

Журнал вычислительной математики и математической физики, 2019, T. 59, № 2, стр. 325-333
О вычислении потенциала в многоатомных системах
О. А. Горкуша 1, *, В. Г. Заводинский 2, **
1 ИПМатем ДВО РАН
680000 Хабаровск, ул. Дзержинского, 56, Россия
2 Ин-т материаловедения ХНЦ ДВО РАН
680042 Хабаровск, ул. Тихоокеанская, 153, Россия
* E-mail: 684bmts@rambler.ru
** E-mail: vzavod@mail.ru
Поступила в редакцию 29.04.2018
После доработки 01.06.2018
Аннотация
Предлагается численный метод нахождения потенциала многоатомной системы в прямом пространстве. Отличительная особенность метода состоит в разделении электронной плотности $\rho $ и потенциала $\varphi $ на две части: $\rho = {{\rho }_{0}} + \hat {\rho }$, $\varphi = {{\varphi }_{0}} + \hat {\varphi }$, где ${{\rho }_{0}}$ – сумма сферических атомных плотностей, а потенциал ${{\varphi }_{0}}$ порождается плотностью ${{\rho }_{0}}$. Потенциал $\hat {\varphi }$ находится путем решения уравнения Пуассона. Граничные условия получены путем разложения обратной величины расстояния между двумя точками в ряд по полиномам Лежандра. Для обеспечения точности метода расчетная область разбивается на многогранники Вороного и применяются асимптотические оценки итераций при замене характеристической функции гладкими приближениями. Для численного решения уравнения Пуассона использованы двухсеточный метод и Фурье-преобразование. Получена оценка точности метода $O({{h}^{{\gamma - 1}}})$, где $h$ – шаг сетки, $\gamma $ – фиксированное число, большее 1. Погрешность метода проанализирована на модельной двухатомной задаче. Библ. 19. Фиг. 2. Табл. 1.
1. ВВЕДЕНИЕ
В задачах квантовой механики и квантовой химии часто возникает необходимость вычисления электростатического потенциала $\varphi (r)$, $r \in {{\mathbb{R}}^{3}}$, формируемого электронной плотностью $\rho (r)$ системы $m$ взаимодействующих атомов, расположенных в точках ${{R}_{1}},{{R}_{2}}, \ldots ,\;{{R}_{m}}$ (см. [1]–[3]). Формула, связывающая потенциал и плотность, выглядит весьма просто:
Однако прямое численное интегрирование с неизбежностью обнажает почти неразрешимую проблему: для достижения разумной точности необходимо разбить интегрируемую область на достаточно малые элементарные ячейки, но время интегрирования при этом катастрофически растет. Проблема эта нетривиальна, и попыткам ее решения посвящено большое число работ, где предлагаются различные подходы, из которых наиболее разработанными являются метод быстрого Фурье-преобразования [4]–[7] и метод мультипольного разложения [8], [9]. Однако метод мультипольного разложения напрямую пригоден лишь для случаев, когда потенциал находится в точках, находящихся за пределами существования электронной плотности, а метод быстрого Фурье-преобразования (БПФ) обеспечивает хорошую точность лишь в обратном пространстве (пространстве волновых векторов). Для вычисления потенциала в прямом пространстве наиболее развит подход, опирающийся на решение уравнения Пуассона [10], [11], но его использование для многоатомных систем требует доработки с целью повышения точности. В нашей работе мы предлагаем комбинацию указанных подходов, а также некоторые другие разработки последних лет.
2. ОПИСАНИЕ ПОДХОДА
Прежде всего обратим внимание на тот факт, что в большинстве задач квантовой механики и квантовой химии расчеты ведутся итерационным способом, и на нулевой итерации электронная плотность многоатомной системы представляет собой сумму электронных плотностей невзаимодействующих (свободных) атомов или ионов. Поскольку электронные плотности свободных атомов (ионов) сферичны, то нулевая плотность системы ${{\rho }^{0}}(r)$ есть сумма сферических плотностей $\rho _{{{\text{sphere}}}}^{a}$, центрированных на атомах (ионах) с координатами ${{R}_{j}}:$
(1)
${{\rho }^{0}}(r) = \sum\limits_{j = 1}^m \,\rho _{{{\text{sphere}}}}^{a}\left( {\left| {r - {{R}_{j}}} \right|} \right).$Сферические плотности заранее вычисляются с помощью подходящего квантово-механического метода, например, в рамках теории функционала плотности [2], и задаются в виде численных функций, зависящих от $R$ – расстояния до центра атома. Шаг величины $R$ обычно весьма мал, поэтому мы можем легко, быстро и достаточно точно вычислить электростатический потенциал отдельного свободного атома в сферических координатах:
(2)
${{\varphi }^{0}}(r) = \sum\limits_{j = 1}^m \,\varphi _{{{\text{sphere}}}}^{a}\left( {\left| {r - {{R}_{j}}} \right|} \right).$3. ВЫЧИСЛЕНИЕ ГРАНИЧНЫХ ЗНАЧЕНИЙ ПОТЕНЦИАЛА
Рассмотрим интеграл (6). Разобьем расчетную область $\Omega $ на многогранники Вороного ${{\nu }^{{(j)}}}$
Далее, с помощью характеристической функции области ${{\nu }^{{(j)}}}$
(7)
$\begin{gathered} \hat {\varphi }(r) = \sum\limits_{j = 1}^m \,{{{\hat {\varphi }}}_{j}}(r), \\ {{{\hat {\varphi }}}_{j}}(r) = \int\limits_{{{\nu }^{{(j)}}}} {\frac{{\mathop {\hat {\rho }}\nolimits_j (r{\kern 1pt} {\text{'}})}}{{\left| {r - r{\kern 1pt} {\text{'}}} \right|}}} dr{\kern 1pt} {\text{'}}{\text{.}} \\ \end{gathered} $Оценим точность этого подхода. Прежде всего заметим, что
(9)
${{\hat {\varphi }}_{j}}(r) = \int\limits_{{{\nu }^{{(j)}}}} {\frac{{\hat {\rho }(r{\kern 1pt} {\text{'}}){{\chi }_{j}}(r{\kern 1pt} {\text{'}})}}{{\left| {r - r{\kern 1pt} {\text{'}}} \right|}}dr{\kern 1pt} {\text{'}}} = \int\limits_{{{\nu }^{{(j)}}}} {\frac{{\hat {\rho }(r{\kern 1pt} {\text{'}}){{{\tilde {\mathcal{P}}}}_{j}}(r{\text{'}};k)}}{{\left| {r - r{\kern 1pt} {\text{'}}} \right|}}} dr{\kern 1pt} {\text{'}} + \varphi _{j}^{'}(r) + {{\varepsilon }_{2}}(r),$(10)
$\varphi _{j}^{'}(r) = \int\limits_{\partial {{\nu }^{{(j)}}}} {\frac{{\hat {\rho }(r{\kern 1pt} ')}}{{\left| {r - r{\kern 1pt} {\text{'}}{\kern 1pt} } \right|}} \cdot \frac{{\mathcal{J}(r{\kern 1pt} {\text{'}}) - 1}}{{\mathcal{J}(r{\kern 1pt} {\text{'}})}}dr{\text{'}}} {\text{,}}$(11)
${{\varepsilon }_{2}}(r) = {{\varepsilon }_{1}}(a) \cdot \int\limits_{{{\nu }^{{(j)}}}\backslash {{O}_{\delta }}({{R}_{j}})} {\frac{{\hat {\rho }(r{\kern 1pt} {\text{'}})}}{{\left| {r - r{\kern 1pt} {\text{'}}{\kern 1pt} } \right|}}} dr{\kern 1pt} {\text{',}}$Обозначим через $R({{\nu }^{{(j)}}})$ радиус минимальной сферы с центром в точке ${{R}_{j}},$ покрывающей область ${{\nu }^{{(j)}}}.$ Будем считать, что все точки, лежащие на границе $\partial \Omega ,$ расположены вне сферы. Тогда величину $\tfrac{1}{{\left| {r - r{\kern 1pt} {\text{'}}} \right|{\kern 1pt} }}$ можно представить в виде сходящегося ряда по системе многочленов Лежандра ${{P}_{l}}(cos\theta )$ [8], [9], [13], [15], [16]
(12)
$\int\limits_{{{\nu }^{{(j)}}}} {\frac{{\hat {\rho }(r{\kern 1pt} {\text{'}}){{{\tilde {\mathcal{P}}}}_{j}}(r{\kern 1pt} {\text{'}};k)}}{{\left| {r - r{\kern 1pt} {\text{'}}{\kern 1pt} } \right|}}} dr{\kern 1pt} {\text{'}} = {{\tilde {\varphi }}_{j}}(r,k) + {{\varepsilon }_{3}}(r,j),$(13)
${{\tilde {\varphi }}_{j}}(r,k) = \frac{1}{{\left| {r - {{R}_{j}}} \right|}}\int\limits_{{{\nu }^{{(j)}}}} {\hat {\rho }} (r{\kern 1pt} {\text{'}}{\kern 1pt} ){{\tilde {\mathcal{P}}}_{j}}(r{\text{'}};k) \cdot \sum\limits_{l = 0}^L {{\left( {\frac{{\left| {r{\kern 1pt} {\text{'}} - {{R}_{j}}} \right|}}{{\left| {r - {{R}_{j}}} \right|}}} \right)}^{l}}{{P}_{l}}(cos\theta )dr{\kern 1pt} {\text{',}}$(14)
${{\varepsilon }_{3}}(r,j) = O\left( {\frac{{\hat {A}}}{{\left| {r - {{R}_{j}}} \right| - R({{\nu }^{{(j)}}})}} \cdot {{{\left( {\frac{{R({{\nu }^{{(j)}}})}}{{\left| {r - {{R}_{j}}} \right|}}} \right)}}^{L}}} \right),\quad L \to \infty .$4. ОЦЕНКА ЧИСЛЕННОГО РЕШЕНИЯ УРАВНЕНИЯ ПУАССОНА
Теперь перейдем непосредственно к решению уравнения Пуассона (5), (6). Учитывая (16), представим решение этой задачи в виде
где $u$ – решение задачи (5) с граничным условием задаваемым соотношением (17), а ${{u}^{0}}$ – решение задачи Дирихле для уравнения Лапласа граничным условием ${{\left. {{{u}^{0}}} \right|}_{{\partial \Omega }}} = {{\varepsilon }_{4}}$. Так как решение ${{u}^{0}}$ достигает максимума на границе, то согласно (15)(20)
$\begin{gathered} \left| {{{u}^{0}}(r)} \right| \ll {{\varepsilon }_{4}}(\hat {A}) + {{\varepsilon }_{1}}(a), \\ {{\varepsilon }_{4}}(\hat {A}) = O\left( {\mathop {max}\limits_{r \in \partial \Omega } \frac{{\hat {A}}}{{\left| {r - {{R}_{j}}} \right| - R({{\nu }^{{(j)}}})}} \cdot {{{\left( {\frac{{R({{\nu }^{{(j)}}})}}{{\left| {r - {{R}_{j}}} \right|}}} \right)}}^{L}}} \right),\quad L \to \infty . \\ \end{gathered} $Обозначим через ${{\Omega }^{h}}$ сетку с шагом $h$ на $\Omega ,$ через ${{\Gamma }^{h}}$ множество узлов $(ih,jh,kh),$ не принадлежащих ${{\Omega }^{h}},$ с условием
Значение функции ${{u}^{h}}$ в точке $(ih,jh,kh)$ обозначим через $u_{{i,j,k}}^{h}.$ Задаче (5), (19) поставим в соответствие разностную задачу
(21)
$\begin{gathered} {{({{L}^{h}}({{u}^{h}}))}_{{i,j,k}}} = \frac{{u_{{2i - 1,2j,2k}}^{{h/2}} - 2\tilde {u}_{{2i,2j,2k}}^{{h/2}} + u_{{2i + 1,2j,2k}}^{{h/2}}}}{{{{{(h{\text{/}}2)}}^{2}}}} + \frac{{u_{{2i,2j - 1,2k}}^{{h/2}} - 2\tilde {u}_{{2i,2j,2k}}^{{h/2}} + u_{{2i,2j + 1,2k}}^{{h/2}}}}{{{{{(h{\text{/}}2)}}^{2}}}} + \\ + \;\frac{{u_{{2i,2j,2k - 1}}^{{h/2}} - 2\tilde {u}_{{2i,2j,2k}}^{{h/2}} + u_{{2i,2j,2k + 1}}^{{h/2}}}}{{{{{(h{\text{/}}2)}}^{2}}}}, \\ \end{gathered} $Известно, что разностная схема (21) сходится и погрешность относительно решения задачи (5), (19) имеет второй порядок аппроксимации. Учитывая представление решения $\tilde {\varphi }$ (18) и оценку (20), получаем погрешность приближенного решения относительно задачи (5), (6):
(22)
$\varphi (r) = {{\varphi }^{0}}(r) + \tilde {u}_{{i,j,k}}^{h} + O\left( {max\left\{ {{{h}^{2}},{{h}^{{\gamma - 1}}}} \right\}} \right),\quad r = (ih,jh,kh) \in {{\bar {\Omega }}^{h}},$5. ЧИСЛЕННАЯ СХЕМА РЕШЕНИЯ УРАВНЕНИЯ ПУАССОНА
Шаг итерационного процесса состоит из следующих этапов.
1. Производим одну итерацию на ${{\bar {\Omega }}^{{h/2}}}$, вычисляя $\tilde {u}_{{2i,2j,2k}}^{{h/2}}$ по формуле (21). Новое приближенное решение к решению на ${{\bar {\Omega }}^{h}}$ на этом этапе определяется формулой
2. Вычисляем параметр сходимости $\mu $ итерационного процесса по формуле
3. Если $\mu $ близко к единице, то итерационный процесс завершен. Иначе мы интерполируем функцию $\tilde {u}_{{i,j,k}}^{h}$ c ${{\bar {\Omega }}^{h}}$ на ${{\bar {\Omega }}^{{h/2}}}$ при помощи оператора интерполяции, точного для многочленов второй степени. И переходим к первому пункту итерационного процесса.
Как и в каждой итерационной процедуре, важную роль играет выбор начальной функции. В нашем случае мы использовали в качестве начального приближения потенциал, вычисленный с помощью БПФ, а именно:
(23)
$\begin{gathered} u(r) = 4\pi \sum\limits_{{{m}_{x}},{{m}_{y}},{{m}_{z}}} \frac{{c({{m}_{x}},{{m}_{y}},{{m}_{z}})}}{{{{\lambda }^{2}} + {{{(2\pi h)}}^{2}} \cdot (m_{x}^{2} + m_{y}^{2} + m_{z}^{2})}}{{e}^{{i({{m}_{x}} \cdot x + {{m}_{y}} \cdot y + {{m}_{z}} \cdot z)}}}, \\ r = (x,y,z) \in \mathop {\overline {Cube(\Omega )} }\nolimits^h . \\ \end{gathered} $6. ЧИСЛЕННЫЕ ЭКСПЕРИМЕНТЫ
Как было сказано во Введении, Фурье-преобразование не дает возможности вычислить потенциал с достаточной точностью. В качестве иллюстрации на фиг. 1 приведены результаты расчета потенциала по формуле (23) при разных значениях параметра $\lambda $ для двухатомной модельной системы $(m = 2)$ с плотностью
(24)
$\rho (r) = \rho (r;\beta ,{{\alpha }_{1}},{{R}_{1}},\; \ldots ,\;{{\alpha }_{m}},{{R}_{m}}) = \sum\limits_{j = 1}^m \left( {{{\alpha }_{j}}{{{\left| {r - {{R}_{j}}} \right|}}^{2}}{{e}^{{ - \beta |r - {{R}_{j}}|}}}} \right),$Фиг. 1.
Зависимость вычисленного потенциала от полной плотности (в сравнении с аналитическим расчетом) от $\lambda .$ Сплошная линия – аналитический расчет, пунктир – расчет с использованием БПФ.

Формула (24) допускает возможность аналитического вычисления потенциала, а именно:
(25)
${{\varphi }_{{{\text{analitic}}}}}(r) = \frac{{4\pi }}{{{{\beta }^{4}}}}\sum\limits_{j = 1}^m \,{{\alpha }_{j}}\left( {\frac{{4!\left( {1 - {{e}^{{ - \beta {{r}_{j}}}}}} \right)}}{{{{r}_{j}}}} - {{e}^{{ - \beta {{r}_{j}}}}}\left( {{{\beta }^{2}}r_{j}^{2} + 6\beta {{r}_{j}} + 18} \right)} \right),\quad {{r}_{j}} = \left| {r - {{R}_{j}}} \right|.$На фиг. 1 представлены для сравнения результаты аналитического вычисления потенциала. Мы видим, что можно подобрать такое значение $\lambda ,$ при котором потенциал, вычисленный с помошью Фурье-преобразования, будет очень близок к аналитическому потенциалу. Однако, как показывают практические расчеты (см. [19]), для вычислений потенциалов, соответствующих реальным атомам, оптимальные значения параметра $\lambda $ необходимо подбирать для каждого типа атомов, кроме того, результат Фурье-преобразования зависит от многих технических параметров (в том числе от размера ячейки, используемой для БПФ, от шага разбиения ячейки, и так далее), что делает применение Фурье-преобразования весьма затруднительным для реальных многоатомных систем.
Ключевым моментом предлагаемого метода является разделение полной плотности $\rho $ системы взаимодействующих атомов на начальную плотность ${{\rho }_{0}},$ состоящую из суммы сферических плотностей, центрированных на отдельных атомах и некой плотности $\hat {\rho }$, образующейся вследствие взаимодействия атомов в системе. Такой подход позволяет ограничить использование Фурье-преобразования лишь применением его для вычисления потенциала $\widehat \varphi $ от плотности $\hat {\rho }$, амплитуда которой, как правило, значительно меньше амплитуды плотности ${{\rho }_{0}}.$
Для численного эксперимента мы рассмотрели модельную двухатомную систему $(m = 2)$ с плотностью ${{\rho }_{0}},$ вычисляемую по формуле (24). Плотность $\widehat \rho $ представим в виде
На фиг. 2 представлено соответствующее распределение плотности. Мы видим, что добавочная плотность $\widehat \rho $ имеет как положительные, так и отрицательные значения, что обеспечивает перетекание плотности из одних областей пространства в другие.
Фиг. 2.
Вид электронной плотности модельного димера вдоль прямой, проходящей через атомные центры. Точки – плотность ${{\rho }^{0}},$ составленная из сферических атомных функций невзаимодействующих атомов, нижняя сплошная кривая – межатомная плотность $\hat {\rho }$, образовавшаяся из-за взаимодействия атомов, верхняя сплошная кривая – суммарная плотность ${{\rho }^{0}} + \hat {\rho }$ атомной системы.
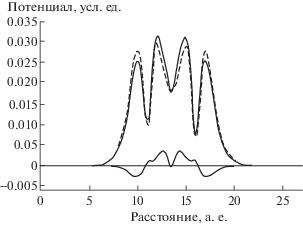
При таком подходе значения потенциалов, вычисленных разными методами, отличаются друг от друга весьма незначительно, и сравнение их в графическом виде малоинформативно. Поэтому мы вычислили среднее отклонение потенциалов от аналитического. В табл. 1 представлены результаты сравнения двух методов. Мы видим, что в данном случае погрешность вычисления потенциала с помощью БПФ весьма мала. Но ее можно еще более уменьшить с помощью метода, предлагаемого в данной работе, где Фурье-преобразование используется только в качестве нулевого приближения двухсеточного метода. Это позволяет почти на порядок уменьшить погрешность численного решения. Следует отметить, что при увеличении параметра $\lambda $ в $100$ раз величины погрешностей и того и другого метода изменяются не более, чем на 20%.
Таблица 1.
Абсолютные и относительные погрешности вычисления потенциала $\lambda = 0.005,$ $K = \frac{{max\hat {\rho }(r)}}{{max{{\rho }_{0}}(r)}}$
| ε | K | Абсолютная погрешность | Относительная погрешность | ||
|---|---|---|---|---|---|
| БПФ | Наш метод | БПФ | Наш метод | ||
| 0.00500 | 0.0018 | 2.53791 × 10–6 | 4.70817 × 10–6 | 4.84735 × 10–6 | 7.75075 × 10–7 |
| 0.02480 | 0.0092 | 1.26257 × 10–5 | 2.34225 × 10–6 | 2.41153 × 10–5 | 3.85499 × 10–6 |
| 0.23512 | 0.0867 | 1.19498 × 10–4 | 2.21687 × 10–5 | 2.28298 × 10–4 | 3.63969 × 10–5 |
| 0.44380 | 0.1636 | 2.25581 × 10–4 | 4.18484 × 10–5 | 4.31078 × 10–4 | 6.85429 × 10–5 |
Представленный нами подход к нахождению потенциала был эффективно применен в работах по развитию нового безорбитального метода моделирования многоатомных систем (их содержание описано в книге [19]).
7. ВЫВОДЫ
1. Разбиение плотности на сумму неизменных сферических плотностей и добавочную плотность, изменяющуюся при взаимодействии атомов, резко увеличивает точность вычисления электростатического потенциала методом Фурье-преобразования.
2. Применение нашего метода позволяет уменьшить погрешность вычисления потенциала еще на порядок.
3. Теоретическая оценка погрешности метода позволяет контролировать параметры численного расчета – амплитуда добавочной плотности должна быть сравнима с величиной ${{h}^{\gamma }},$ с $\gamma \in (1,\;2)$.
Список литературы
Ландау Л.Д., Лифшиц Е.М. Квантовая механика. Нерелятивисткая теория. М.: Физматгиз, 1963.
Kohn W., Sham J.L. Self-consistent equations including exchange and correlation effects // Phys. Rev. 1965. V. 140. A1133.
Заводинский В. Компьютерное моделирование наночастиц и наносистем. М.: Физматлит, 2013.
Sköllermo G. A fourier method for the numerical solution of Poisson’s equation // Math. Comput. 1975. V. 29. P. 697.
Chun-Min Chang, Yihan Shao, Jing Kong. Ewald mesh method for quantum mechanical calculations // J. Chem. Phys. 2012. V. 136. 114112.
Бобров В.Б., Загородний А.Г., Тригер С.А. // Физ. низких температур. 2015. Т. 41. С. 1154.
Chelikowsky J.R., Troullier N., Saad Y. Finite-difference-pseudopotential method: Electronic structure calculations without a basis // Phys. Rev. Lett. 1994. V. 72. P. 1240.
Kleinman L., Bylander D.M. Efficacious form for model pseudopotentials // Phys. Rev. Lett. 1982. V. 48. P. 1425–1428.
Cheng H., Greebgard L., Rokhlin V. A fast adaptive multipole algorithm in three dimensions // J. Comput. Phys. 1999. V. 155. P. 468.
Mortensen J.J., Hansen L.B., Jacobsen K.W. Real-space grid implementation of the projector augmented wave method // Phys. Rev. B Condensed Matter. 2005. V. 71. 035109.
Chelikowsky J.R., Wu K., Troullier N., Saad Y. Higher-order finite-difference pseudopotential method: An application to diatomic molecules // Phys. Rev. B. 1994. V. 50. 11355.
Becke A.D. A multicenter numerical integration scheme for polyatomic molecules // J. Chem. Phys. 1988. V. 88. 2547.
Hiroshe Kikuji, Ono Tomoya, Fujimoto Yoshitaka, Tsukamoto Shigeru. First-Principles Calculations in Real-Space Formalism. London: Imperial College Press, 2005.
Okabe A., Boots B., Sugihara K., Chiv S.N., Kendall D.G. Spatial Tesselations: Concepts and Applications of Voronoi Diagrams. New York: Wiley, 2000.
Gonze X., Stumpf R., Scheffler M. Analysis of separable potentials // Phys. Rev. B. 1991. V. 44. 8503.
Troullier N., Martins J.L. Efficient pseudopotentials for plane-wave calculation // Phys. Rev. B. 1991. V. 43. 1993.
Brandt A. Multi-level adaptive solutions to boundary-value problems // Math. Comput. 1977. V. 31. P. 333.
Харрисон У. Электронная структура и свойства твердых тел. Т. 2 / Под ред. Ж.И. Алферова. М.: Мир, 1983.
Заводинский В. Квантовое моделирование многоатомных систем без волновых функций. Saarbrucken, Deutschland: LAP LAMBERT Academic Publishing, 2017.
Дополнительные материалы отсутствуют.
Инструменты
Журнал вычислительной математики и математической физики