

Высокомолекулярные соединения (серия С), 2022, T. 64, № 2, стр. 206-217
САМООРГАНИЗАЦИЯ МОЛЕКУЛЯРНЫХ ЩЕТОК НА ОСНОВЕ ПОЛИФЛУОРЕНА С БОКОВЫМИ ЦЕПЯМИ ПОЛИМЕТАКРИЛОВОЙ КИСЛОТЫ В ЭТАНОЛЕ И ВОДЕ
М. А. Симонова a, *, Д. М. Ильгач a, К. И. Каскевич a, М. И. Непомнящая a, Л. С. Литвинова a, А. П. Филиппов a, А. В. Якиманский a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
* E-mail: mariasimonova1983@mail.ru
Поступила в редакцию 16.04.2022
После доработки 13.05.2022
Принята к публикации 07.07.2022
- EDN: DGFMYR
- DOI: 10.31857/S2308114722700145
Аннотация
Методом радикальной полимеризации с переносом атома синтезированы привитые сополимеры, основной цепью которых служил полифлуорен, а боковыми цепями поли-трет-бутилметакрилат и полиметакриловая кислота. Поведение синтезированных щеток в растворах в хлороформе, этаноле и в воде изучено методами молекулярной гидродинамики и оптики. Привитые сополимеры характеризуются высокой плотностью прививки. Щетки с боковыми цепями полиметакриловой кислоты образуют унимолекулярные мицеллы, способные инкапсулировать куркумин в водных растворах. Плотность прививки боковых цепей влияет на свойства комплексов щеток с куркумином.
ВВЕДЕНИЕ
Современные методы синтеза позволяют получать привитые сополимеры с заданной молекулярной архитектурой и молекулярной массой. Отдельный интерес представляют амфифильные полимерные щетки, основная и боковые цепи которых сильно различаются по сродству к растворителю [1–8]. При получении таких щеток можно в широких пределах варьировать химическое строение компонентов, а также структурные параметры, такие как длина основной и боковых цепей, плотность прививки последних, что является объектом пристального внимания ученых как при отработке методов синтеза, так и при исследовании образующихся в растворе моно- и надмолекулярных структур.
Ранее были получены и исследованы полимерные щетки с основной полиимидной цепью и боковыми цепями полиметилметакрилата [9–11] и полиметакриловой кислоты (ПМАК) [4, 12]. Показано, что амфифильные полимерные щетки с боковыми цепями ПМАК могут использоваться в медицине, поскольку, например, способны образовывать мицеллы с инкапсулированным люминофором для фотодинамической терапии рака [12].
Особое внимание привлекают полимерные щетки с основной сопряженной цепью, которая обладает таким полезным свойством, как люминесценция. В таких щетках в качестве основной цепи используются полипарафенилен, политиофен и полифлуорен (ПФ) [13, 14]. Последний характеризуется синей люминесценцией с высокими значениями квантового выхода. С помощью коммерчески доступных люминофоров можно управлять флуоресценцией, вводя их в состав полифлуореновой цепи [15–17]. Легкость модифицирования положения С9 флуорена позволяет получать мономеры для дальнейшего синтеза молекулярных щеток. Материалы на основе люминесцирующих полимерных щеток находят применение в разных областях, например, в качестве сенсоров и покрытий для электролюминесценции [18–23].
Активное исследование полифлуореновых щеток связано с их перспективами в медицине и биотехнологии, в частности для направленной доставки лекарств и диагностики различных заболеваний [24–31]. Однако для применения в биомедицине требуется хорошая растворимость в воде, что достигается путем синтеза амфифильных полимерных щеток с гидрофильными боковыми цепями. Кроме того, растворимость в воде может быть обеспечена созданием надмолекулярных структур, таких как мицеллы, везикулы, полимерные наночастицы и т.п. [32–34]. Известно, что на конформацию молекулярных щеток в растворе влияет плотность прививки боковых цепей, их длина, природа и селективность растворителя. Благодаря высокой плотности прививки боковых цепей щетки склонны в селективных растворителях к образованию мономолекулярных мицелл (конформационным перестройкам на молекулярном уровне), а не больших агрегатов – надмолекулярных структур [35–37]. Способность молекулярных щеток к мицеллообразованию позволяет получать на их основе наноконтейнеры для инкапсулирования гидрофобных лекарств [12, 25, 27, 38, 39].
Для полимерных щеток, плохо растворяющихся в воде, существует метод получения мицелл, заключающийся в том, чтобы растворить полимер в хорошем растворителе и добавить раствор к деионизованной воде. Например, в работе [30], полимерную щетку растворяли в ТГФ, выливали раствор в воду и выдерживали на воздухе для избавления от органического растворителя.
В работе [25] были получены щетки с основной цепью поли(флуорен-альт-(4,7-бис-(гексилтиен)-2,1,3-бензотиадиазол))а и боковыми цепями поликапролактон–блок–полиолиго(этиленгликоль)метакрилат метиловый эфир. Они обладали флуоресценцией в ближней инфракрасной области и могли образовывать мицеллы с инкапсулированным внутрь доксорубицином. Такое сочетание свойств дало возможность создать материалы, потенциально применимые в тераностике. Размер и морфологию унимолекулярных мицелл изменяли путем варьирования длины боковых цепей. Присутствие в полимерной щетке гидрофобного блока позволяло увеличить фотостабильность в водной среде и обеспечить более эффективную загрузку лекарства.
Используется также стратегия тандемной доставки малых интерфирирующих РНК и апконверсионных наночастиц фотодинамической терапии с помощью нанокомпозитов [40]. Данные наночастицы были стабилизированы оболочкой из полимерной щетки с цвиттер-ионными боковыми цепями кватернизованного поли-диметиламиноэтилметакрилата. В работе [27] также были получены нанокомпозиты для тандемной доставки лекарств фотодинамической терапии, которые состояли из pH-чувствительной оболочки, про-лекарства, светочувствительного агента и полифлуореновой молекулярной щетки с кватернизованными пропансультоном боковыми цепями полидиметиламиноэтилметакрилата, придающими композиту растворимость в воде за счет образования цвиттер-ионного сопряженного полиэлектролита.
В зависимости от типа доставляемого вещества могут быть применены различные методы его инкапсулирования в полимерные системы, например наноосаждение, эмульсионная диффузия, распылительная сушка, ионное гелеобразование и другие [41]. Для доставки доксорубицина использовали метод рН градиента [42], при инкапсулировании галоперидола в сополимер – метод эмульгирования путем обработки ультразвуком для выпаривания растворителя [43].
Солюбилизирующую способность полимерных систем можно оценить с помощью модельного вещества куркумина [44]. Несмотря на фармакологическую активность потенциального лекарства, куркумин имеет ряд недостатков: нерастворимость в воде, низкая биодоступность и быстрый метаболизм. Для преодоления этих недостатков куркумин инкапсулируют в полимерные мицеллы [45, 46]. В работе [47] методом мембранной гидратации были получены мицеллы из термочувствительных щеткоподобных триблок-сополимеров с инкапсулированным куркумином. Мицеллы получали также путем растворения амфифильного блок-сополимера и куркумина в ДМФА [48]. К раствору в ДМФА медленно добавляли деионизованную воду при непрерывном перемешивании, после чего подвергали диализу. С помощью амфифильного п-сульфонато-каликс [4]арен-О-гексилового эфира была увеличена растворимость и уменьшена скорость деградации куркумина в воде [49]. Изучена загрузка куркумина в производное карбоксиметилхитозана [50]. Наночастицы получали путем ионотропного гелеобразования. При перемешивании растворяли карбоксиметилхитозан в деионизованной воде, в раствор по каплям вводили раствор куркумина в этаноле, после чего добавляли трифосфат натрия и перемешивали, что приводило к образованию наночастиц. Для солюбилизации куркумина также могут быть использованы полимерные щетки. Нагруженные наноконтейнеры на основе поликапролактон–прив–олигокаррагинана получают испарением ацетона [51]. Методом динамического рассеяния света зафиксировано небольшое уменьшение размера наночастиц по сравнению с пустыми мицеллами, обусловленное, вероятно, высоким сродством гидрофобного ядра полимерной щетки и гидрофобного лекарства. Сравнение спектров флуоресценции свободного куркумина и нагруженных им мицелл показало, что происходит смещение максимума эмиссии c 558 до 517 нм.
В нашей предыдущей работе [52] были исследованы полифлуореновые полимерные щетки c боковыми цепями ПМАК, обладающие сравнительно высокой степенью функционализации – 75%. Щетки были получены радикальной полимеризацией с переносом атома (ATRP) и полимеризацией с активатором, генерируемым переносом электрона, (AGET ATRP) трет-бутилметакрилата на полифлуореновом многофункциональном макроинициаторе с последующим протонолизом трет-бутильных групп. Показано, что благодаря своей амфифильной природе щетки могут формировать мицеллы, состоящие из 2 или 10 макромолекул, которые сохраняются и при инжектировании в воду.
Цель настоящей работы – синтез амфифильных полифлуореновых щеток с плотностью прививки боковых цепей, приближающейся к 100%, а также сопоставление характеристик мицелл на основе щеток с разной степенью прививки боковых цепей в разных растворителях (спирт, вода) и мицелл с включенным куркумином.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы исследования
В работе использовали 2-бромизобутироил бромид (98%, “Acros Organics”), KI (99%, “Acros Organics”), CuCl2 (99%, “Acros Organics”), 4,4'-динонил-2,2'-бипиридил (dNbpy, 97%, “Sigma-Aldrich”), 2-этилгексаноат олова (II) (Sn(EH)2, 95%, “Alfa-Aesar”), куркумин (98%, “Sigma-Aldrich”). Анизол (99%, “Sigma-Aldrich”) перегоняли над натрием дважды. Тетрагидрофуран (99%, “Вектон”) перегоняли над гидридом кальция, трет-бутил метакрилат (ТБМА, 98%, “Sigma-Aldrich”) – под вакуумом. Метанол (98%, “Вектон”), триэтиламин (99%, “Sigma-Aldrich”), хлористый метилен (98%, “Вектон”), трифторуксусную кислоту (99%, “Sigma-Aldrich”) применяли без дополнительной очистки.
Спектры ЯМР записывали на приборе “Bruker AVANCE-400 SB” (400 МГц), спектры поглощения в видимой УФ-области – на спектрофотометре “Shimadzu UV-1900”, спектры фотолюминесценции – на спектрофлуориметре “Shimadzu RF-6000”. Кинетику реакции изучали с помощью газового хроматографа “Shimadzu GC-2010Plus”. ИК-фурье-спектры регистрировали на приборе “Shimadzu IRAffinity-1S” с приставкой “Quest single ATR” (Specac), диапазон частот 7800–400 см–1.
Средневесовую молекулярную массу Mw и полидисперсность Ð полимеров определяли методом ГПХ на жидкостном хроматографе “Agilent Technologies 1260 Infinity” (Мультидетекторная система GPC/SEC Agilent 1260 Infinity), оснащенном тремя детекторами: рефрактометрическим (DRI, длина волны лазера 660 нм), вискозиметрическим (VS) и светорассеивающим (LS, углы рэлеевского рассеяния 15° и 90°; длина волны/мощность лазера: 660 нм/50 МВт). Установка включала набор хроматографических колонок, соединенных последовательно: предколонку PLgel 5 мкм Guard размером 50 × 7.5 мм и две колонки Agilent Technologies PLgel 5 мкм MIXED-C размером 300 × 7.5 мм. Температура детекторов и колонок составляла 40°C. Подвижной фазой служил ТГФ, перегнанный над КОН и стабилизированный 2,6-ди-трет-бутилгидрокситолуолом (0.02%); скорость элюирования 1.0 мл/мин. Концентрация введенного образца не превышала 2 мг/мл. Объем образца был равен 100 мкл.
Синтез макроинициатора (МИ-2)
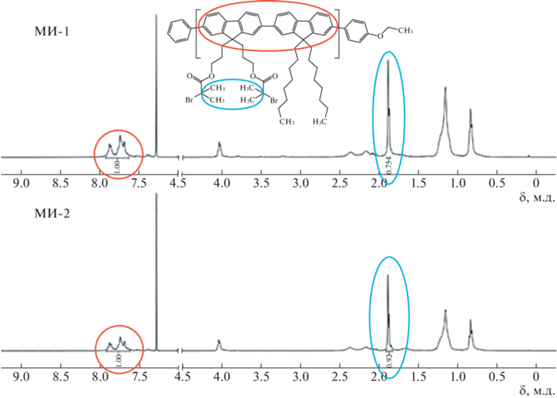
В 24 мл сухого ТГФ растворяли 0.470 г поли[(9,9'-бис-(3-гидроксипропил)-флуорен)-альт-(9,9'-диоктилфлуорена)] (ранее получен по методике [52]), затем добавляли 1.41 мл (10.1 ммоля) триэтиламина и 20 мг иодида калия. Реакционную массу перемешивали в атмосфере аргона, охлаждая на ледяной бане. Далее прибавляли по каплям 0.94 мл (7.6 ммоля) 2-бромизобутирил бромида, после чего реакционную смесь нагревали до комнатной температуры и перемешивали в течение 24 ч. После окончания реакции смесь фильтровали от соли, отгоняли растворитель и высаждали в метиловый спирт. Для переосаждения полимера его растворяли в 3–4 мл ТГФ и медленно прикапывали раствор в 50 мл метанола, активно перемешивая. Осажденный продукт снова фильтровали на фильтре Шотта, промывая метанолом, а затем сушили в вакууме при 50°С. Полимер был охарактеризован методом ГПХ в ТГФ (Mn = 21030, Ð = 1.5) и тройным ЯМР 1Н (400 МГц, CDCl3, δ) 8.13–7.51 (уш., 12H, Ar H), 4.04 (уш., J = 5.9 Гц, 4H, 2CH2), 2.43–2.03 (уш., 8H, 4CH2), 1.88 (с, 12H, 4CH3), 1.31–1.01 (м, 28H, 14CH2), 0.96–0.71 (м, 6H, 2CH3).
Было найдено, что добавление небольшого количества иодида калия при получении мультицентрового макроинициатора приводит к увеличению степени функционализации с 75% (макроинициатор МИ-1 [52]) до 92% (МИ-2, настоящая работа). Степень функционализации макроинициатора – мольную долю прореагировавших гидроксильных групп определяли методом ЯМР 1Н, измеряя соотношения интегральных интенсивностей ароматических протонов и протонов бромэфирных групп (рис. 1). Добавление KI, вероятно, приводит к галогенообмену, и равновесие смещается в сторону образования продукта реакции, так как I– является лучшей уходящей группой, чем Br–. В табл. 1 представлены характеристики полученного ранее и нового мультицентровых макроинициаторов – МИ-1 и МИ-2 соответственно.
Синтез щеток полифлуорен–прив–поли-трет-бутилметакрилат (ПФ-ПБМА) по механизму ATRP AGET
В колбу Шленка вводили МИ-2 (30 мг, 0.029 ммоля), CuCl2 (7.9 мг, 0.059 ммоля), dNbpy (50.2 мг, 0.123 ммоля). Колбу закрывали септой, вакуумировали и трижды заполняли аргоном. Через септу добавляли 7.6 мл (80 об. %) анизола. После растворения компонентов добавляли 1.9 мл (0.0117 моля) ТБМА. Затем в реакционную смесь через септу вводили 0.5 мл раствора Sn(EH)2 (48 мг, 0.118 ммоля в 1 мл анизола). Реакцию проводили при непрерывном перемешивании при 80°С. Анизол, ТБМА и раствор Sn(EH)2 продували аргоном в течение 30 мин перед добавлением в систему.
По окончании реакции реакционную массу охлаждали и разбавляли ТГФ до образования раствора зеленого цвета. Раствор пропускали через колонку с нейтрально активированной окисью алюминия, промывая ТГФ. Далее отгоняли растворитель и продукт высаживали в систему МеОН–вода (10 : 1). Выпавший полимер выделяли на фильтре Шотта, промывали 3 раза смесью МеОН–вода, затем переосаждали. Для этого растворяли порошок полимера в 7–10 мл ТГФ и медленно прикапывали раствор в 100 мл смеси МеОН–вода, активно перемешивая. Осажденный продукт снова фильтровали на фильтре Шотта, промывая той же смесью осадителя, а затем сушили в вакууме при 50°С.
Для определения конверсии мономера из реакционной смеси отбирали аликвоты (10 мкл), разбавляли их ТГФ (200 мкл) и анализировали с помощью газовой хроматографии. Внутренним стандартом служил анизол.
На рис. 2 видно, что анаморфоза кинетической кривой первого порядка линейна, и имеется индукционный период, равный 14 мин, который, вероятно, соответствует времени восстановления комплекса Cu(II) в Cu(I).

Получение щеток полифлуорен–прив–полиметакриловая кислота (ПФ-ПМАК)
В колбе растворяли 0.1 г ПФ-ПБМА в 2.5 мл хлористого метилена и добавляли 0.8 г трифторуксусной кислоты. Реакцию проводили в колбе с магнитной мешалкой при комнатной температуре и оставляли на ночь. После реакции растворитель удаляли при пониженном давлении, полученный порошок сушили под вакуумом при 50°С.
Получение наночастиц
Для получения наночастиц образец ПФ-ПБМА (8 мг) растворяли в этаноле (0.4 мл). Затем раствор (0.1 мл) инжектировали в деионизованную воду (2.5 мл) при обработке ультразвуком (частота 35 кГц) в течение 1 мин.
Инкапсулирование куркумина в щетке ПФ-ПМАК
Перед получением наноконтейнеров с куркумином раствор ПФ-ПМАК подвергали диализу, воду меняли 3 раза каждый час и оставляли на ночь. Далее раствор лиофилизировали. Приготовленный порошок ПФ-ПМАК и куркумин растворяли по отдельности в этиловом спирте, смешивали в различных соотношениях, разбавляли спиртом до необходимого объема. Полученный раствор инжектировали в деионизованную воду под действием ультразвука (частота 35 кГц) в течение 1 мин.
Определение молекулярно-массовых и гидродинамических характеристик щеток ПФ-ПБМА
Молекулярную массу и гидродинамический радиус Rh-D макромолекул ПФ-ПБМА – молекулярных щеток с боковыми цепями ПБМА определяли методами статического и динамического рассеяния света в разбавленных растворах в хлороформе (ρ = 1.486 г/см3, η0 = 0.57 сП и n0 = 1.443). Светорассеяние изучали на установке “Photocor Complex” (“Photocor Instruments Inc.”, Россия), источником света служил диодный лазер Photocor-DL (мощность от 5 до 30 мВт, длина волны λ = 659.1 нм). Калибровку приборa проводили по бензолу (RV = 2.32 × 10–5 см–1). Корреляционную функцию интенсивности рассеянного света получали на корреляторе “Photoсor-PC2” с числом каналов 288 и обрабатывали с помощью программного обеспечения Dynal S. Опыты проводили при 21.0°С. По данным динамического светорассеяния в исследованной области концентраций раствора с распределение рассеивающих частиц по размеру является унимодальным. Гидродинамический радиус Rh-D не зависит от c. Гидродинамические радиусы макромолекул, найденные при разных концентрациях, усредняли; среднее по концентрациям значение Rh-D приведено в табл. 1.
Инкремент показателя преломления dn/dc в хлороформе измеряли на рефрактометре “Refractometer RA-620” (КЕМ, Япония). При исследовании растворов наблюдалась асимметрия светорассеяния, поэтому Мw полимера определяли по методу Зимма [53–57]. Полученные величины второго вириального коэффициента показывают, что хлороформ является термодинамически хорошим растворителем. Характеристическую вязкость измеряли в вискозиметре Оствальда.
Исследование мицеллообразования в растворах ПФ-ПМАК в этаноле
Поведение растворов ПФ-ПМАК в этаноле исследовали методами светорассеяния и вискозиметрии на описанной выше установке “Photocor Complex”. Растворы фильтровали через фильтры Chromafil Xtra PA c диаметром пор 0.45 мкм. Процедура измерений подробно описана в работах [23, 24]. Чтобы доказать диффузионный характер мод, для растворов ПФ-ПМАК в этаноле также исследовали угловые зависимости I и Rh в интервале углов рассеяния света 40°–140°. Результаты обрабатывали по методу Зимма. Отметим, что этанол для щеток с боковыми цепями ПМАК являлся термодинамически плохим растворителем.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез полимерных щеток
Амфифильные полимерные щетки полифлуорен-прив-полиметакриловая кислота были получены в несколько стадий [52]:
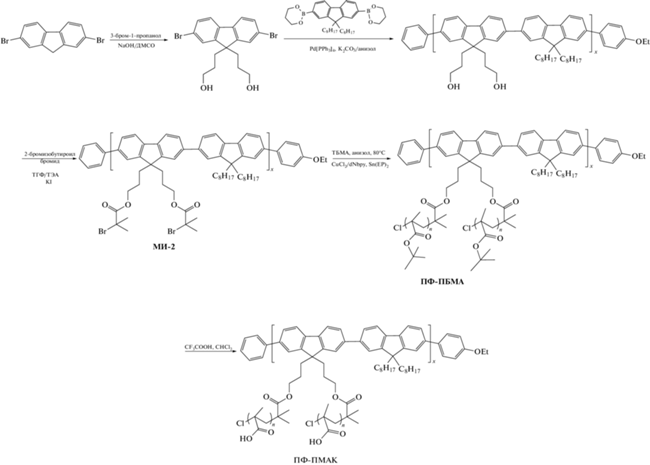
Сначала был синтезирован полифлуорен с боковыми гидроксильными группами, который затем модифицирован 2-бромизобутироил бромидом для введения инициирующих ATRP-групп. Далее были синтезированы полимерные щетки с боковыми цепями поли-трет-бутилметакрилата путем ATRP AGET трет-бутилметакрилата на полифлуореновом макроинициаторе в анизоле при 80°C. Амфифильные полимерные щетки были получены путем снятия защиты карбоксильных групп кислотой в мягких условиях.
Полимерные щетки с полифлуореновой основной цепью и боковыми цепями поли-трет-бутилметакрилата были синтезированы методом контролируемой радикальной полимеризации по механизму ATRP AGET. В качестве восстановителя в системе использовали Sn(EH)2. Было приготовлено два образца (ПФ-ПБМА-1 и ПФ-ПБМА-2) с соотношением МИ-2 : мономер 1 : 200. Образцы ПФ-ПБМА-1 и ПФ-ПБМА-2 различались конверсией мономера, она составляла 29 и 50% соответственно.
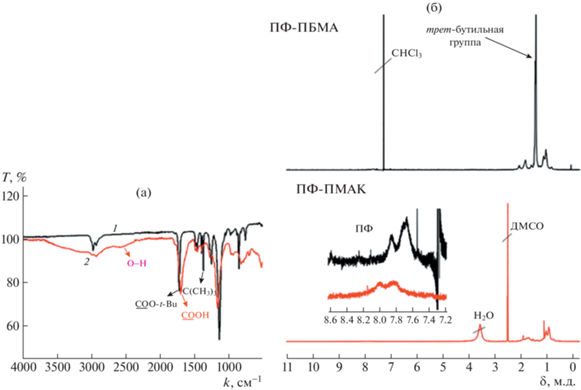
Снятие трет-бутильной защиты с боковых цепей щеток ПФ-ПБМА контролировали методами ИК- и ЯМР-спектроскопии. На рис. 3 видно, что после протонолиза не наблюдаются сигналы трет-бутильных групп.
Характеристики макроинициатора и ПФ-ПБМА в хлороформе
Молекулярную массу макроинициатора МИ-2 определяли в двух растворителях: ТГФ (ГПХ с тройным детектированием) и хлороформе (методом статического рассеяния света). Хроматограмма в ТГФ является унимодальной, степень полидисперсности низкой, Mw = 7 × 104. Соответственно степень полимеризации макроинициатора NMИ = = 38, а контурная длина его цепей LMИ = 57 нм, принимая во внимание, что длина макромономера макроинициатора составляет 1.5 нм. Так как степень функционализации z* = 0.92, каждая молекула полифлуорена содержит в среднем 70 функциональных групп. Отметим, что растворы макроинициатора в хлороформе были молекулярно-дисперсными, что, видимо, связано с высокой степенью функционализации полимера.
Растворы образцов ПФ-ПБМА в хлороформе также были молекулярно-дисперсными. Это позволило оценить их молекулярно-массовые и гидродинамические характеристики (табл. 2). Для определения ММ использовали метод Зимма (рис. 4). Отметим, что фактор формы – отношение среднеквадратичного радиуса инерции Rg к гидродинамическому размеру Rh-D – соответствует теоретическим значениям для линейной макромолекулы в хорошем растворителе.
Таблица 2.
Молекулярно-массовые и гидродинамические характеристики ПФ-ПБМА в хлороформе и ПФ-ПМАК в этаноле
| Образец | Mw × 10–3 | Mcal × 10–3 | [η], см3/г | Rg, нм | Rh-D, нм | Rg/Rh |
|---|---|---|---|---|---|---|
| ПФ-ПБМА -1 | 1280 | 1300 | 33 | 70 | 35 | 2.0 |
| ПФ-ПБМА -2 | 2100 | 2040 | 43 | 104 | 50 | 2.1 |
| ПФ-ПМАК-1 | 850 | 790 | 36 | – | 30 | – |
| ПФ-ПМАК-2 | 1200 | 1270 | 48 | 91 | 39 | 2.3 |
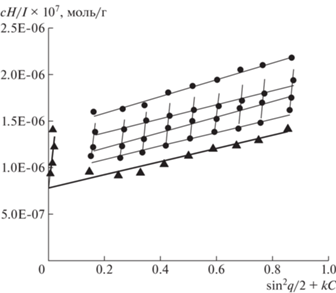
Используя данные табл. 1 и учитывая, что степень полимеризации привитых цепей составляет 58 и 100 для ПФ-ПБМА-1 и ПФ-ПБМА-2 соответственно, можно рассчитать максимально возможную молекулярную массу Mcal полученных щеток. Действительно,
(1)
${{M}^{{{\text{cal}}}}} = {{N}_{{{\text{MИ}}}}} \times z \times {{M}_{{{\text{s}}{\text{.ch}}}}}_{.} + {{M}_{{{\text{MИ}}}}},$Средняя линейная плотность прививки ΔL боковых цепей равна отношению длины основной цепи LMИ к числу привитых цепей fs.ch., и для обеих щеток ΔL = 57/70 = 0.8 нм. Параметр Ls.ch/ΔL, где Ls.ch – длина привитых цепей, характеризует стерические взаимодействия боковых цепей: чем больше Ls.ch/ΔL, тем сильнее эти взаимодействия. Отношение Ls.ch/ΔL равно 18 и 30 для ПФ-ПБМА-1 и ПФ-ПБМА-2 соответственно. Отсюда следует, что рассматриваемые привитые сополимеры являются очень плотными молекулярными щетками с сильным взаимодействием боковых цепей, что определяет поведение образцов ПФ-ПМАК в этаноле и в водных растворах.
Характеристики ПФ-ПМАК в растворах в этаноле
Для ПФ-ПМАК-1 и ПФ-ПМАК-2, полученных из щеток ПФ-ПБМА-1 и ПФ-ПБМА-2, распределение интенсивности I по гидродинамическим радиусам Rh рассеивающих объектов в растворе в этаноле является унимодальным. Молекулярные массы щеток ПФ-ПМАК заметно меньше, чем ММ образцов ПФ-ПБМА. Это различие обусловлено изменением молекулярной массы мономерного звена при переходе от ПФ-ПБМА к ПФ-ПМАК. Действительно, вычисленные с учетом различия в ММ мономерных звеньев молекулярные массы образцов ПФ-ПМАК (Mcal) с хорошей точностью совпадают со значениями Mw, определенными для данных полимеров в этаноле. Иными словами, в растворах ПФ-ПМАК в этаноле рассеивающими объектами являются макромолекулы. Учитывая, что основная цепь ПФ не растворяется в этаноле, можно предположить, что макромолекулы ПФ-ПМАК в нем напоминают по форме цилиндрические унимолекулярные мицеллы, в которых основная цепь экранируется от растворителя плотной короной из боковых цепей ПМАК.
Качественно в пользу такого заключения свидетельствует изменение гидродинамического радиуса и радиуса инерции при переходе от ПФ-ПБМА-2 к ПФ-ПМАК-2 (К сожалению, для образца ПФ-ПМАК-1 величину Rg надежно определить не удалось). Как видно из табл. 2, значения Rg и Rh-D для ПФ-ПМАК-2 меньше, чем для ПФ-ПБМА-2, что свидетельствует о небольшом изменении размеров макромолекул ПФ-ПМАК-2 в этаноле по сравнению с размерами молекул ПФ-ПБМА-2. Кроме того, параметр формы Rg/Rh-D несколько увеличивается при переходе от ПФ-ПБМА-2 к ПФ-ПМАК-2, что указывает на вытянутую форму макромолекул последнего. Другими словами, можно предположить, что цепи ПМАК-2 более “поджаты” по сравнению с цепями ПБМА в молекулах ПФ-ПБМА-2, т.е. диаметр молекул ПФ-ПБМА-2 больше диаметра молекул ПФ-ПМАК-2. Для обеих серий образцов характеристическая вязкость [η] для ПФ-ПМАК несколько больше, чем для ПФ-ПБМА, что может быть связано с уменьшением толщины макромолекул при переходе от ПФ-ПБМА к ПФ-ПМАК.
В работе [52] были исследованы сополимеры ПФ-ПМАК, полученные на макроинициаторе со степенью функционализации 0.75. Соответственно плотность прививки боковых цепей в щетках была гораздо ниже, чем у исследованных в данной работе образцов ПФ-ПМАК, и отношение Ls.ch./ΔL было меньше 18. Это приводило к тому, что в случае сополимеров с низкой плотностью прививки в растворах в этаноле формировались палочкообразные надмолекулярные структуры, состоящие из 2–10 макромолекул ПФ-ПМАК [52]. Иными словами, плотность прививки сильно влияет на поведение щеток ПФ-ПМАК в этаноле: в растворах очень плотных щеток формируются унимолекулярные мицеллы, а снижение плотности прививки приводит к агрегации.
Люминесцентные свойства мицелл
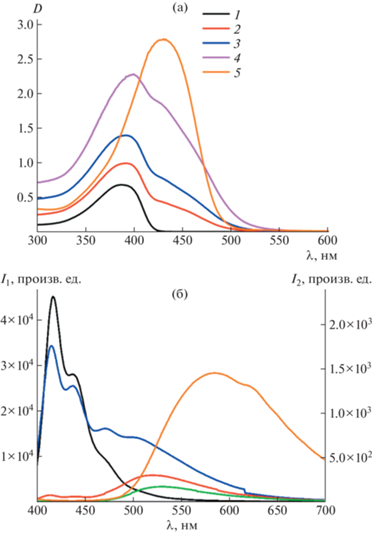
Несомненным достоинством исследуемых мицелл являются их люминесцентные свойства. Были изучены спектры поглощения (рис. 5а) и люминесценции в растворах (рис. 5б) с разным соотношением полимерная щетка : куркумин. Из спектров видно, что при образовании мицелл с куркумином происходит резкое тушение люминесценции, что свидетельствует о специфическом взаимодействии амфифильных полимерных щеток с куркумином. Кроме того, заметен перенос энергии люминесценции с амфифильных полимерных щеток (синяя область) на куркумин (зеленая область). Причем чем выше концентрация куркумина в мицеллах, тем перенос значительней. Таким образом, образование мицелл с куркумином подтверждено методом люминесценции.
Рис. 5.
Спектры поглощения (а) и спектры флуоресценции (б) куркумина, “пустых” и “нагруженных” куркумином мицелл амфифильных полимерных щеток. 1 – ПФ-ПМАК, 2 – ПФ-ПМАК + куркумин-1, 3 – ПФ-ПМАК + куркумин-2, 4 – ПФ-ПМАК + куркумин-3, 5 – куркумин; I1 – интенсивность люминесценции мицелл ПФ-ПМАК, I2 – интенсивность люминесценции композитов и куркумина.
Поведение ПФ-ПМАК и комплексов ПФ-ПМАК с куркумином в водной среде
Гидродинамический размер частиц в водном растворе ПФ-ПМАК-2 совпадает с радиусом Rh-D рассеивающих объектов в растворах этого сополимера в этаноле (табл. 3). Соответственно можно предположить, что при переходе из этанола в водную среду конформация ПФ-ПМАК-2 сохраняется, т.е. водные растворы ПФ-ПМАК-2 также молекулярно-дисперсны, а форма молекул ПФ-ПМАК-2 приближается к форме палочкообразной мицеллы типа ядро–оболочка.
Таблица 3.
Гидродинамические размеры растворенных комплексов с куркумином
| Образец | СПФ-ПМАК, г/дл | Скуркумин, г/дл | Rh-D, нм | Массовое соотношение куркумин : полимер |
|---|---|---|---|---|
| ПФ-ПМАК-2 | 0.019 | 0 | 39 | – |
| 0.019 | 0.0013 | 55 | 0.060 | |
| 0.019 | 0.0025 | 50 | 0.130 | |
| 0.019 | 0.0038 | 50 | 0.200 | |
| ПФ-ПМАК-3 | 0.022 | 0.0021 | 52 | 0.097 |
| 0.019 | 0.0022 | 71 | 0.110 | |
| 0.017 | 0.0023 | 71 | 0.136 | |
| 0.016 | 0.0024 | 71 | 0.155 | |
| 0.014 | 0.0025 | 78 | 0.174 | |
| 0.013 | 0.0026 | 96 | 0.193 | |
| – | 0 | 24 | – |
Иная ситуация наблюдается для образца с низкой плотностью прививки ПФ-ПМАК-3, исследованного в работе [52] (Mw = 3.75 × 105, Rh-D = 17 нм в хлороформе). Гидродинамический радиус Rh-D частиц ПФ-ПМАК-3 в этаноле был вследствие агрегации на 20% больше, чем Rh-D, определенный в хлороформе, а ММ агрегатов составляла 7 × 105 (средняя степень агрегации равна 2) [52]. В водной среде размер частиц ПФ-ПМАК-3 увеличивается еще на 20% (табл. 3), т.е. степень агрегации возрастает. Таким образом, сопоставление данных для ПФ-ПМАК-2 и ПФ-ПМАК-3 показывает, что плотность прививки боковых цепей влияет на поведение ПФ-ПМАК в воде. В табл. 3 и на рис. 6 также представлены размеры “пустых” и “нагруженных” мицелл в водно-спиртовых растворах с соответствующими концентрациями ПФ-ПМАК и куркумина.
Рис. 6.
Распределение по гидродинамическим размерам “пустых” (а) и “нагруженных” куркумином (б–г) мицелл амфифильных полимерных щеток ПФ-ПМАК-3 в водных растворах. Iмакс – максимальная интенсивность рассеянного света при данной концентрации. Содержание куркумина 0.0021 (б), 0.00250 (в) и 0.0026 г/дл (г).
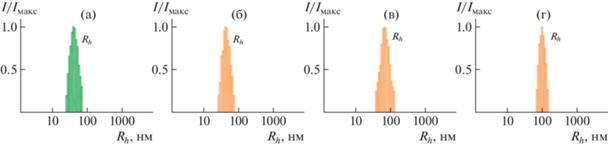
Влияние плотности прививки проявляется и при добавлении куркумина в водные растворы образцов ПФ-ПМАК-2 и ПФ-ПМАК-3. В случае ПФ-ПМАК-2 при добавлении куркумина увеличивается гидродинамический размер Rh-D рассеивающих объектов (табл. 3). Следовательно, куркумин снижает растворимость ПФ-ПМАК-2 и приводит к агрегации цепей. При этом значение Rh-D не зависит от массовой доли куркумина в растворе, т.е. в исследованном интервале содержания куркумина степень агрегации в растворах ПФ-ПМАК-2 не изменяется, чему, вероятно, способствует плотная оболочка из боковых цепей полиметакриловой кислоты.
При добавлении небольшого количества куркумина в раствор ПФ-ПМАК-3 гидродинамический радиус увеличивается в два раза, т.е. гораздо сильнее, чем в случае ПФ-ПМАК-2. Кроме того, увеличение относительного содержания куркумина сопровождается быстрым возрастанием размеров агрегатов, что также отличает поведение более редкой щетки ПФ-ПМАК-3 от очень плотной щетки ПФ-ПМАК-2.
ЗАКЛЮЧЕНИЕ
Методом контролируемой радикальной полимеризации синтезирована серия амфифильных молекулярных щеток с полифлуореновой основной цепью и боковыми цепями ПБМА и ПМАК с высокой плотностью прививки. Как растворы ПФ-ПБМА в хлороформе, так и растворы ПФ-ПМАК в этаноле являются монодисперсными по данным динамического светорассеяния. В этаноле макромолекулы ПФ-ПМАК по форме близки к унимолекулярным мицеллам, в которых нерастворимая основная цепь экранируется от растворителя плотной оболочкой из боковых цепей ПМАК. Снижение плотности прививки боковых цепей приводит к агрегации макромолекул ПФ-ПМАК в этаноле.
В водных растворах даже плотных щеток ПФ-ПМАК наблюдается агрегация макромолекул. Характер поведения щеток ПФ-ПМАК в водных растворах при добавлении куркумина зависит от плотности прививки боковых цепей. Размер агрегатов в растворах комплексов куркумин–плотная щетка не зависит от содержания куркумина, а в случае менее плотных щеток степень агрегации увеличивается при повышении относительного содержания куркумина.
Работа выполнена при финансовой поддержке мегагранта Министерства науки и высшего образования Российской Федерации (госконтракт № 14.W03.31.0022).
Список литературы
Cheng G., Böker A., Zhang M., Krausch G., Müller A.H.E. // Macromolecules. 2001. V. 34. № 20. P. 6883.
Lu X., Wei A., Fan Q., Wang L., Chen P., Dong X., Huang W. // Mater. Res. Bull. 2012. V. 47. № 12. P. 4335.
Zhou L., Geng J., Wang G., Liu J., Liu B. // Polym. Chem. 2013. V. 4. № 20. P. 5243.
Meleshko T.K., Ivanov I.V., Kashina A.V., Bogorad N.N., Simonova M.A., Zakharova N.V., Filippov A.P., Yakimansky A.V. // Polymer Science B. 2018. V. 60. № 1. P. 35.
Borodinov N., Gil D., Savchak M., Gross C.E., Yadavalli N.S., Ma R., Tsukruk V.V., Minko S., Vertegel A., Luzinov I. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 16. P. 13941.
Ivanov I.V., Meleshko T.K., Kashina A.V., Yakimansky A.V. // Russ. Chem. Rev. 2019. V. 88. № 12. P. 1248.
Lian X., Wu D., Song X., Zhao H. // Macromolecules. 2010. V. 43. № 18. P. 7434.
Teulère C., Ben-Osman C., Barry C., Nicolaÿ R. // Eur. Polym. J. 2020. V. 141. P. 110080.
Filippov A.P., Belyaeva E.V., Krasova A.S., Simonova M.A., Meleshko T.K., Ilgach D.M., Bogorad N.N., Yakimansky A.V., Larin S.V., Darinskii A.A. // Polymer Science A. 2014. V. 56. № 4. P. 393.
Filippov A.P., Belyaeva E.V., Krasova A.S., Simonova M.A., Tarabukina E.B., Meleshko T.K., Ilgach D.M., Bogorad N.N., Yakimansky A.V. // Polymer Science. A. 2014. V. 56. № 1. P. 1.
Tarabukina E., Amirova A., Belyaeva E., Krasova A., Simonova M., Filippov A., Meleshko T., Ilgach D., Bogorad N., Yakimansky A. // J. Macromol. Sci. B. 2013. V. 52. № 11. P. 1545.
Yakimansky A.V., Meleshko T.K., Ilgach D.M., Bauman M.A., Anan’Eva T.D., Klapshina L.G., Lermontova S.A., Balalaeva I.V., Douglas W.E. // J. Polym. Sci., Polym. Chem. 2013. V. 51. № 20. P. 4267.
Grimsdale A.C., Chan K.L., Martin R.E., Jokisz P.G., Holmes A.B. // Chem. Rev. 2009. V. 109. № 3. P. 897.
Zhu C., Liu L., Yang Q., Lv F., Wang S. // Chem. Rev. 2012. V. 112. № 8. P. 4687.
Nosova G.I., Ilgach D.M., Berezin I.A., Zhukova E.V., Kopylova T.N., Nikonova E.N., Gadirov R.M., Smyslov R.Y., Yakimansky A.V. // Mendeleev Commun. 2017. V. 27. № 3. P. 265.
Ilgach D.M., Nosova G.I., Kopylova T.N., Nikonova E.N., Gadirov R.M., Smyslov R.Y., Litvinova L.S., Yakimansky A.V. // Mendeleev Commun. 2017. V. 27. № 4. P. 357.
Simonova M., Filippov A., Nosova G., Zhukova E., Litvinova L., Berezin I., Yakimansky A. // Mater. Today Chem. 2021. V. 22. P. 100553.
Zhang Z., Fan Q., Sun P., Liu L., Lu X., Li B., Quan Y., Huang W. // Macromol. Rapid Commun. 2010. V. 31. № 24. P. 2160.
Zhang Z., Lu X., Fan Q., Hu W., Huang W. // Polym. Chem. 2011. V. 2. № 10. P. 2369.
Liu X., Shi L., Zhang Z., Fan Q., Huang Y., Su S., Fan C., Wang L., Huang W. // Analyst. 2015. V. 140. № 6. P. 1842.
Gu P., Liu X., Tian Y., Zhang L., Huang Y., Su S., Feng X., Fan Q., Huang W. // Sensors Actuators, Chem. 2017. V. 246. P. 78.
Balcı Leinen M., Klein P., Sebastian F.L., Zorn N.F., Adamczyk S., Allard S., Scherf U., Zaumseil J. // Adv. Electron. Mater. 2020. V. 6. № 11. P. 1.
Zhou M., He Z., Chen Y., Zhu L., Li L., Li J. // Macromol. Rapid Commun. 2021. V. 42. № 4. P. 2.
Müllner M. // Macromol. Chem. Phys. 2016. V. 217. № 20. P. 2209.
Yang C., Huang S., Wang X., Wang M. // Polym. Chem. 2016. V. 7. № 48. P. 7455.
Liu F., Zhao X., Zhang X., Zhang X., Peng J., Yang H., Deng K., Ma L., Chang C., Wei H. // Polym. Chem. 2018. V. 9. № 39. P. 4866.
Ji Y., Lu F., Hu W., Zhao H., Tang Y., Li B., Hu X., Li X., Lu X., Fan Q., Huang W. // Biomaterials. 2019. V. 219. P. 119393.
Yang Z., Li L., Jin A.J., Huang W., Chen X. // Mater. Horizons. 2020. V. 7. № 6. P. 1474.
Xu Q., Lv F., Liu L., Wang S. // Macromol. Rapid Commun. 2020. V. 41. № 15. P. 1.
Yang C., Liu H., Zhang Y., Xu Z., Wang X., Cao B., Wang M. // Biomacromolecules. 2016. V. 17. № 5. P. 1673.
Zhang Z., Tan M., Kong L., Lu X., Sun P., Mo H., Fan Q., Huang W. // J. Chem. 2020. V. 2020.
Zhu Y., Yang B., Chen S., Du J. // Prog. Polym. Sci. 2017. V. 64. P. 1.
Blanazs A., Armes S.P., Ryan A.J. // Macromol. Rapid Commun. 2009. V. 30. № 4–5. P. 267.
Deshmukh A.S., Chauhan P.N., Noolvi M.N., Chaturvedi K., Ganguly K., Shukla S.S., Nadagouda M.N., Ami-nabhavi T.M. // Int. J. Pharm. 2017. V. 532. № 1. P. 249.
Simonova M., Ivanov I., Meleshko T., Kopyshev A., Santer S., Yakimansky A., Filippov A. // Polymers. 2020. V. 12. № 12. P. 1.
Krasova A., Belyaeva E., Tarabukina E., Filippov A., Meleshko T., Ilgach D., Bogorad N., Yakimansky A. // Macromol. Symp. 2012. V. 316. № 1. P. 32.
Simonova M., Kamorin D., Kazantsev O., Nepomnyashaya M., Filippov A. // Polymers. 2021. V. 13. № 16. P. 2715.
Meng Z., Hou W., Zhou H., Zhou L., Chen H., Wu C. // Macromol. Rapid Commun. 2018. V. 39. № 5. P. 1.
Wu Y., Xiao Y., Huang Y., Xu Y., You D., Lu W., Yu J. // Biomacromolecules. 2019. V. 20. № 3. P. 1167.
Zhao H., Hu W., Ma H., Jiang R., Tang Y., Ji Y., Lu X., Hou B., Deng W., Huang W., Fan Q. // Adv. Funct. Mater. 2017. V. 27. № 44. P. 1.
Froiio F., Lammari N., Tarhini M., Alomari M., Louaer W., Meniai A.H., Paolino D., Fessi H., Elaissari A. // Micro and Nano Technologies, Smart Nanocontainers. Amsterdam: Elsevier. 2020. P. 271.
Zou Y., Meng F., Deng C., Zhong Z. // J. Control. Release. 2016. V. 239. P. 149.
Budhian A., Siegel S.J., Winey K.I. // Int. J. Pharm. 2007. V. 336. № 2. P. 367.
Blokhin A.N., Razina A.B., Bursian A.E., Ten’kov-tsev A.V. // Polymer Science B. 2021. V. 63. № 1. P. 52.
Farhoudi L., Kesharwani P., Majeed M., Johnston T.P., Sahebkar A. // Int. J. Pharm. 2022. V. 617. P. 121622.
Qiu N., Du X., Ji J., Zhai G. // Drug Dev. Ind. Pharm. 2021. V. 47. № 6. P. 839.
Hu Y., Darcos V., Monge S., Li S. // Int. J. Pharm. 2015. V. 491. № 1–2. P. 152.
Chang T., Trench D., Putnam J., Stenzel M.H., Lord M.S. // Mol. Pharm. 2016. V. 13. № 3. P. 924.
Migliore R., D’antona N., Sgarlata C., Consoli G.M.L. // Nanomaterials. 2021. V. 11. № 11. P. 2930.
Anitha A., Maya S., Deepa N., Chennazhi K.P., Nair S.V., Jayakumar R. // J. Biomater. Sci. Polym. Ed. 2012. V. 23. № 11. P. 1381.
Youssouf L., Bhaw-Luximon A., Diotel N., Catan A., Giraud P., Gimié F., Koshel D., Casale S., Bénard S., Meneyrol V., Lallemandc L., Meilhaca O., D’Hellencourta C.L., Jhurryb D., Couprie J. // Carbohydr. Polym. 2019. V. 217. P. 35.
Simonova M., Ilgach D., Kaskevich K., Nepomnyashaya M., Litvinova L., Filippov A., Yakimansky A. // Polymers. 2021. V. 13. № 24. P. 4429.
Kratohvil P. Classical Light Scattering from Polymer Solution. Amsterdam: Elsevier, 1987.
Schärtl W. Light Scattering from Polymer Solutions and Nanoparticle Dispersions. Berlin: Springer, 2007.
Kratohvil J.P., Aminabhavi T.M. // J. Phys. Chem. 1982. V. 86. № 8. P. 1254.
Gasilova E.R., Koblyakova M.A., Filippov A.P., Zakharova O.G., Zaitsev S.D., Semchikov Yu.D. // Polymer Science A. 2006. V. 48. № 9. P. 989.
Tsvetkov V.N. Rigid-Chain Polymers. New York: Plenum, 1989.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)