

Химия высоких энергий, 2019, T. 53, № 3, стр. 243-249
Плазмоэлектрохимический синтез малослойных графеновых структур для модификации эпоксидного связующего
А. Г. Кривенко a, *, Р. А. Манжос a, В. К. Кочергин b, Г. В. Малков a, А. Е. Тарасов a, Н. П. Пивень c
a Институт проблем химической физики Российской академии наук
142432 Московская обл., г. Черноголовка, пр. акад. Семенова, 1, Россия
b Московский государственный университет им. М.В. Ломоносова, Химический факультет
119992 Москва, Россия
c Институт энергетических проблем химической физики Российской академии наук
142432 Московская обл., г. Черноголовка, пр. акад. Семенова, 1, Россия
* E-mail: krivenko@icp.ac.ru
Поступила в редакцию 21.12.2018
После доработки 28.12.2018
Принята к публикации 28.12.2018
Аннотация
Малослойные графеновые структуры, синтезированные плазмоэлектрохимическим методом, были использованы в качестве модификаторов эпоксидных связующих. Показано, что максимальный положительный эффект полученные структуры оказывают на прочностные характеристики отверждeнной эпоксиаминной композиции при концентрации 0.2 мас. %.
ВВЕДЕНИЕ
Эпоксидные смолы являются одними из наиболее распространенных термоотверждаемых полимеров, используемых в промышленности. Благодаря технологичности и относительно низкой стоимости они находят применение в качестве адгезивов, герметиков, покрытий, а также в качестве матриц композиционных материалов. Недостатком этого типа материалов является их хрупкость и низкая трещиностойкость, которые возникают из-за густой трехмерной сетки, образующейся в процессе отверждения. В настоящий момент общепринято, что возможным путем преодоления этого недостатка является введение в эпоксидную смолу в качестве наполнителя углеродных наночастиц, в частности, малослойных графеновых структур (МГС) см. например обзоры [1–3]. Однако использование МГС в качестве модификаторов термореактивных полимерных связующих (эпоксидные, цианэфирные смолы, полиимиды и т.д.) сталкивается с серьезными затруднениями, обусловленными низкой полярностью их поверхности и малым сродством между матрицей и нанонаполнителем. Известно, что микро- и макроскопические свойства композиционного материала зависят не только от характеристик каждого из компонентов, но и от свойств границы раздела между ними. Свойства интерфейса определяются наличием и силой адгезионных взаимодействий между поверхностью наполнителя и полимерной матрицей, которые представляют собой комбинацию химических (ковалентных) и межмолекулярных (Ван-дер-Ваальса, водородные и т.п.) связей. В связи с этим модифицирование поверхности наполнителя реакционноспособными и/или полярными группами, позволяющими образовываться межмолекулярным связям, заведомо должно способствовать улучшению распределения наполнителя в полимерной матрице, увеличению адгезии матрицы к наполнителю и улучшению передачи напряжений с матрицы на наполнитель и обратно. Поэтому для углеродных волокон, разработаны и применяются различные подходы функционализации исходно инертной поверхности – окисление в газовой фазе, обработка плазмой или концентрированными кислотами и т.д. [4].
По-видимому, одним из наиболее простых методов получения МГС с заданной степенью декорирования поверхности функциональными группами представляется электрохимическое расщепление графита, что обусловлено рядом преимуществ данного подхода по сравнению с другими распространенными способами синтеза углеродных наноструктур: экологичность (использование нейтральных растворов с умеренной концентрацией солей, в отличие от всех химических методов, использующих концентрированные кислоты), одностадийность, простота очистки синтезируемого материала, возможность получения как практически неокисленных и бездефектных графеновых частиц, так и структур с высоким содержанием кислородсодержащих функциональных групп [5]. Наиболее перспективным в этом плане, по мнению авторов, является плазмоэлектрохимический способ синтеза МГС, который был подробно описан в [6].
Целью настоящей работы было получение электрохимическим расщеплением графита малослойных графитовых структур в условиях катодной и анодной электролизной плазмы и исследование их влияния в качестве наполнителя на физико-механические свойства отвержденной эпоксидной композиции.
МЕТОДИКА
Плазмоэлектрохимический синтез МГС осуществляли путем подачи на электроды из промышленно выпускаемого графита ГР-280, погруженные в раствор водного электролита, импульсов напряжения различной полярности амплитудой до 300 В и длительностью нарастания <0.5 мкс. При уменьшении размеров одного из электродов до величин, обеспечивающих величину плотности тока >20 А/см2, вокруг этого электрода происходило образование электролизной плазмы и наблюдали взрывное испарение раствора, сопровождавшееся интенсивной световой и акустической генерацией рис. 1 [6]. Для очистки от следов электролита полученной суспензии МГС применяли многократное центрифугирование и декантирование. После отделения водного раствора на центрифуге к осадку добавлялся ацетон, и проводилось еще два цикла центрифугирования. В результате получали устойчивую на временном интервале более 8 месяцев суспензию МГС в ацетоне с концентрацией ~2.0–15 мг/мл.
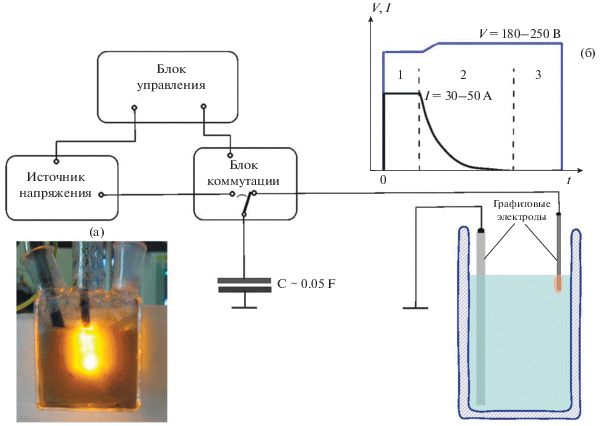
Рис. 1.
Схема установки для плазмоэлектрохимического расщепления графита. Фотография ячейки в момент плазменной вспышки (а), характерный вид временных зависимостей напряжения и тока электрохимической ячейки во время синтеза МГС (б).
Для изготовления полимерных нанокомпозитов использовали эпоксидную смолу ЭД-20 (ГОСТ 10587-84, производитель ФКП “Завод имени Я.М. Свердлова”, 22.8–23 мас. % эпоксидных групп) и низкотемпературный отвердитель ТЭТА, представляющий собой смесь этиленовых аминов с преимущественным содержанием линейного изомера триэтилентетрамина (Huntsman).
Синтезированные малослойные графеновые структуры в виде дисперсии в ацетоне (концентрация до 2 мас. %) смешивали с эпоксидной смолой в соотношении 1 : 1, подвергали УЗВ-обработке в течение 10–15 мин, ацетон затем удаляли при длительном нагревании в течение 10–12 ч при 100°С при интенсивном перемешивании. После остывания необходимое количество эпоксидной смолы, содержащей МГС, смешивали с нативной смолой, а затем готовили связующее смешением компонентов (при эквимольном соотношении эпоксидных и аминных групп) в течение не менее чем 15 мин, после чего смесь вакуумировали в течение 20–25 мин при остаточном давлении не выше 0.1 мм. рт. ст. и выливали в силиконовые формы. Отвержение композиций проводили в несколько этапов – сначала в течение суток при комнатной температуре, затем температуру увеличивали до 80°С и выдерживали еще сутки. После отверждения полученные образцы доводили до размеров, соответствующих размерам Type 5, ASTM D638. Физико-механические испытания на растяжение проводили на разрывной машине Zwick Z010 TC-FR010TH, скорость деформирования 1 мм/мин. ДСК-анализ образцов проводили на приборе METTLER TOLEDO DSC822e в температурном интервале 0–200°C, в токе азота, со скоростью сканирования 10°C/мин. ИК-спектры регистрировали на FTIR-спектрофотометре “BRUKER ALPHA” с использованием приставки нарушенного полного внутреннего отражения. Условия регистрации: шаг сканирования 2 см–1, диапазон измерения 4000–360 см–1, 56 сканов для снятия спектров образца и фона. Спектры комбинационного рассеяния (КР) регистрировались при длине возбуждения 976 нм с использованием спектрометра “Nicolet 9810 FRS”. ЭПР спектры регистрировались с использованием “SE/X-2544” спектрометра Х диапазона. РФЭ-спектры получали с помощью прибора Specs (Германия) с полусферическим анализатором Phoibos 150 с использованием MgKα излучения (1253.6 эВ). Давление в рабочей камере спектрометра не превышало 5 × 10–8 Па.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЯ
Не вызывает сомнения, что строгая кинетическая формализация описания наблюдаемых при электрохимическом расщеплении графита временных зависимостей напряжения и тока вряд ли возможна вследствие многообразия и сложности физико-химических процессов, протекающих вблизи границы раздела при используемом нами способе воздействия. К таковым, в первую очередь, можно отнести взрывное выделение газообразных продуктов электродных реакций и инжекцию активных интермедиатов, вскипание перегретой жидкости и образование парогазовой оболочки, ионизацию газа в этой оболочке и создание вокруг электрода неравновесной низкотемпературной плазмы (рис. 1а) [7], а также вполне вероятное наличие значимых кавитационных, электрофлотационных и гидродинамических эффектов. Тем не менее, по мнению авторов, возможна качественная трактовка характерной формы наблюдаемых зависимостей V(t) и I(t), приведенных на рис. 1б. Первый участок соответствует зарядке ДЭС графитового электрода до потенциала заведомого протекания электродных реакций. С учетом высокой пористости используемой марки графита время этого процесса можно оценить как ~L2/D, составляющее 1–10 мс, при характерной глубине пор L ~ 1–10 мкм и коэффициенте диффузии ионов электролита D ~ 10–9 м2/с. Постоянство тока заряжения обеспечивается схемными решениями, использованными при конструировании генератора напряжения. Участок спада 2 обусловлен расширением области существования плазмы и как следствие уменьшением напряженности электрического поля, приводящего к прекращению электрического разряда, и созданием вокруг электрода изолирующей парогазовой оболочки, а участок 3 соответствует полной изоляции электрода от раствора. В результате электродные реакции и газовыделение прекращаются, изолирующая оболочка разрушается и контакт с электролитом восстанавливается. Порождаемая этими процессами генерация гидравлических ударных волн, подобно ультразвуковой обработке, приводит к механическому разрыхлению поверхности графита и отщеплению от нее МГС.
На рис. 2 приведены СЭМ изображения (a, в) и РФЭ спектры высокого разрешения С1s (б, г) осадков суспензий, полученных при воздействии катодной (а, б) и анодной (в, г) плазмы. Осадки выглядят как совокупность слипшихся тонких и гибких графеноподобных структур с латеральными размерами от 0.05 до 0.3 мкм, частично агломерированные из более мелких составляющих при высушивании. Данные оценки подтверждаются результатами измерений распределения по размерам частиц водной дисперсии полученных МГС (д). Из рисунка видно, что для дисперсии, полученной при воздействии катодной плазмы, распределение по размерам частиц имеет явный бимодальный характер (со средними латеральными размерами 0.11 и 0.18 мкм). Можно предположить, что это связано с особенностью расщепления графита катодной плазмой. В этом случае было установлено уменьшение массы, как электродa подвергающeгoся воздействию плазмы, так и графитовoгo противоэлектродa с существенно большей поверхностью и, таким образом, бимодальность дисперсии обусловлена одновременным расщеплением двух графитовых электродов. По сравнению с катодной плазмой воздействие анодной плазмой приводит к образованию дисперсии, для которой центр распределения по размерам частиц несколько сдвинут в сторону больших латеральных размеров ~0.19 мкм, возможной причиной чего могут быть более мягкие условия образования анодной плазмы.
Рис. 2.
СЭМ изображения (a, в) и РФЭ спектры высокого разрешения С1s (б, г) осадков суспензий, полученных при воздействии катодной (а, б) и анодной (в, г) плазмы; распределение по размерам водной дисперсии МГС (д).
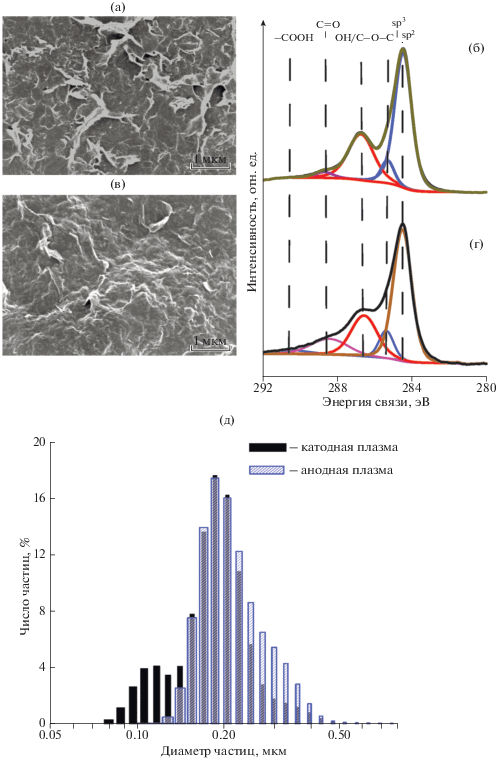
Результаты декомпозиции РФЭ-спектров суспензии МГС, проведенные в согласии с данными [8], так же указывают на существенную зависимость степени декорирования поверхности графеновых структур кислородсодержащими функциональными группами от режима синтеза. Основными линиями, присутствующими в РФЭ-спектрах образцов, являются линии с энергиями 284.5 и 285.5 эВ, относящиеся к атомам углерода в состоянии sp2 и sp3 гибридизации см. напр. [9]. Что касается кислородсодержащих функциональных групп, то из рис. 2 видны существенные различия высокоэнергетической части спектра осадков МГС в зависимости от использованного варианта синтеза. Это отличие заключается в различной интенсивности составляющих с энергиями связей 286.4, 288.5 и 290.6 эВ, которые относятся к гидроксильным и эпоксидным группам (OH/C–O–C), карбонильным фрагментам (C=O) и карбоксильным группам (COOH), соответственно [8]. Количественный и качественный состав кислородсодержащих функциональных групп поверхностного слоя осадков МГС для всех использованных вариантов электрохимического расщепления, полученный из анализа данных РФЭС, приведен в табл. 1. Из таблицы видно, что наибольшая степень декорирования поверхности МГС кислородсодержащими функциональными группами, такими как (OH/C–O–C) и (C=O), достигается при анодном плазмохимическом варианте расщепления.
Таблица 1
| Тип воздействия | С, ат. % | О, ат. % | –OH/C–O–C, ат. % | –COOH, ат. % | C=O, ат. % |
|---|---|---|---|---|---|
| Катодная плазма | 79.8 | 18 | 16.0 | – | 2 |
| Анодная плазма | 67.5 | 26 | 18.0 | 0.1 | 7.5 |
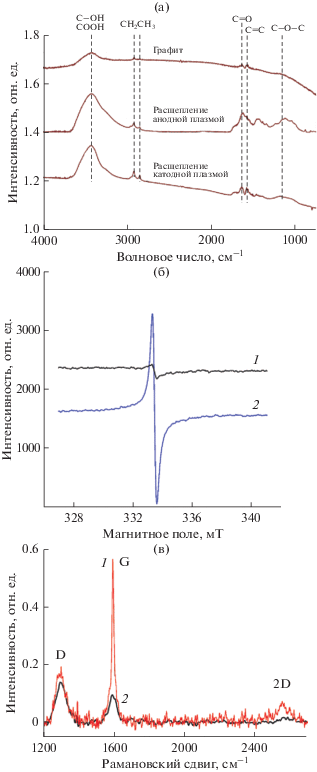
На рис. 3 приведены данные по исследованию МГС различными спектральными методами, подтверждающие результаты приведенные в табл. 1. ИК спектры порошков, рис. 3а, указывают на заметно большее количество C=O- и OH/C–O–C-групп в МГС, получаемых при воздействии анодной плазмой, по сравнению с катодной. Спектры ЭПР порошков суспензий, получаемых при расщеплении графита катодной и анодной плазмой, приведены на рис. 3б кривые 1 и 2 соответственно. Как следует из рисунка, спектры 1 и 2 характеризуются одиночным узким сигналом, расположенным вблизи значений g-тензора g = 2. Концентрация спинов составляет 1.5 × 1017 и 2 × 1018 спинов/г для 1 и 2 соответственно, т.е. для образца 2 эта величина превышает более чем на порядок концентрацию спинов в образце 1. Это свидетельствует о существенно большем количестве дефектов, к которым можно отнести функциональные группы на поверхности МГС, синтезированных при расщеплении графита анодной плазмой, по сравнению с катодным воздействием [10]. Спектры КР водных суспензий МГС, синтезированных в режиме катодной 1, анодной 2 плазмы приведены на рис. 3д. Вид спектра 1 соответствует умеренно дефектным малослойным графеновым структурам: отношение интенсивностей D/G линий составляет ~0.3, а широкая 2D линия смещена в сторону меньших значений волнового числа по сравнению с графитом [11]. Другая ситуация наблюдается для МГС, полученных при расщеплении графита анодной плазмой. В этом случае отношение интенсивностей D/G линий составляет ~1.3, что указывает на меньшую среднюю толщину (~3–5 слоев) МГС и большую степень их дефектности (функционализации) по сравнению с катодным способом синтеза [12], что подтверждается и анализом данных РФЭ и ЭПР спектроскопии см. рис. 2, 3. Таким образом, представляется очевидным, что использованный нами плазмоэлектрохимический подход позволяет контролируемым образом варьировать содержание кислородсодержащих фукциональных групп в поверхностном слое МГС в диапазоне от 18 до 27 ат. %.
Рис. 3.
ИК спектры порошков МГС (а). ЭПР спектры (б) и спектры КР (в) порошков МГС, полученных при расщеплении графита катодной 1 и анодной 2 плазмой.
Как было сказано выше, наиболее перспективным наполнителем для увеличения физико-механических характеристик полимерного композиционного материала являются МГС, поверхность которых декорирована реакционноспособными и полярным группами, способствующими образованию межмолекулярных связей. По этой причине в данной работе для модификации эпоксидного связующего были использованы МГС, полученные при расщеплении графита при воздействии анодной плазмы. Малослойные графеновые структуры вводились в полимерную матрицу через предварительно приготовленную дисперсию (masterbatch) в эпоксидной смоле. Полученная дисперсия МГС в эпоксидной смоле не склонна к агрегации и имеет значительно меньшую вязкость, чем дисперсии УНТ близкой концентрации, что является положительной особенностью синтезированных наночастиц, и позволяет проводить быстрое смешение с высокоактивными низкотемпературными отвердителями с удовлетворительным распределением наночастиц в полимерной матрице. Использование двухстадийного режима отверждения позволило добиться максимальной конверсии функциональных групп в отвержденном эпоксиаминном связующем. Показано, что введение МГС в эпоксидное связующее не приводит к существенному изменению температуры стеклования (97–99°С).
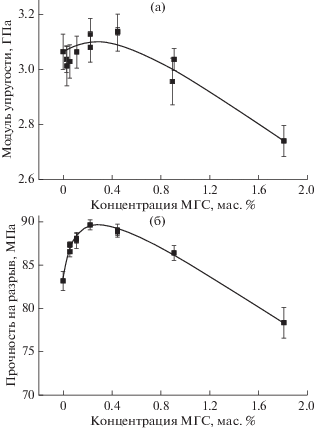
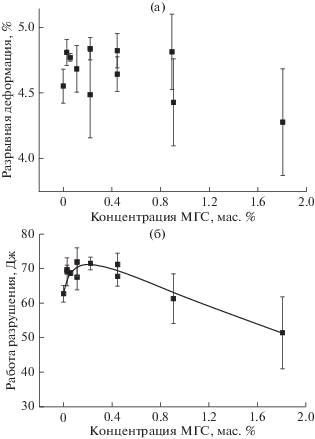
Изменение модуля упругости (Ераст) с увеличением содержания МГС в отвержденной эпоксидной композиции представлено на рис. 4а. Видно, что зависимость Ераст от содержания МГС носит экстремальный характер с максимальным значением при содержании МГС 0.2 мас. %. Модуль упругости при этом выше, чем у ненаполненного образца на 2.4%. Дальнейшее увеличение концентрации МГС приводит к монотонному снижению Ераст вплоть до значения 2.75 Гпа (ниже, чем у ненаполненного образца на 10.6%) при содержании МГС 1.8 мас. %. Зависимость предела прочности от содержания МГС подобна таковой для модуля упругости (рис. 4б). Также, как и в случае Ераст, максимальное значение предела прочности наблюдается при содержании МГС 0.2 мас. %. Прочность при этом выше, чем у ненаполненного образца на 8.0%. Прочность монотонно понижается при дальнейшeм увеличении концентрации МГС, но не так интенсивно (на 5.8% относительно ненаполненного образца), как происходит снижение модуля упругости. Значения деформации при разрушении у отвержденной эпоксидной композиции не имеют четко выраженной зависимости от содержания МГС (рис. 5а) и ее изменение с увеличением концентрации МГС не превышаeт типичной ошибки измерения этой величины, поэтому можно сказать, что наполнение эпоксиаминной композиции частицами МГС не оказывает влияния на разрывную деформацию нанокомпозита. В результате, максимальное положительное влияние МГС проявляется в изменении работы разрушения отвержденной эпоксиаминной композиции. Характер зависимости работы разрушения от содержания МГС в нанокомпозите повторяет зависимости модуля упругости и предельной прочности (рис. 5б) – работа разрушения максимальна при содержании МГС в нанокомпозите 0.2 мас. % (на 15.0% выше, чем у ненаполненного образца). При дальнейшeм увеличении его содержания понижается и значение работы разрушения (минимальное значение, наблюдаемое при 1.8% МГС, на 18.0% ниже чем у ненаполненного образца).
ЗАКЛЮЧЕНИЕ
Отработана методика синтеза малослойных графеновых структур в условиях электрохимического расщепления графита. Полученные МГС охарактеризованы комплексом методов физико-химического анализа и опробованы в качестве модификаторов эпоксидных связующих. Результаты испытаний нанокомпозитов свидетельствуют об увеличении физико-механических характеристик у отвержденной эпоксиаминной композиции при введении 0.2 мас. % МГС. Развитие работы может быть направлено на изменение условий электрохимического синтеза с целью значительного увеличения степени функционализации поверхности МГС для создания более прочного интерфейса в эпоксидных нанокомпозитах.
Выражаем благодарность за РФЭС-исследования, выполненные с использованием оборудования центра коллективного пользования НЦЧ РАН Е.Н. Кабачковым.
Работа выполнена при финансовой поддержке гранта РФФИ 16-03-00475а.
Список литературы
Domun N., Hadavinia H., Zhang T., Sainsbury T., Liaghat G.H., Vahid S. // Nanoscale. 2015. V. 7. P. 10294.
Liu S., Chevali V.S., Xu Z., Hui D., Wang H. // Compos. Part B Eng. 2018. V. 136. P. 197.
Badamshina E.R., Gafurova M.P., Estrin Y.I. // Russ. Chem. Rev. 2010. V. 79. P. 945.
Fitzer E., Geigl K.-H., Hüttner W., Weiss R. // Carbon. 1980. V. 18. P. 389.
Chae S., Hashimi K., Bratescu M.A., Saito N. // Nanoscience and Nanotechnology Letters. 2016. V. 10. P. 784.
Krivenko A.G., Manzhos R.A., Kotkin A.S. // High Energy Chem. 2018. V. 52. P. 272.
Belkin P.N., Kusmanov S.A. // Review Surface Engineering and Applied Electrochemistry. 2016. V. 52. P. 531.
Li O.L., Chiba S., Wada Y. // J. Materials Chemistry A. 2017. V. 5. P. 2073.
Gardner S.D., Singamsetty C.S.K., Booth G.L., He G-R. // Carbon. 1995. V. 33. P. 587.
Zaka M., Ito Y., Wang H., Yan W., Robertson A., Wu Y.A., Rummeli M.H., Staunton D., Hashimoto T., Morton J.J.L., Ardavan A., Briggs G.A.D., Warner J.H. // ACS Nano. 2010. V. 4. P. 7708.
Kannan M.V., Kumar G.G. // Biosensors & Bioelectronics. 2016. V. 77. P. 1208.
Song Y., Liu T.-Y., Xu G.-L., Feng D.-Y., Yao B., Kou T.-Y., Liu X.-X., Li Y. // J. Mater. Chem. A. 2016. V. 4. P. 7683.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий