

Высокомолекулярные соединения (серия С), 2022, T. 64, № 2, стр. 176-191
ИОННАЯ ПОЛИМЕРИЗАЦИЯ С РАСКРЫТИЕМ ЦИКЛА В СИНТЕЗЕ ЗВЕЗДООБРАЗНЫХ ПОЛИМЕРОВ
А. Н. Блохин a, *, М. М. Дудкина a, А. В. Теньковцев a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
* E-mail: 44stuff44@gmail.com
Поступила в редакцию 05.04.2022
После доработки 26.07.2022
Принята к публикации 09.08.2022
- EDN: YIJRJY
- DOI: 10.31857/S2308114722700182
Аннотация
Рассмотрены особенности получения звездообразных полимеров методом ионной полимеризации с раскрытием цикла. Продемонстрированы возможности контролируемого синтеза звездообразных структур с использованием циклических мономеров различных классов. Систематизированы и обобщены данные по получению звездообразных полимеров с применением подходов “прививка от” мультифункционального инициатора, “прививка через” макромономер, “прививка на” мультифункциональный связующий агент.
ВВЕДЕНИЕ
Звездообразные полимеры занимают особое место в науке о полимерах благодаря своим свойствам и характеристикам. Активный интерес к звездообразным полимерам различного состава связан с возможностью их практического использования в разных областях науки и техники. Так, звездообразные полимеры могут выступать в роли мицеллообразующих агентов, служить для изготовления материалов с нелинейными оптическими свойствами, жидкокристаллических и электропроводящих полимерных материалов, высокоупорядоченных пористых пленок и мембран, пленок Лэнгмюра–Блоджетт [1–7]. Звездообразные гибридные макромолекулы могут выполнять функции наноконтейнеров и нанореакторов [8, 9].
Приоритетными направлениями использования звездообразных полимеров являются медицина, биотехнология и биоинженерия. В настоящее время объектами особого интереса стали звездообразные системы на основе биосовместимых и стимул-чувствительных полимеров, поскольку они могут служить для разработки “умных” полимерных материалов, выполняющих роль носителей лекарственных препаратов с функцией их контролируемого выделения.
ПОЛИМЕРИЗАЦИЯ С РАСКРЫТИЕМ ЦИКЛА
В последнее десятилетие внимание ученых привлекают процессы полимеризации с раскрытием цикла различных гетероциклических соединений [10]. Живой характер, отсутствие реакций необратимого обрыва цепей и низкая вероятность протекания реакций передачи цепей позволяют задействовать полимеризации с раскрытием цикла для синтеза полимеров сложной архитектуры с заданными характеристиками и узким молекулярно-массовым распределением в том числе и для звездообразных полимеров. Биосовместимость материалов на основе гетероцепных полиэфиров, полиамидов и поликарбонатов, полученных данным методом, дает возможность применять их в системах доставки лекарственных препаратов, в тканевой инженерии и других биомедицинских приложениях [11–13], а уникальные механические свойства позволяют использовать, например, алифатические полиэфиры в качестве компонентов в материалах с памятью формы [14, 15]. Полимеризацию с раскрытием цикла можно применять для широкого спектра циклических мономеров, таких как лактоны, лактамы, лактиды, циклические карбонаты, оксазолы, силоксаны и простые эфиры, что дает возможность получать звездообразные полимеры с лучами различного химического строения [16]. Движущей силой полимеризации в большинстве случаев (3–8-атомные циклы) является внутренняя напряженность цикла, которая обусловливает термодинамическую выгоду в результате его раскрытия. При этом ненапряженные шестичленные циклы не должны полимеризоваться. Однако в некоторых случаях, например для циклических силоксанов, дисульфидов и карбонатов, движущей силой полимеризации служит энтропийный выигрыш в результате формирования дополнительных вращательных степеней свободы при раскрытии цикла [17]. В зависимости от используемого инициатора, каталитической системы и мономера полимеризация с раскрытием цикла может протекать по различным механизмам.
Катионный механизм
В качестве инициатора здесь выступает соединение с электрофильным центром, способствующим образованию в кольце положительного заряда. Далее, реакция бимолекулярного нуклеофильного замещения с участием другого мономера приводит к раскрытию цикла. На конце цепи образуется электрофильный центр, в результате чего процесс присоединения молекул мономера повторяется [18].
Анионный механизм
Инициатор анионного типа атакует α-атом углерода в мономере, что приводит к раскрытию цикла и образованию анионного центра полимеризации на конце цепи. Процесс роста цепи повторяется путем нуклеофильной атаки со стороны растущей цепи на другую молекулу мономера [19].
Ионно-координационный механизм
Реализация в данном случае происходит в присутствии металлорганических катализаторов. Мономер координируется к катализирующему фрагменту, после чего происходит перегруппировка электронов, вызывающая раскрытие цикла. Конец растущей цепи координируется к атому металла в комплексе, после чего процесс повторяется с другой молекулой мономера [20].
Метод активированного мономера
На первой стадии проводится протонирование мономера (активация), после чего цикл становится более восприимчивым к нуклеофильной атаке гидроксильной группой инициатора, что способствует раскрытию цикла, а затем процесс повторяется [21]. Данный механизм является частным случаем катионной полимеризации с раскрытием цикла.
ЦИКЛИЧЕСКИЕ СЛОЖНЫЕ ЭФИРЫ
К одному из наиболее активно исследуемых классов звездообразных полимеров, получаемых методом полимеризации с раскрытием цикла, относятся алифатические полиэфиры. В качестве мономеров для получения полиэфиров могут применяться разнообразные циклические эфиры карбоновых кислот, эфиры фосфорной кислоты, а также карбонаты. В числе наиболее часто используемых мономеров можно указать L-лактид (1), ε-капролактон (2), гликолид (3), β-бутиролактон (4) и триметиленкарбонат (5) [22]:

Полимеризация с раскрытием цикла этих мономеров инициируется гидроксилсодержащими соединениями в присутствии кислот Льюиса (катионный механизм), протонных доноров (метод активированного мономера), или протонных акцепторов (анионный механизм), а рост цепей протекает в контролируемом режиме без необратимого обрыва цепей.
В качестве инициаторов полимеризации используют полифункциональные гидроксилсодержащие соединения различной природы, в том числе макроциклические соединения, позволяющие получать звездообразные алифатические полиэфиры по схеме “прививка от” с функциональными центрами ветвления – циклодекстрины (1), дендримеры на основе пентаэритрита (2, 3), каликсарены (4), аннулены (5) и циклические силоксаны (6) [23]:

Опубликовано большое число работ, посвященных синтезу и исследованию свойств звездообразных систем на основе поли-ε-капролактона, обладающего биосовместимостью и стабильностью в биологических средах [24]. Это сделало полимер пригодным для широкого применения: от биомедицинских приложений до производства упаковки и микроэлектроники [25]. Для синтеза звездообразных полимеров были задействованы полифункциональные инициаторы различного типа. Например, четырех- и шестилучевые звездообразные поли-ε-капролактоны получены с применением соответствующих полифункциональных иницицаторов каликс[n]аренового типа методом ионно-координационной полимеризации с раскрытием цикла [26]. Инициирующие гидроксильные группы были внедрены в нижний обод каликсаренового макроцикла через короткий алифатический спейсер (C2), катализатором служил трис-(2,6-ди-трет-бутил-4-метилфенолят)иттрия. В работе [27] синтезирован звездообразный амфифильный блок-сополимер с центром ветвления дендримерного типа и лучами поли-ε-капролактон-блок-полиэтиленгликоля.
Настоящая работа представляет собой пример совместного использования подходов “прививка от” и “прививка на” в синтезе звездообразных блок-сополимеров. В качестве инициатора полимеризации ε-капролактона использован полиамидоаминовый дендример с концевыми гидроксильными группами. На заключительной стадии к образовавшимся лучам поли-ε-капролактона реакцией этерификации были привиты гидрофильные блоки полиэтиленгликоля. Шестилучевой звездообразный поли-ε-капролактон синтезирован с применением в качестве инициатора D-сорбита [28], что привело к получению полимеров с хиральным центром ветвления. Кроме того, в качестве инициаторов полимеризации ε-капролактона использованы такие соединения, как пентаэритрит, порфирины, циклодекстрины и глицерин [29–32].
В табл. 1 приведены характеристики звездообразных систем, полученных на основе поли(ε-капролактон)а с использованием подхода “прививка от мультифункционального инициатора” при различных способах проведения полимеризации ε-капролактона. Как видно из таблицы, наибольший контроль достигается в случае полимеризации по катионному механизму. Кроме того, узким молекулярно-массовым распределением характеризуются лишь образцы с небольшим числом лучей (N = 3–6), в то время как для сильноразветвленных систем такого контроля достичь не удается.
Таблица 1.
Молекулярно-массовые характеристики звездообразных поли(ε-капролактон)ов
| Инициатор/катализатор | Механизм | Число лучей, N | Mn × 103 | Ð |
|---|---|---|---|---|
| Триметилолпропан/фосфазеновое основание [33] | анионный | 3 | 12.0 | 1.48 |
| Триметилолпропан/фумаровая кислота [34] | катионный | 3 | 15.4 | 1.14 |
| Пентаэритрит/фумаровая кислота [34] | катионный | 4 | 15.8 | 1.10 |
| Каликс[4]арен/трис-(дитретбутилфенолят)иттрия [26] | ионно-координационный | 4 | 18.3 | 1.41 |
| Каликс[6]арен/трис-(дитретбутилфенолят)иттрия [26] | ионно-координационный | 6 | 36.7 | 1.42 |
| D-сорбит/этилгексаноат олова [28] | катионный | 6 | 6.1 | 1.11 |
| D-сорбит/ферментативный катализ [28] | катионный | 6 | 10.1 | 1.23 |
| Полиамидоамин дендример [35] | анионный | 4 | 14.0 | 1.51 |
| Окта-(2-гидроксиэтилтио)порфирин/октаноат олова [30] | катионный | 8 | 24.9 | 1.78 |
| Силсесквиоксан полиол/октаноат олова [36] | катионный | 29 | 106.7 | 3.13 |
Примечание. Здесь и в табл. 2, 3 молекулярные массы определены методом гель-проникающей хроматографии.
Кроме поли-ε-капролактона, популярность приобрели и другие биоразлагаемые полиэфиры, среди которых полигликолид и полилактид [37]. Полилактид представляет собой биоразлагаемый полимер, мономерным звеном является молочная кислота. Альтернативный поликонденсационный метод получения полилактида из молочной кислоты имеет ряд недостатков, в том числе низкую степень контроля над структурой продукта, в связи с чем полимер традиционно синтезируют полимеризацией с раскрытием цикла димера молочной кислоты (лактида), которая позволяет получать продукт с заданной молекулярной массой и узким молекулярно-массовым распределением (Đ = 1.1–1.5). Первый звездообразный полилактид был получен в 1989 г. с использованием макроинициатора – четырехлучевого звездообразного полиэтиленгликоля с центром ветвления типа пентаэритрита [38]. В качестве катализатора был использован октаноат олова. Синтезированный звездообразный блок-сополимер предполагалось задействовать в качестве наноконтейнеров для лекарственных препаратов.
Для синтеза звездообразных полилактидов с применением подхода “прививка от” применялись различные полифункциональные гидроксилсодержащие соединения. Среди них стоит выделить наиболее часто применяемые простые полиолы – пентаэритрит и дипентаэритрит, которые дают четырех- и шестилучевые звездообразные структуры соответственно [29, 39], а также макроциклические (β-циклодекстрин) и металлокомплексные центры ветвления [40–42]. Полимерными макроинициаторами для получения звездообразных полилактидов служили системы на основе полиэтиленимина, полисилоксана и полиамина [43–45].
В табл. 2 приведены молекулярно-массовые характеристики звездообразных полилактидов, полученных методом катионной полимеризации с раскрытием цикла на основе полифункциональных гидроксилсодержащих инициаторов типа глицерина и пентаэритрита. Как видно, низкомолекулярные образцы полилактида характеризуются достаточно узким молекулярно-массовым распределением в диапазоне Ð = 1.05–1.20, что свидетельствует о быстром и одновременном инициировании полимеризации по всем группам центра ветвления. Тем не менее при многократном увеличении длины полимерных лучей дисперсность аномально возрастает. Также отметим, что количество гидроксильных групп, участвующих в инициировании полимеризации, не влияет существенно на молекулярно-массовое распределение полимера.
Таблица 2.
Молекулярно-массовые характеристики звездообразных полилактидов
АНИОННО-ПОЛИМЕРИЗУЕМЫЕ МОНОМЕРЫ
Полимеризацию с раскрытием цикла, протекающую по анионному механизму, также применяют для получения ряда полиэфиров, политиоэфиров и полисилоксанов, в том числе промышленного назначения [49–53]:
Мономер 




Растущая цепь 




Например, полимеризация ε-капролактама по анионному механизму является важным производственным процессом получения нейлона-6 [54]. Для инициирования анионной полимеризации с раскрытием цикла использовались различные металлорганические соединения с нуклеофильными центрами, среди них классические алкиллитиевые инициаторы, алкоголяты и карбоксилаты щелочных металлов, алкоксиды алюминия.
В ряду полимеризующихся по анионному механизму циклических мономеров особое место занимают этиленоксид, а также его ближайший гомолог пропиленоксид. Получаемые на их основе гомополимерные полиэтиленоксиды, полипропиленоксиды и сополимеры (плюроники, полоксамеры) нашли широчайшее применение в различных областях науки, промышленности и медицины [55]. Нетоксичность и устойчивость к распознаванию иммунной системой полиэтиленоксида закрепили за ним статус “золотого стандарта” среди полимеров медицинского применения [56]. В многочисленных работах по синтезу разветвленных структур, содержащих гидрофильные полиэфирные фрагменты, для внедрения блоков полиэтилен–пропилен-оксидов чаще всего применяют подходы “прививка на” и “прививка через”. Предпочтение таким подходам отдается по причине наличия концевых гидроксильных групп у промышленно доступных полиэтилен- и полипропиленгликолей разнообразной массы, а также их сополимеров. Известны работы, посвященные синтезу разветвленнных, в том числе звездообразных, полиэфиров данного типа с использованием полифункциональных инициаторов. В 1988 г. P. Rempp с соавторами [57] впервые получили образцы звездообразных полиэтиленоксидов методом анионной полимеризации с раскрытием цикла. Инициатором служил алкоголят триметилолпропана для трехлучевого полиэтиленоксида, а также макроинициатор анионного типа на основе сшитого ядра дивинилбензола для получения мультилучевого звездообразного полиэтиленоксида. Однако стоит отметить, что образцы полимеров были синтезированы с довольно значительным разбросом функциональности. Большего контроля над структурой удалось достичь J. Roovers и соавторам [58], которые синтезировали анионной полимеризацией четырех-, восьми- и шестнадцатилучевые полиэтиленоксиды на основе макроинициаторов дендримерного типа – карбосилановых дендримеров нулевой, первой и второй генерации соответственно. Молекулярно-массовые и структурные характеристики синтезированных образцов приведены в табл. 3. Из данных таблицы видно, что дисперсность полимерных систем слабо зависит от числа и длины лучей, и это характерно для процесса истинно живой анионной полимеризации. Синтезированные образцы отличаются чрезвычайно узким молекулярно-массовым распределением (Ð < 1.1).
Таблица 3.
Молекулярно-массовые характеристики зведообразных полиэтиленгликолей [58]
| Центр ветвления/инициирующая группа | Число лучей, N | Mn × 103 | Ð |
|---|---|---|---|
| Карбосилановый дендример (G0)/алкоголят калия | 4 | 9.8 | 1.07 |
| 4 | 16.8 | 1.06 | |
| 4 | 30.1 | 1.09 | |
| Карбосилановый дендример (G2) | 16 | 30.5 | 1.07 |
В работе [59] впервые была применена комбинация подходов “прививка через” и “прививка от” в синтезе сверхразветвленного полиэтиленоксида:

Здесь в качестве сшивателя растущих линейных цепей полиэтиленоксида выступал бис-эпоксид, после чего анионные центры в составе ядра звездообразного полимера были использованы для инициирования полимеризации мономерного этиленоксида. Также методом анионной полимеризации с раскрытием цикла были синтезированы полиэтиленгликоли с функциональными макроциклическими центрами ветвления. Например, Y. Gnanou с соавторами [60] продемонстрировали возможность синтеза восьмилучевого полиэтиленоксида с каликс[8]ареновым ядром в соответствии с подходом “прививка от”. Показано, что с использованием полифункциональных анионных инициаторов на основе каликс[8]арена могут быть получены звездообразные полимеры с заданной молекулярной массой и узким молекулярно-массовым распределением (Đ < 1.2).
Анионная полимеризация пропиленоксида с раскрытием цикла также была использована в синтезе амфифильных звездообразных полимеров, в том числе, в рамках последовательной сополимеризации с другими мономерами [61, 62].
КАТИОННО-ПОЛИМЕРИЗУЕМЫЕ МОНОМЕРЫ
Многие гетероциклические соединения способны к катионной полимеризации с раскрытием цикла, в связи с чем данный процесс используют в синтезе ряда полимеров промышленного значения, таких как полисилоксаны, полиэтиленимины или полиацетали [63]. Однако лишь ограниченное количество мономеров полимеризуется в контролируемом режиме, позволяющем применять их в синтезе сложноразветвленных систем, – это тетрагидрофуран (1), 3,3-диметилтиетан (2), 2-метил-2-оксазолин (3) и 1,3-диоксолан (4):
Мономер 



Растущая цепь 



Среди гетероциклических соединений по катионному механизму способны полимеризоваться простые и сложные циклические эфиры и тиоэфиры, циклические карбонаты, оксазолы, этиленимин, диоксолан, триоксан, а также циклические силоксаны. В качестве инициаторов катионной полимеризации с раскрытием цикла могут выступать кислоты Бренстеда, кислоты Льюиса в присутствии доноров протонов (вода, спирты, галогеноводороды) или карбокатионов (алкилгалогениды, простые эфиры), а также некоторые ковалентные соединения, обладающие выраженными электрофильными свойствами.
Катионная полимеризация некоторых гетероциклов, в частности циклических простых эфиров, в определенных диапазонах температуры и концентрации мономера протекает в условиях близких к “живым”. Побочные реакции необратимого обрыва и передачи цепей отсутствуют или их влиянием на ход процесса можно пренебречь. Одна из наиболее успешно изученных катионных “живых” систем – полимеризация тетрагидрофурана [64]. Инициирование полимеризации тетрагидрофурана обычно происходит путем протонирования кислорода в гетероцикле или путем нуклеофильной атаки кислорода на электрофильный атом углерода в молекуле инициатора. Затем рост цепи продолжается путем нуклеофильной атаки мономера на электрофильный атом углерода в α-положении к гетероатому:
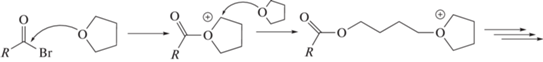
Последующее добавление нуклеофильных терминирующих агентов (вода, спирты, тиолы, амины) в систему приводит к обрыву цепей и завершению процесса полимеризации. Использование функциональных терминирующих агентов в этом случае позволяет получать полимеры с концевыми группами заданного строения с целью их дальнейшей модификации, сополимеризации или анализа структурных особенностей и молекулярно-массовых характеристик.
Известны два принципиально различающихся между собой метода получения звездообразных систем на основе политетрагидрофурана. Первый метод впервые был представлен в 1997 г. как пример применения подхода “прививка на” в условиях “живой” катионной полимеризации [65]. Звездообразные полимеры с разным числом лучей получены реакцией терминирования “живых” цепей политетрагидрофурана диэтилентриамином в присутствии 2,2,6,6-тетраметилпиперидина, выполняющего роль ловушки для протонов:
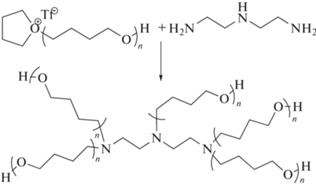
Теоретическая функциональность выбранного терминирующего агента равна f = 7, однако вследствие стерических эффектов, а также электростатического отталкивания достичь такой степени присоединения лучей представляется затруднительным. При этом было показано, что число привитых к ядру лучей сильно зависит от их молекулярной массы. Аналогичный подход был представлен для синтеза звездообразного политетрагидрофурана с функциональными алкеновыми концевыми группами, но в качестве мультифункционального терминирующего агента использован три-2-аминоэтиламин с теоретической функциональностью f = 10 [66]. Получены звездообразные макромолекулы с переменным числом лучей от трех до шести.
Большего контроля над структурой звездообразных политетрагидрофуранов удалось добиться в 2000 г. группе авторов [67], которые предложили использовать мультифункциональные инициаторы трифлатного типа (эфиры трифторметансульфокислоты), приготовленные in situ непосредственно перед проведением катионной полимеризации тетрагидрофурана. С использованием подхода “прививка от”, были получены звездообразные полимеры с заданным числом лучей и узким молекулярно-массовым распределением (Đ = 1.05–1.12). Авторы работы [68] использовали тетрафункциональный инициатор этилендиаминтетра(этил-(трифторметансульфонат)) для блок-сополимеризации тетрагидрофурана и 2-метил-2-оксазолина. Полученный четырехлучевой амфифильный звездообразный блок-сополимер был предложен в качестве альтернативы полимерам на основе полиэтиленгликоля для трансфекции генов.
По аналогии с тетрагидрофураном, катионно полимеризуется с раскрытием цикла и четырехчленный кислородсодержащий гетероцикл – оксетан. Полимеризация оксетана и его производных в присутствии трифторида бора была впервые описана в 1971 году [69]. Показано, что инициирование происходит быстро, а полимеризация носит “живой” характер. До настоящего момента полиоксетанам не уделялось достаточного внимания, несмотря на возможность их функционализации или прививки боковых цепей, позволяющей использовать полиоксетаны в различных областях материаловедения [70]. Одной из возможных причин данного явления может быть наличие побочного процесса образования циклических олигомеров оксетана в процессе его полимеризации. Известен лишь один случай синтезрования звездообразных полиоксетанов с применением подхода “прививка от” [71]. Остальные работы из области химии полиоксетанов в своем большинстве посвящены синтезу сверхразветвленных сополимеров на основе функционального мономера 3-этил-3-гидроксиметилоксетана [72].
В табл. 4 приведены молекулярно-массовые характеристики звездообразных полимерных систем, полученных на основе катионно-полимеризуемых мономеров. Синтезированные полимеры обладают узким молекулярно-массовым распределением в диапазоне Ð = 1.1–1.4, однако, как видно из представленных данных, существуют значительные ограничения по молекулярной массе таких звездообразных макромолекул, обусловленные описанными выше особенностями процесса полимеризации таких мономеров [65, 70].
Таблица 4.
Молекулярно-массовые характеристики звездообразных полимеров, синтезированных на основе катионно-полимеризуемых мономеров
| Инициатор/центр ветвления | Мономер | Число лучей, N | Mn × 103 | Ð |
|---|---|---|---|---|
| Диэтилентриамин (“прививка на”) [65] | ТГФ | 7 | 7.3* | 1.35 |
| 1,3,5-Три-(гидроксиметил)бензол трифлат [67] | ТГФ | 3 | 12.6* | 1.12 |
| Тетра-(гидроксиметилдиэтиленамина трифлат [68] | ТГФ/MeOx | 4 | 3.9* | 1.29 |
| Тетра-[((карбокси)этокси)метил]метан [71] | Диметилоксетан | 4 | 7.2** | 1.38 |
ЦИКЛИЧЕСКИЕ ИМИНОЭФИРЫ
Среди гетероциклических соединений, полимеризующихся по катионному механизму, интерес представляют оксазолины – пятичленные N,O-содержащие гетероциклы, а также их близкие гомологи – шестичленные 2-алкил-5,6-дигидрооксазины, относящиеся к классу циклических иминоэфиров. Наибольшее практическое применение в полимерной химии представляют замещенные во втором положениии гетероцикла производные оксазолинов и оксазинов, которые могут быть получены с использованием ряда синтетических методов [73].
Катионная полимеризация с раскрытием цикла 2-оксазолинов и 2-оксазинов была открыта и впервые изучена в середине 60-х годов ХХ века [74–77]. В результате исследования процессов катионной полимеризации 2-оксазолинов под действием кислот Бренстеда и кислот Льюиса было показано, что рост полиоксазолиновых цепей протекает в “живом” режиме без необратимого обрыва цепей, давая возможность получать поли-N-ацилэтиленимины с заданной молекулярной массой при варьировании мольного соотношения инициатора и мономера в системе, а также блок-сополимеры [78, 79]:
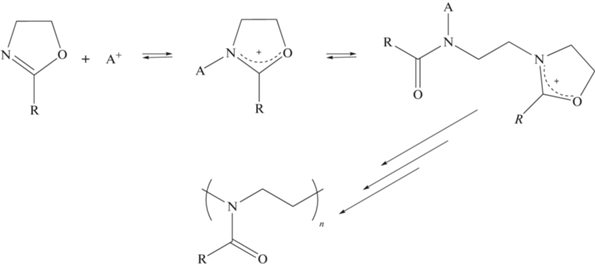
Живой характер катионной полимеризации с раскрытием цикла 2-алкил-2-оксазолинов в совокупности с низкой вероятностью протекания побочных процессов позволяют получать полимеры сложной архитектуры с заданными характеристиками и узким молекулярно-массовым распределением, используя функциональные инициаторы и обрыватели цепи. Разнообразные заместители могут быть введены во второе положение оксазолинового кольца, вследствие чего открываются широкие возможности для варьирования свойств синтезируемых поли-2-оксазолинов.
Интерес к химии полиоксазолинов, возникший начиная с 90-х годов прошлого века, вызван открытием ряда их практически важных свойств, таких как термочувствительность и биосовместимость [80]. Это послужило стимулом к развитию методов синтеза сложноструктурированных полимерных материалов на их основе, предназначенных для использования в различных сферах медицины и биотехнологии, в том числе для систем доставки лекарственных препаратов. Первые звездообразные трех- и четырехлучевые структуры поли-2-алкил-2-оксазолинов с простейшими центрами ветвления бензильного и аллильного типа были получены в 1989–1992 гг. с применением подхода “прививка от” [81–83]. Для синтеза звездообразных полимеров использованы три- и тетрафункциональные инициаторы с бензил- и аллилбромидными функциональными группами соответственно, а также трифункциональные инициаторы с бензилйодидными и бензилтолуолсульфонатными функциональными группами:

Впоследствии данный подход был активно использован для синтеза разнообразных звездообразных поли-2-алкил-2-оксазолинов с функциональными центрами ветвления (табл. 5). Преимущественно применяли полифункциональные инициаторы с алкилгалогенидными и сульфонатными функциональными группами.
Таблица 5.
Молекулярно-массовые характеристики звездообразных поли(2-алкил-2-оксазолин)ов, полученных с применением подхода “прививка от”
| Инициатор/центр ветвления | Мономер | Число лучей, N | $M_{n}^{{{\text{луч}}}}$ × 103 | $M_{n}^{{{\text{зв}}}}$ × 103 | Ð |
|---|---|---|---|---|---|
| Циклотрифосфазена алкил бромид [84] | MeOx | 6 | 1.3*** | 10.1* | 1.30 |
| Алкил иодид/Рутений трис-бипиридил [85] | EtOx | 6 | – | 26.2* | 1.07 |
| Тетрафенилпорфирина алкил хлорид [86] | MeOx | 4 | 3.7*** | 15.4* | 1.38 |
| Тетрафенилпорфирина тозилат [87] | EtOx | 4 | 4.6*** | 10.7* | 1.18 |
| Пентаэритрита трифторметансульфонат [88] | EtOx | 4 | – | 21.8* | 1.34 |
| Дипентаэритрита нозилат [89] | EtOx | 6 | 5.9** | 35.6** | 1.32 |
| Полиглицидола тозилат [89] | EtOx | 13 | 1.7** | 23.2** | 1.11 |
| Каликс[4]арена сульфонилхлорид [90] | EtOx | 4 | 2.1*** | 9.5** | 1.17 |
| Каликс[8]арена сульфонилхлорид [91] | EtOx | 8 | 2.5*** | 22.2* | 1.55 |
В 1994 г. был описан синтез шестилучевого звездообразного поли-2-метил-2-оксазолина с макроциклическим циклотрифосфазеновым центром ветвления на основе гексафункционального инициатора алкилбромидного типа [84]:

В 1997 г. был предложен способ синтеза звездообразных поли-2-алкил-2-оксазолинов с металлокомплексным центром ветвления [85], при этом в качестве полифункциональных инициаторов применены комплексы переходных металлов железа(II) и рутения(II) c галогенсодержащими лигандами на основе 2,2-бипиридина. Позднее в качестве полифункциональных инициаторов полимеризации 2-алкил-2-оксазолинов были использованы соединения на основе тетрафенилпорфирина и силсесквиоксана [86, 92].
Применение алкилгалогенидов для инициирования катионной полимеризации 2-алкил-2-оксазолинов обусловлено легкостью введения галогенидных групп в структуру различных соединений. Однако нуклеофильность галогенидных противоионов приводит к тому, что рост полиоксазолиновых цепей протекает в бóльшей степени по механизму “обрыв–реинициирование” с низкой скоростью. В связи с этим, предложено использовать соли трифторметансульфоновой кислоты в качестве сокатализаторов катионной полимеризации 2-оксазолинов, инициируемой галогенидами [93]. Показано, что скорость полимеризации значительно возрастает при добавлении в реакционную смесь трифторметансульфоната калия или серебра в результате обмена галогенидного противоиона на сульфонатный, который не проявляет нуклеофильных свойств. Это наблюдение привело к широкому использованию для инициирования катионной полимеризации 2-алкил-2-оксазолинов полифункциональных эфиров сульфокислот (п-толуолсульфонаты, п-нитробензолсульфонаты, трифторметансульфонаты), обеспечивающих быстрое инициирование полимеризации и высокую скорость роста цепей на активных центрах ионного типа [87–89]. Анализ данных молекулярно-массовых характеристик звездообразных поли(2-алкил-2-оксазолин)ов, приведенных в табл. 5, демонстрирует, что полифункциональные иницииирующие системы на основе эфиров сульфокислот позволяют получать звездообразные макромолекулы с разнообразными центрами ветвления и заданными структурными характеристиками, при сохранении узкого молекулярно-массового распределения в пределах Ð = 1.2–1.3.
Использование функциональных терминирующих агентов в комбинации с полифункциональными инициаторами катионной полимеризации 2-оксазолинов позволяет получать “умные” разветвленные системы, способные к образованию супрамолекулярных ансамблей. В работе [94] синтезирован звездообразный семилучевой поли-2-этил-2-оксазолин с β-циклодекстриновым центром ветвления, с использованием пер-7-йод-β-циклодекстрина в качестве инициатора и анилина в роли терминирующего агента.
Развитие методов “клик-химии”, получивших в последнее десятилетие широкое распространение в синтезе разветвленных полимерных систем, открыло доступ к альтернативным способам синтеза звездообразных поли-2-алкил-2-оксазолинов сложной архитектуры. Предложен эффективный способ синтеза звездообразных поли-2-алкил-2-оксазолинов со сшитым полимерным ядром по схеме “прививка через” [95]. На первой стадии сополимеризацией 2-этил-2-оксазолина и функционального мономера 2-(3-бутинил)-2-оксазолина был получен линейный блок-сополимер. Далее методом самосборки в водной среде была сформирована структура макромолекул, после чего в присутствии функциональных тиолов осуществлена сшивка поли-2-(3-бутинил)-2-оксазолинового блока:

Синтез звездообразных полимеров с лучами-щетками на основе поли-2-алкил-2-оксазолинов был осуществлен с применением подхода “прививка через” [96]. При этом использована комбинация катионной полимеризации 2-алкил-2-оксазолинов, полимеризация олефинов в условиях реакции метатезиса и метод азид-алкинового [3+2]-циклоприсоединения.
Известны работы по синтезу звездообразных поли-2-алкил-2-оксазолинов с применением подхода “прививка на”. В работе [97] для получения звездообразных структур была использована реакция обрыва “живых” цепей поли-2-этил-2-оксазолина на дендримерах первой и второй генераций, содержащих концевые аминогруппы:

Исследование кинетики терминирования катионной полимеризации 2-этил-2-оксазолина показало, что эффективность реакции понижается при увеличении длины полиоксазолиновых лучей. Этот же подход был применен для синтеза четырехлучевых поли-2-этил-5,6-дигидрооксазинов путем прививки “живых” цепей полиимина на гидразиде тетракис-карбоксиметилкаликс [4]арена [98].
Для синтеза звездообразного поли-2-алкил-2-оксазолина с функциональным тетрафенилпорфириновым центром ветвления [99] задействована реакция азид-алкинового [3+2]-циклоприсоединения:

Полифункциональный связывающий агент был получен алкилированием тетрафенилпорфирина и его металлокомплекса пропаргилбромидом, в то время как полимеризация 2-этил-2-оксазолина была терминирована азидом натрия. Высокоэффективно протекающая реакция циклоприсоединения позволила получить образцы четырехлучевых звездообразных полимеров с узким молекулярно-массовым распределением (Đ < 1.2).
Анализ публикаций, посвященных синтезу звездообразных поли-2-алкил-2-оксазолинов показывает, что в большинстве работ авторы не приводят достаточно убедительных доказательств количества лучей, приходящихся на звездообразную макромолекулу. Как правило, априори принимается, что количество лучей равно числу инициирующих групп в структуре полифункционального инициатора при условии, что инициирование полимеризации происходит быстро и одновременно по всем группам. Однако известны случаи, когда использование, например, октафункционального инициатора приводило к образованию лишь четырехлучевого звездообразного полимера из-за стерических препятствий [100]. Обнаружено, что стерические препятствия можно преодолеть путем введения спейсеров в структуру инициатора, пространственно разделяющих центр ветвления и инициирующие группы. Так, α-бромуксусный эфир трет-бутилкаликс[8]арена не может инициировать полимеризацию 2-изопропил-2-оксазолина, вместе с тем аналогичный 11-бромундекановый эфир позволяет получить восьмилучевые звездообразные поли-2-алкил-2-оксазолины с количественным выходом [101].
Производные каликс[4]-, каликс[8]-, а также тиакаликс [4]арена широко использованы в качестве центра ветвления для синтеза звездообразных полиоксазолинов и полиоксазинов разнообразного строения. Для синтеза полимеров с центральным каликсареновым центром ветвления, функционализированным по нижнему ободу, в нижний кольцевой обод соответствующих каликс[n]аренов вводились спейсеры с алкилбромидными и алкилсульфонилхлоридными инициирующими группами, позволяющими проводить катионную полимеризацию 2-алкил-2-оксазолинов с раскрытием цикла. Полученные инициаторы были использованы в синтезе звездообразных поли-2-алкил-2-оксазолинов, с лучами гомо-, блок- и градиентных сополимеров 2-этил-2-оксазолина и 2-изопропил-2-оксазолина [102–104], а также поли-2-этил-5,6-дигидрооксазинов [105].
Методом селективной деструкции продемонстрировано, что структура звездообразных полимеров соответствует заявленной восьмилучевой (для полимеров с каликс[8]ареновым центром ветвления) или четырехлучевой (для полимеров с каликс[4]ареновым и тиакаликс[4]ареновым центром ветвления). При этом полимерные лучи характеризуются сравнительно узким молекулярно-массовым распределением Đ = 1.4–1.5, что подтверждает псевдоживой механизм протекания полимеризации.
Для получения звездообразных полиоксазолинов на основе каликсаренов, функционализированных по верхнему ободу, в качестве инициаторов полимеризации использовались п-хлорсульфонилкаликс[4, 8]арены, полученные прямым сульфохлорированием соответствующих макроциклов [90, 91, 106]:

При этом было показано [91, 107], что как ароматические, так и алифатические сульфонилгалогениды являются эффективными инициаторами катионной полимеризации оксазолинов, позволяющими проводить процесс в условиях отсутствия необратимого обрыва и получать полимеры с узким молекулярно-массовым распределением.
ЗАКЛЮЧЕНИЕ
Последние достижения в области органической и полимерной химии создали широкий набор инструментов, который включает различные методы контролируемой “живой” ионной полимеризации, позволяющие синтезировать звездообразные макромолекулы с беспрецедентным контролем их структуры и молекулярно-массовых характеристик. Существующие и описанные в литературе подходы позволяют получать звездообразные макромолекулы с заданным числом лучей (N = 3–8), и хорошо контролируемой в диапазоне 102–104 молекулярной массой полимерных лучей при сохранении узкого молекулярно-массового распределения (Ð = 1.1–1.5).
Тем не менее в настоящее время существует ряд проблем, ограничивающих применение метода ионной полимеризации с раскрытием цикла в синтезе звездообразных систем. Получение звездообразных полимеров со строго заданным числом лучей все еще остается нетривиальной задачей, требующей тщательного выбора инициирующих полимеризацию функциональных групп, подбора необходимых условий для полимеризации и разработки методик синтеза звездообразных полимеров с заданными параметрами.
Процессы создания звездообразных систем с большим числом лучей (N > 8) являются довольно трудоемкими из-за возникновения сильного стерического фактора, а также необходимости получения центров ветвления с заданной конфигурацией функциональных групп, ответственных за формирование звездообразной структуры. Тем не менее такие разветвленные полимерные системы представляют значительный интерес в полимерной химии, вследствие чего должны предприниматься шаги по разработке методов получения таких систем.
Синтез звездообразных полимеров с высокомолекулярными лучами, масса которых превышает (10–20) × 103, является также одним из приоритетных направлений для развития метода ионной полимеризации с раскрытием цикла. Полимерные объекты с такими параметрами описаны лишь в малом числе работ, что свидетельствует о наличии существенных ограничений по длине полимерных лучей в звездообразных молекулах.
Ионная полимеризация в настоящее время является наиболее перспективным методом синтеза биосовместимых и биодеградируемых макромолекул, в том числе стимул-чувствительных, что особенно важно для использования полимеров в биомедицинских приложениях. В дополнение к физико-химическим свойствам, обусловленным сильно разветвленной архитектурой, звездообразные полимеры дали возможность разработать новые функциональные материалы, применение которых в различных областях науки и техники увеличивается с каждым годом. Можно полагать, что в недалеком будущем звездообразные полимеры, полученные методами ионной полимеризации, будут играть все более важную роль в материаловедении, нанотехнологии и медицине.
Работа выполнена при финансовой поддержке гранта Правительства РФ в рамках государственной поддержки ведущих научных школ (грант № 14.W03.31.0022).
Список литературы
Tsitsilianis C., Voulgaris D., Stepanek M., Podhajecka K., Prochazka K., Tuzar Z., Brown W. // Langmuir. 2000. V. 16. P. 6868.
Ishizu K., Ichimura A., Ono T. // Polymer. 1998. V. 39. P. 2579.
Tsitsilianis C., Alexandridis P., Lindman B. // Macromolecules. 2001. V. 34. P. 5979.
Rougier A., Rauh D., Nazrieds G. // Proc. Electrochem. Soc. PV2003-17. Electrochromic Materials and Applications. 2003. P. 176.
Widawski G., Rawiso M., François B. // Nature. 1994. V. 369. P. 378.
François B., Ederle Y., Mathis C. // Synth. Met. 1999. V. 103. P. 2362.
Xu H., Erhardt R., Abetz V., Mueller A.H.E., Goedel W.A. // Langmuir. 2001. V. 17. P. 6787.
Beil J.B., Zimmerman S.C. // Macromolecules. 2004. V. 37. P. 778.
Youk J.H., Park M.-K., Locklin J., Advincula R., Yang J., Mays J. // Langmuir. 2002. V. 18. P. 2455.
Nuyken O., Pask S.D. // Polymers. 2013. V. 5. P. 361.
Albertsson A.-C., Varma I.K. // Biomacromolecules. 2003. V. 4. P. 1466.
Tian H., Tang Z., Zhuang X., Chen X., Jing X. // Prog. Polym. Sci. 2012. V. 37. P. 237.
Wang Y.-C., Yuan Y.-Y., Du J.-Z., Yang X.-Z., Wang J. // Macromol. Biosci. 2009. V. 9. P. 1154.
Bellin I., Kelch S., Langer R., Lendlein A. // Proc. Natl. Acad. Sci. A. 2006. V. 103. P. 18043.
Knight P.T., Lee K.M., Qin H., Mather P.T. // Biomacromolecules. 2008. V. 9. P. 2458.
Dubois P., Coulembier O., Raquez J.M. Handbook of Ring-Opening Polymerization. Weinheim: Wiley, 2009.
Brunelle D.J. Introduction in Ring-Opening Polymerization. Munich: Hanser Publ., 1993.
Kricheldorf H.R., Dunsing R. // Makromol. Chem. 1986. V. 187. P. 1611.
Jedliński Z., Walach W., Kurcok P., Adamus G. // Makromol. Chem. 1991. V. 192. P. 2051.
Ryner M., Stridsberg K., Albertsson A.-C., H. von Schenck, Svensson M. // Macromolecules. 2001. V. 34. P. 3877.
Shibasaki Y., Sanada H., Yokoi M., Sanda F., Endo T. // Macromolecules. 2000. V. 33. P. 4316.
Cameron D.J.A., Shaver M.P. // Chem. Soc. Rev. 2011. V. 40. P. 1761.
Ren J.M., McKenzie T.G., Fu Q., Wong E.H.H., Xu J., An Z., Shanmugam S., Davis T.P., Boyer C., Qiao G.G. // Chem. Rev. 2016. V. 116. P. 6743.
Cama G., Mogosanu D.E., Houben A., Dubruel P. Science and Principles of Biodegradable and Bioresorbable Medical Polymers. Cambridge: Woodhead Publ., 2017. P. 79.
Hedrick J.L., Magbitang T., Connor E.F., Glauser T., Volksen W., Hawker C.J., Lee V.Y., Miller R.D. // Chemistry. 2002. V. 8. P. 3308.
Gou P., Zhu W., Shen Z. // Front. Chem. China. 2008. V. 3. P. 330.
Wang F., Bronich T.K., Kabanov A.V., Rauh R.D., Roovers J. // Bioconjugate Chem. 2005. V. 16. P. 397.
Baheti P., Gimello O., Bouilhac C., Lacroix-Desmazes P., Howdle S.M. // Polym. Chem. 2018. V. 9. P. 5594.
Biela T., Duda A., Rode K., Pasch H. // Polymer. 2003. V. 44. P. 1851.
Celik A., Kemikli N., Ozturuk R., Muftuoglu A.E., Yilmaz F. // React. Funct. Polym. 2009. V. 69. P. 705.
Xu J., Shi W. // Polymer. 2006. V. 47. P. 5161.
Hao Q., Li F., Li Q., Li Y., Jia L., Yang J., Fang A., Cao A. // Biomacromolecules. 2005. V. 6. P. 2236.
Li H., Zhao N., Ren C., Liu S., Li Z. // Polym. Chem. 2017. V. 8. P. 7369.
Sanda F., Sanada H., Shibasaki Y., Endo T. // Macromolecules. 2002. V. 35. P. 680.
Oledzka E., Kaliszewska D., Sobczak M., Raczak A., Nickel P., Kolodziejski W. // J. Biomater. Sci. 2012. V. 23. P. 2039.
Xu J., Shi W. // Polymer. 2006. V. 47. P. 5161.
Woodruff M.A., Hutmacher D.W. // Progr. Polym. Sci. 2010. V. 35. P. 1217.
Zhu K.J., Song B., Yang S. // J. Polym. Sci., Polym. Chem. 1989. V. 27. P. 2151.
Kim E.S., Kim B.C., Kim S.H. // Polym. Sci., Polym. Phys. 2004. V. 42. P. 939.
Adeli M., Zarnegar Z., Kabiri R. // Eur. Polym. J. 2008. V. 44. P. 1921.
Gorczynski L., Chen J., Fraser C.L. // J. Am. Chem. Soc. 2005. V. 127. P. 14956.
Fiore G.L., Klinkenberg J.L., Fraser C.L. // Macromo-lecules. 2008. V. 41. P. 9397.
Adeli M., Haag R. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 5740.
Ni C., Zhu G., Zhu C., Yao B., Kumar D.N.T. // Coll. Polym. Sci. 2010. V. 288. P. 1193.
Kowalski A., Libiszowski J., Biela T., Cypryk M., Duda A., Penczek S. // Macromolecules. 2005. V. 38. P. 8170.
Lee S.H., Kim S.H., Han Y.K., Kim Y.H. // J. Polym. Sci., Polym. Chem. 2001. V. 39. P. 973.
Wang L., Dong C.M. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 2226.
Lee J.S., Choo D.J., Kim S.H., Kim Y.H. // Polymer. 1998. V. 22. P. 880.
Zuh K.J., Song B., Yang S. // J. Polym. Sci., Polym. Chem. 1989. V. 27. P. 2151.
Nicco A., Boucheron R.G. // Eur. Polym. J. 1970. V. 6. P. 1477.
Richards D.H., Eastmond G.C., Stewart M.J. Telechelic Polymers: Synthesis and Applications. Boca Rotan: CRC Press, 1989.
Duda A., Penczek S. // Macromolecules. 1990. V. 23. P. 1636.
Лебедев Б.В., Мухина Н.Н., Кулагина Т.Г. // Высокомолек. cоед. А. 1978. Т. 20. № 6. P. 1297.
Moody V., Needles H.L. Major Fibers and Their Properties. Norwich: Tufted Carpet, 2004.
Ma L., Deng L., Chen J. // Drug. Dev. Ind. Pharm. 2014. V. 40. P. 845.
Sedlacek O., Monnery B.D., Filippov S.K., Hoogenboom R., Hruby M. // Macromol. Rapid Commun. 2012. V. 33. P. 1648.
Gnanou Y., Lutz P., Rempp P. // Makromol. Chem. 1988. V. 189. P. 2885.
Comanita B., Noren B., Roovers J. // Macromolecules. 1999. V. 32. P. 1069.
Lapienis G., Penczek S. // Macromolecules. 2000. V. 33. P. 6630.
Taton D., Saule M., Logan J., Duran R., Hou S., Chaikof E.L., Gnanou Y. // J. Polym. Sci., Polym. Chem. 2003. V. 41. P. 1669.
Zhu W., Ling J., Shen Z. // Macromol. Chem. Phys. 2006. V. 207. P. 844.
Sunder A., Mulhaupt R., Frey H. // Macromolecules. 2000. V. 33. P. 309.
Vairon J.-P., Spassky N. // Cationic Polymerizations. New York: Marcel Dekker, 1996. P. 683.
Piotti M.E. Encyclopedia of Materials: Science and Technology. Oxford: Pergamon Press, 2001.
P. Van Caeter, Goethals E.J. // Macromol. Rapid Commun. 1997. V. 18. P. 393.
van Renterghem L.M., Goethals E.J., Du Prez F.E. // Macromolecules. 2006. V. 39. P. 528.
Oike H., Yoshioka Y., Kobayashi S., Nakashima M., Tezuka Y., Goethals E.J. // Macromol. Rapid Commun. 2000. V. 21. P. 1185.
Rasolonjatovo B., Pitard B., Haudebourg T., Bennevault V., Guégan P. // Eur. Polym. J. 2017. V. 88. P. 689.
Saegusa T., Hashimoto Y., Matsumoto S. // Macromolecules. 1971. V. 4. P. 1.
Parzuchowski P., Maminski M.L. // Polymers. 2020. V. 12. P. 222.
Guo Y.-M., Zou Y.-F., Pan C.-Y. // Macromol. Chem. Phys. 2001. V. 202. P. 1094.
Sharma K., Zolotarskaya O.Yu., Wynne K.J., Yang H. // J. Bioactive Comp. Polym. 2012. V. 27. P. 525.
Gant T.G., Meyers A.I. // Tetrahedron. 1994. V. 50. P. 2297.
Tomalia D.A., Sheetz D.P. // J. Polym. Sci. A. 1966. V. 4. P. 2253.
Kagiya T., Narisawa S., Maeda T., Fukui K. // J. Polym. Sci., Polym. Lett. 1966. V. 4. P. 441.
Seeliger W., Aufderhaar E., Diepers W., Feinauer R., Nehring R., Thier W., Hellmann H. // Angew. Chem., Int. Ed. Engl. 1966. V. 78. P. 875.
Bassiri T.G., Levy A.J., Litt M.H. // J. Polym. Sci., Polym. Lett. 1967. V. 5. P. 871.
Kagiya T., Matsuda T. // J. Macromol. Sci. A. 1971. V. 5. P. 1265.
Saegusa T., Ikeda H., Fuji H. // Polym. J. 1972. V. 3. P. 176.
Hoogenboom R. // Angew. Chem. Int. Ed. 2009. V. 48. P. 7978.
Cai G., Litt M. H. // J. Polym. Sci., Polym. Chem. 1989. V. 27. P. 3603.
Kobayashi S., Uyama H., Narita Y. // Macromolecules. 1992. V. 25. P. 3232.
Chujo Y., Sada K., Kawasaki T., Saegusa T. // Polym. J. 1992. V. 24. P. 1301.
Chang J.Y., Ji H.J., Han M.J., Rhee S.B., Cheong S., Yoon M. // Macromolecules. 1994. V. 27. P. 1376.
Lamba J.J.S., Fraser C.L. // J. Am. Chem. Soc. 1997. V. 119. P. 1801.
Jin R.-H., Motoyoshi K.-I. // J. Porphyrins Phthalocyanines. 1999. V. 3. P. 60.
Hoogenboom R., Fijten M.W.M., Kickelbick G., Schubert U.S. // Beilstein J. Org. Chem. 2010. V. 6. P. 773.
Plet L., Delecourt G., Hanafi M., Pantoustier N., Pembouong G., Midoux P., Bennevault V., Guegan P. // Eur. Polym. J. 2020. V. 122. P. 109323.
Kowalczuk A., Kronek J., Bosowska K., Trzebickaa B., Dworak A. // Polym. Int. 2011. V. 60. P. 1001.
Blokhin A.N., Kirila T.Yu., Kozina N.D., Razina A.B., Filippov A.P., Ten’kovtsev A.V. // Mendeleev Commun. 2022. V. 32. P. 247.
Blokhin A.N., Razina A.B., Bursian A.E., Ten’kovtsev A.V. // Polymer Science B. 2021. V. 63. P. 52.
Kim K.-M., Ouchi Y., Chujo Y. // Polym. Bull. 2003. V. 49. P. 341.
Weberskirch R., Hettich R., Nuyken O., Schmaljohann D., Voit B. // Macromol. Chem. Phys. 1999. V. 200. P. 863.
Adeli M., Kalantari M., Zarnega Z., Kabiri R. // RSC Advances. 2012. V. 2. P. 2756.
Brummelhuis N., Schlaad H. // Polym. Chem. 2011. V. 2. P. 1180.
Alvaradejo G.G., Nguyen H.V.-T., Harvey P., Gallag-her N.M., Le D., Ottaviani M.F., Jasanoff A., Delaittre G., Johnson J.A. // ACS Macro Lett. 2019. V. 8. P. 473.
Lambermont-Thijs H.M.L., Fijten M.W.M., Schubert U.S., Hoogenboom R. // Aust. J. Chem. 2011. V. 64. P. 1026.
Smirnova A., Kirila T., Blokhin A., Kozina N., Kurlykin M., Tenkovtsev A., Filippov A. // Eur. Polym. J. 2021. V. 156. P. 110637.
Rudolph T., Crotty S., Schubert U.S., Schacher F.H. // e-Polymers. 2015. V. 15. P. 227.
Stradman S., Pulkkinen P., Tenhu H. // J. Polym. Sci., Polym.Chem. 2005. V. 43. P. 3349.
Tenkovtsev A.V., Trofimov A.E., Shcherbinskaya L.I. // Polymer Science B. 2012. V. 54. P. 142.
Kurlykin M.P., Bursian A.E., Dudkina M.M., Tenkov-tsev A.V. // Fibre Chem. 2015. V. 47. P. 291.
Kirila T., Smirnova A., Kurlykin M., Tenkovtsev A., Filippov A. // Coll. Polym. Sci. 2020. V. 298. P. 535.
Lezov A.A., Gubarev A.S., Podsevalnikova A.N., Senchukova A.S., Lebedeva E.V., Dudkina M.M., Tenkov-tsev A.V., Nekrasova T.N., Andreeva L.N., Smyslov R.Yu., Gorshkova Yu.E., Kopitsa G.P., Rădulescu A., Pipich V., Tsvetkov N.V. // Coll. Polym. Sci. 2019. V. 297. P. 285.
Kurlykin M.P., Dudkina M.M., Tenkovtsev A.V. // Polymer Science B. 2019. V. 61. № 1. P. 51.
Blokhin A.N., Razina A.B., Ten’kovtsev A.V. // Polymer Science B. 2018. V. 60. № 3. P. 307.
Blokhin A.N., Kurlykin M.P., Razina A.B., Dudkina M.M., Ten’kovtsev A.V. // Polymer Science B. 2018. V. 60. № 4. P. 421.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)