao B., Zhang X., Lu Zh. // RSC Adv. 2016. V. 6. № 2. P. 1589.
Li J., Tan S., Ding S., Li H., Yang L. // J. Mater. Chem. 2012. V. 22. № 44. P. 23468.
Hu P., Gao S., Zhang Y., Zhang L., Wang C. // Compos. Sci. Technol. 2018. V. 156. P. 109.
Wanga J., Xiea Y., Liua J., Zhanga Z., Zhang Y. // Appl. Surf. Sci. 2019. V. 469. P. 437.
Valiyaneerilakkal U., Singh A., Subash C.K., Singh K., Abbas S.M., Varghese S. // Polym. Compos. 2015. V. 38. P. 1.
Li J., Gong H., Yang Q., Xie Y., Yang L., Zhang Z. // Appl. Phys. Lett. 2014. V. 104. P. 263901.
Matyjaszewski K., Tsarevsky N.V. // J. Am. Chem. Soc. 2014. V. 136. № 18. P. 6513.
Hu X., Cui G., Zhu N., Zhai J., Guo K. // Polymers. 2018. V. 10. № 1. P. 68.
Hu X., Li J., Li H., Zhang Z. // J. Polym. Sci. A. 2012. V. 50. P. 3126.
Pan X., Tasdelen M.A., Laun J., Junkers T., Yagci Y., Matyjaszewski K. // Progr. Polym. Sci. 2016. V. 62. P. 73.
Matyjaszewski K., Tsarevsky N.V. // J. Am. Chem. Soc. 2014. V. 136. P. 6513.
Frick E., Anastasaki A., Haddleton D.M., Barner-Kowollik C. // J. Am. Chem. Soc. 2014. V. 137. № 21. P. 6889.
Anastasaki A., Nikolaou V., Zhang Q., Burns J., Samanta S.R., Waldron C., Haddleton A.J., McHale R., Fox D., Percec V., Wilson P., Haddleton D.M. // J. Am. Chem. Soc. 2014. V. 136. P. 1141.
Hu X., Li J., Li H., Zhang Z. // J. Polym. Sci. A. 2013. V. 51. P. 4378.
Khudyshkina A.D., Luponosov Yu.N., Shevchenko V.G., Ponomarenko S.A. // EXPRESS Polym. Lett. 2021. V. 15. № 10. P. 957.
Zhang M.F., Russell T.P. // Macromolecules. 2006. V. 39. P. 3531.
Hu X., Li J., Li H., Zhang Z. // J. Polym. Sci. A. 2012. V.50. P. 3126.
Gong H., Li J., Di D., Lib N., Zhang Zh. // RSC Adv. 2015. V. 5. P. 19117.
Tan S., Xiong J., Zhao Y., Liu J., Zhang Z. // J. Mater. Chem. C. 2018. V. 6. № 15. P. 1.
Zhu N., Hu X., Zhang Y., Zhang K., Li Z., Guo K. // Polym. Chem. 2016. V. 7. P. 474.
Tan S., Xiong J., Zhao Y., Liu J., Zhang Z. // J. Mater. Chem. C. 2013. V. 6. № 15. P. 1.
Gaur U., Wunderlich B.B., Wunderlich B. // J. Phys. Chem. Ref. Data. 1983. V. 12. P. 29.
Gaur U., Lau S.-F., Wunderlich B.B., Wunderlich B. // J. Phys. Chem. Ref. Data. 1982. V. 11. P. 1065.
Gregorio R.Jr. // J. Appl. Polym. Sci. 2006. V. 100. № 4. P. 3272.
Li J., Tan S., Ding S., Li H., Yang L., Zhang Z. // J. Mater. Chem. 2012. V. 22. P. 23468.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)