

Физикохимия поверхности и защита материалов, 2019, T. 55, № 1, стр. 3-10
Применение шкалы абсолютных поверхностных потенциалов к реакциям хемосорбции и электрокатализа на металлах. Часть 2
Ю. Я. Андреев 1, *, И. А. Сафонов 1, А. В. Дуб 1
1 Национальный исследовательский технологический университет “МИСиС”
119049 Москва, Ленинский проспект, 4, Россия
* E-mail: yuandr@rambler.ru
Поступила в редакцию 10.11.2017
После доработки 19.01.2018
Принята к публикации 15.02.2018
Аннотация
Ранее [1] изложены представления об абсолютном поверхностном потенциале (ASP) ES грани (hkl) кристалла металла с величиной ES = ΔGs/zF, непосредственно связанной c поверхностной энергией Гиббса ΔGS и с равновесием между (авто) адсорбцией собственных атомов на этой грани Nad и отрицательно заряженными вакансиями (ОЗВ) NV(S). На этой основе, шкала ASP рассматривается как шкала адсорбционных потенциалов точечных дефектов – ОЗВ и адатомов, определяющей их статистическое распределение на поверхности как результат совместного влияния поверхностных и электростатических сил на эти квазичастицы. Этот дуализм направлен на преодоление различия в понимании поверхностного потенциала в электростатической теории Гельмгольца и в теории Гиббса. Адсорбционная шкала сингулярной грани металла имеет особую точку - потенциал нулевого заряда (ПНЗ) электрода $E_{{\text{N}}}^{0}{\text{ = }}-\Delta G_{{\text{S}}}^{0}{{(hkl)} \mathord{\left/ {\vphantom {{(hkl)} {zF}}} \right. \kern-0em} {zF}}$ с минимумом адсорбции атомов и ОЗВ. Точка абсолютного ПНЗ разделяет шкалу адсорбции в области катодной и анодной поляризации (рис. 4.1, ч. 1) с преимущественной адсорбцией ОЗВ или адатомов, достигающей максимума при потенциале второй особой точки $E_{{\text{S}}}^{0}$ в каждой из областей идеальной поляризации электрода. В части 2 рассматривается переход от шкалы ASP к – водородной, используя принятое IUPAC соотношение между стандартной и абсолютной величиной водородного электрода. Совмещение шкалы ASP со шкалой абсолютных потенциалов электродных реакций позволило провести расчет электродных потенциала хемосорбционной электрокаталитической реакции выделения водорода на различных металлах, а также – потенциала образования пассивирующегого оксида на металлах (Ni, Cr), известного как Фладе-потенциал.
5. АБСОЛЮТНАЯ И ВОДОРОДНАЯ ШКАЛА ПОВЕРХНОСТНЫХ ПОТЕНЦИАЛОВ МЕТАЛЛОВ. ЭЛЕКТРОКАТАЛИТИЧЕСКАЯ РЕАКЦИЯ ВЫДЕЛЕНИЯ АТОМАРНОГО ВОДОРОДА НА МЕТАЛЛАХ
Как отмечено ранее (часть 1 [1]), совмещенная шкала абсолютных потенциалов может иметь двойственный характер. Она допускает совместное использование не только величин абсолютных (а) поверхностных потенциалов $E_{{\text{S}}}^{{\text{a}}},$ относящихся к существованию промежуточных частиц – адатомов и вакансий, но и абсолютных потенциалов $E_{i}^{{\text{a}}}$ любых обратимых электродных реакций. Здесь особо будет рассмотрено использование абсолютного потенциала водородного электрода $E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{{0,{\text{a}}}} = 4.44\;{\text{В }}$ [2] и ПНЗ $E_{{\text{N}}}^{0}$ металла, на котором выделяется водород. Переход от шкалы абсолютных поверхностных потенциалов (ASP) к водородной шкале (SHS) необходим для практического применения шкалы ASP. Этот переход произведен на основе анализа механизма выделения водорода, в котором выделяют две основные стадии:
(5.1)
${{{\text{H}}}^{ + }} + \quad{{{\text{e}}}^{ - }}\quad = {{{\text{H}}}_{{{\text{ad}}}}}\,\,\left( {{\text{р е а к ц и я }}\,\,{\text{Ф о л ь м е р а }}} \right),$(5.2)
${{{\text{H}}}_{{{\text{ad}}}}} + {{{\text{H}}}_{{{\text{ad}}}}} = {{{\text{H}}}_{{\text{2}}}}\,\,\left( {{\text{р е а к ц и я }}\,\,{\text{Т а ф е л я }}} \right).$В настоящее время наибольший интерес вызывают теории, в которых обсуждаются связи между электрохимическими аспектами и физикой металлов. Большое распространение получила теория, рассматривающая корреляцию между перенапряжением водорода и работой выхода электрона в металлах по Bockris [3], развитая Trasatti [4–6].
Андреев [7], используя известные экспериментальные данные, собранные Trasatti [4] для величин перенапряжения H2 на различных металлах в растворах H2SO4 с концентрацией 0.5–1.0 моль/л при T = 293–298 K и приведенные к виду уравнения Тафеля
(5.3)
$\begin{gathered} {{\eta }_{{{{{\text{H}}}_{2}}}}} = --\left( {a + b{\text{lg}}{{i}_{k}}} \right), \\ {\text{г д е }}a = \eta _{{{{{\text{H}}}_{2}}}}^{0} = - 0.118\lg {{i}_{0}}, \\ \end{gathered} $показал, что для большинства металлов с ГЦК структурой и близкой к ней – ГПУ существует линейная зависимость (рис. 5.1) между экспериментальными величинами $a = {\eta }_{{{{{\text{H}}}_{2}}}}^{0},$ взятыми у Trasatti [4] и величинами минимальной поверхностной энергии $\Delta U_{{\text{S}}}^{0}\left( {hkl} \right)$ сингулярной грани, относящейся к кристаллической структуре данного металла, взятыми из работы Vitos с соавт. [8]. Для ГЦК металла – это грань (111), для ГПУ (0001) и для ОЦК (110). Из графика рис. 5.1 получена зависимость для ГЦК и ГПУ металлов [7]:
(5.4)
$a = {\eta }_{{{{{\text{H}}}_{2}}}}^{0} = \quad\left( { - 1.97 \pm 0.02} \right) + 0.02017\left[ {\Delta U_{{\text{S}}}^{0}\left( {hkl} \right)} \right].$

Близкая к этой получается корреляция, если использовать экспериментальные данные по Антропову [9] (см. в [7], но где точка для Pt принадлежит ур. (5.4). Что касается результатов аналогичной корреляции для ОЦК металлов, то на графике (рис. 5.1), из-за недостатка экспериментальных данных, она представлена приблизительно и обозначена штриховой линией. Обращает на себя внимание, что все приведенные данные по величинам ${\eta }_{{{{{\text{H}}}_{2}}}}^{0}$ относятся к металлам с исходной поликристаллической структурой, тогда как наблюдаемая корреляция (5.4) реализуется при условии, если только использовать величины минимальной поверхностной энергией $\Delta U_{{\text{S}}}^{0}$ (hkl) для низкоиндексных грани ГЦК (111) или ГПУ(0001), взятыми из [8]. Это объясняется электрохимической реконструкцией ПС металлов (при поляризации электрода), которая состоит в образовании низкоиндексных граней (hkl) с минимальной поверхностной энергией.
При трактовке корреляции (5.4) возьмем за основу известное представление о двух энергетических формах существования атомов водорода: 1) хемосорбированных атомов H-ad на поверхности металла и 2) атомарного водорода ${{{\text{H}}}^{0}}.$ Важно дать оценку величинам абсолютных потенциалов обеих форм существования атомов водорода. Очевидно, энергия образования молекулы ${{U}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ равна сумме удвоенной энергии образования атома ${{U}_{{{{{\text{H}}}^{0}}}}}$ и энергии рекомбинации ${{U}_{{{{{\text{H}}}^{0}} - {{{\text{H}}}^{0}}}}}$ этих атомов в молекулу в виде
(5.5)
$2{{U}_{{{{{\text{H}}}^{0}}}}} = {{U}_{{{{{\text{H}}}_{{\text{2}}}}}}} - {{U}_{{{{{\text{H}}}^{0}} - {{{\text{H}}}^{0}}}}}.$Далее, реализуем соотношение (5.5), обычное для диссоциативной хемосорбции газообразного
водорода на металле [10], применительно к электрохимическим реакциям, и поэтому рассматриваем абсолютный
потенциал реакции ${{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}^{0}}}}} \right. \kern-0em} {{{{\text{H}}}^{0}}}}$ на инертном электроде в растворе ионов ${{{\text{H}}}^{ + }}$ с учетом энергии их гидратации как $E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}^{0}}}}} \right. \kern-0em} {{{{\text{H}}}^{0}}}}}}^{a}$ = – (${{U}_{{{{{\text{H}}}^{0}}}}}$/F + + ΔGгидр). Аналогично принимая  как абсолютный потенциал образования Н2 в растворе ионов H+ с той же энергией их гидратации, получим соотношение между абсолютными потенциалами
образования атома и молекулы водорода в водном растворе H+ ионов:
как абсолютный потенциал образования Н2 в растворе ионов H+ с той же энергией их гидратации, получим соотношение между абсолютными потенциалами
образования атома и молекулы водорода в водном растворе H+ ионов:
(5.6)
$ - E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}^{0}}}}} \right. \kern-0em} {{{{\text{H}}}^{0}}}}}}^{a} = - 0.5E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{a} + {{{{U}_{{{{{\text{H}}}^{0}} - \,{{{\text{H}}}^{0}}}}}} \mathord{\left/ {\vphantom {{{{U}_{{{{{\text{H}}}^{0}} - \,{{{\text{H}}}^{0}}}}}} {2F}}} \right. \kern-0em} {2F}}.$Здесь, согласно IUPAC [2] на основе работы Trasatti [5], численную величину абсолютного потенциала стандартного H2-электрода рекомендуется рассматривать как величину при T = 298.15 K, равную
(5.7)
$E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}_{{\text{2}}}}}}} \right. \kern-0em} {{{{\text{H}}}_{{\text{2}}}}}}}}^{{0,\,a}} = 4.44 \pm 0.02{\;В }{\text{.}}$Согласно [5], эта величина относится к суммарной энергии окисления молекулы H2, включающей ее диссоциацию (атомизацию), ионизацию атома H и гидратацию иона H+. Тогда как реакция образования H2 имеет противоположное направление с обратным (reversed) знаком энергии Гиббса и, следовательно, с величиной ${\;}E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{{{\text{0,a}}}} = - 4.44{\;В }.$ Эта величина относится к катодной поляризации относительно ПНЗ, тогда как первая по (5.7) к – анодной (см. рис. 4.1 ). Именно в этом состоит главное различие между абсолютной и условной водородной шкалой потенциалов, так как в последней стандартный потенциал любой реакции согласно IUPAC относится равно как к ее катодному, так и к анодному направлению.
Следуя абсолютной шкале на рис. 4.1 , выделение газа H2 должно происходить при смещении потенциала относительно ПНЗ металла, взятой за ноль как EN (0), до величины (минус) 4.44 В, что соответствует ${\;}E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{0} = 0$ по водородной шкале. Однако, как показывает график рис. 5.1 и экспериментальная зависимость (5.4) для большинства металлов, выделение водорода в отсутствии влияния подложки (при $\Delta {{U}_{{\text{S}}}} = 0$) происходит при значительно более положительном потенциале, равном E = –1.97 (SHE), по сравнению с (минус) 4.44 В по абсолютной шкале. В общем случае соотношение между потенциалом электрода Ea по абсолютной шкале и потенциалом по водородной шкале E (SHE) можно представить по аналогии с формулой связи между термодинамической (абсолютной) температурой Т и температурой t по шкале Цельсия как T = 273.15 + t в виде
где знак минус относится к катодной области абсолютных потенциалов.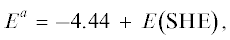
Подставляя в (5.7а) величину E = 1.97 (SHE) из ур. (5.4), получим ту же самую экспериментальную величину потенциала
выделения водорода, но теперь по абсолютной шкале, равную Ea = 2.47 В, которая близка к величине $0.5E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{{0,a}}$ = –2.22 B в формуле (5.6). Разницу между ними мы относим приближенно к величине $E_{{{{{\text{H}}}^{{\text{0}}}} - {{{\text{H}}}^{{\text{0}}}}}}^{a} = \quad - 0.25\,\,{\text{В
}},$ определяющей среднюю энергию рекомбинации атомов ${{{\text{H}}}^{0}}$ как ${{U}_{{{{{\text{H}}}^{0}} - {{{\text{H}}}^{0}}}}}$ = 48 кДж/моль для диссоциативной физической адсорбции ${{{\text{H}}}^{0}}$ на металлах [10], которая мало зависит от природы металла. При этих допущениях принимаем, что в формуле
(5.6) численная величина  = –2.47 B по абсолютной шкале или Е = –1.97 В (SHE) соответствует катодной реакции образования атомарного водорода H0, а не молекулы H2. Этот важный вывод подтверждается расчетами для выделения водорода на металлах (ниже).
= –2.47 B по абсолютной шкале или Е = –1.97 В (SHE) соответствует катодной реакции образования атомарного водорода H0, а не молекулы H2. Этот важный вывод подтверждается расчетами для выделения водорода на металлах (ниже).
С этой целью перейдем непосредственно к шкале ASP для некоторой грани (hkl) кристалла, где имеется нулевая точка $E_{{\text{S}}}^{{\text{'}}} = E_{{\text{N}}}^{{0,a}},$ а теперь добавлена точка $E_{{\text{S}}}^{{{\text{''}}}} = - E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}^{{\text{0}}}}}}} \right. \kern-0em} {{{{\text{H}}}^{{\text{0}}}}}}}}^{a}.$ Очевидно сдвиг катодного потенциала от величины ПНЗ $E_{{\text{S}}}^{'} = E_{{\text{N}}}^{{{\text{0,}}a}}$ (точка минимальной адсорбции атомов металла (см. ур. (4.6)) посредством идеальной поляризации должен привести к росту адсорбции ${{{\text{N}}}_{{{\text{ad}}}}}$ атомов металла Me-ad, не вызывая образования атомов водорода ${{{\text{H}}}^{0}}.$ И только при достижении обратимого потенциала образования атома водорода $E_{{\text{S}}}^{{{\text{''}}}} = - E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}^{{\text{0}}}}}}} \right. \kern-0em} {{{{\text{H}}}^{{\text{0}}}}}}}}^{a}$ становится термодинамически возможным его образование на металле и последующая адсорбция в виде адатома H-ad по реакции Фольмера (5.1). Тогда, определяя сдвиг потенциала, необходимый для образования адатомов водорода как перенапряжение электрода ${\eta }_{{{\text{H}} - {{{\text{ad}}} \mathord{\left/ {\vphantom {{{\text{ad}}} {{\text{Me}}}}} \right. \kern-0em} {{\text{Me}}}}}}^{a}$ относительно потенциала $E_{{\text{N}}}^{{{\text{0,}}a}},$ запишем:
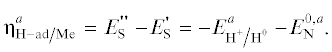
В дополнение, будем также учитывать, что замещение одного адатома металла одним адатомом водорода на поверхности грани должно удовлетворять условию нулевого баланса электронов в валентной зоне поверхности металла. Так как согласно формуле (4.3) величина ПНЗ $E_{{\text{N}}}^{{\text{0}}}$ = –$\Delta G_{S}^{0}$/zF относится к единичной валентной связи металла, то для металла с валентностью z формула (5.8) принимает вид:
(5.9)
${\eta }_{{{\text{H}} - {{{\text{ad}}} \mathord{\left/ {\vphantom {{{\text{ad}}} {{\text{Me}}}}} \right. \kern-0em} {{\text{Me}}}}}}^{a} = - E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}^{{\text{0}}}}}}} \right. \kern-0em} {{{{\text{H}}}^{{\text{0}}}}}}}}^{a} - zE_{{\text{N}}}^{{{\text{0,}}a}}.$Теперь перейдем от шкалы абсолютных потенциалов к стандартной водородной (SHE), заменяя величину $E_{{{{{{{\text{H}}}^{{\text{ + }}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{{\text{ + }}}}} {{{{\text{H}}}^{{\text{0}}}}}}} \right. \kern-0em} {{{{\text{H}}}^{{\text{0}}}}}}}}^{a}$ в ур. (5.9) ее численной экспериментальной величиной Е = –1.97 в ур. (5.4) в виде
(5.10)
${{{\eta }}_{{{\text{H}} - {{{\text{ad}}} \mathord{\left/ {\vphantom {{{\text{ad}}} {{\text{Me}}}}} \right. \kern-0em} {{\text{Me}}}}}}} = - 1.97 - zE_{{\text{N}}}^{0}\left( {{\text{SHE}}} \right)$Мы относим формулу (5.10) к реакции (5.1) Фольмера, т.е. полагаем, что она подтверждает реакцию образования адатомов водорода как реакцию, лимитирующую суммарную (overall) реакцию выделения молекул H2 на различных металлах. Это означает, что измеряемая величина ${{{\eta }}_{{{{{{{\text{H}}}_{{\text{2}}}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}_{{\text{2}}}}} {{\text{Me}}}}} \right. \kern-0em} {{\text{Me}}}}}}},$ под которой обычно подразумевают перенапряжение выделения водорода (ПВВ) в виде молекул Н2 на металле, в действительности относится к образованию атомарного водорода, т.е. сохраняя принятое обозначение, представляем ур. (5.10) как
(5.11)
${\eta }_{{{{{\text{H}}}_{2}}}}^{0} = - 1.97 - zE_{{\text{N}}}^{0}\left( {{\text{SHE}}} \right)$Чтобы получить теоретический аналог экспериментального уравнения (5.4), заменим в формуле (5.11) величину $E_{{\text{N}}}^{0}$ на термодинамическую формулу (4.3) для ПНЗ металлов при Т = 0 K в виде $E_{{\text{N}}}^{0}$ = $ - \Delta U_{S}^{0}$|zF.
Отсюда следует формула:
В ур. (5.12) реализуется исходная идея о замещении адатомов металла (автоадсорбция атомов металла с поверхностной энергией $\Delta U_{S}^{0}$) адатомами водорода на металле с энергией адсорбции $\Delta {{U}_{{{\text{ads}}}}} = - \Delta {{U}_{{\text{S}}}}.$ Еще раньше Фрумкин в теории замедленного разряда ПВВ (в виде альтернативы теории замедленной рекомбинации) качественно объяснил зависимость величины перенапряжения водорода от материала электрода, допустив, что перенапряжение катодной реакции определяется величиной энергии адсорбции водорода qads на данном металле как [11]
(5.13)
${{\eta }_{K}} = - {\text{const}} + \frac{{{{q}_{{{\text{ads}}}}}}}{F} + \frac{{RT}}{{\alpha F}}{\text{ln}}{{i}_{K}}.$Хотя очевидна близость формул (5.12) и (5.13), одинаково определяющих большую роль адсорбции в процессе ПВВ, в литературе отсутствует объяснение причин различной способности поверхности отдельных металлов адсорбировать водород. Известные теории ПВВ не раскрывают смысл так называемого каталитического ряда металлов по Bounhoffer (1924):
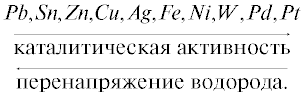
Из сравнения (5.14) с формулой (5.12) и рис. 5.1 следует, что этот ряд можно заменить на ряд металлов с ГЦК или ГПУ структурой и ряд с ОЦК структурой, в каждом из которых перенапряжение водорода определяется минимальной величиной поверхностной энергии Гиббса $\Delta U_{{\text{S}}}^{0}$ низкоиндексной грани металла (наблюдаемые отклонения от этой закономерности для некоторых металлов (Pt, Pd, Rh, Ir, Co, Fe), вероятно, связаны с их наводораживанием).
Имея в виду важное практическое значение электрокаталитической реакции выделения водорода, мы предлагаем рассмотреть другую сторону этой реакции, связанную непосредственно с адсорбцией атомов Had и промежуточных частиц – поверхностных вакансий V(S) на металле, в виде квазиэлектрохимической реакции
(5.15)
${{{\text{H}}}^{ + }} + \quad{{{\text{e}}}^{ - }}\left( {{\text{М е }}} \right) + {{V}_{{\left( {\text{S}} \right)}}}\quad = {{H}_{{{\text{ad}}}}}{{V}_{{\left( {\text{S}} \right)}}}.$В терминах кинетики электролитического выделения водорода, реакция (5.15) лимитируется замедленным переходом электрона от металла к иону Н+, но, с другой стороны, в литературе [12] указывают на возможность “захвата” образующегося атома водорода подповерхностной вакансией. В нашей трактовке, образование адатома Нad протекает как хемосорбционная реакция (5.15) непосредственно между ионом Н+ в водном растворе и вакансией V(S) в ПС металла, которая, во-первых, подчиняется формальной кинетике химической реакции 1-го порядка и, во-вторых, несет в себе характерные черты электрокаталитической реакции, так как зависит от поверхностного потенциала ES металла. Последний является мерой поверхностной энергии ES = GS/zF металла, но при идеальной поляризации металла его поверхности навязывается потенциал электрода Ei = ES (см. ур. (2.8), ч. 1). Отсюда с позиций электродной кинетики, скорость образования адатомов Had, выраженную в токовых единицах как iH–ad, оценим, используя закон действующих масс для реагирующих частиц, в виде
(5.16)
${{i}_{{{\text{H}} - {\text{ad}}}}} = {{K}_{{{\text{ch}}}}}{{C}_{{{{{\text{H}}}^{ + }}}}}\left[ {{{N}_{{V\left( {\text{S}} \right)}}}\left( {{{E}_{{\text{S}}}}} \right)} \right],$(5.17)
$\begin{gathered} - RT{\text{ln}}{{N}_{{V\left( {\text{S}} \right)}}} = - RT{\text{ln}}{{N}_{{{\text{ad}}}}} = \\ = {\Delta }G_{{\text{S}}}^{0} - zF\left| {{{E}_{{\text{S}}}} - E_{{\text{N}}}^{0}} \right|. \\ \end{gathered} $Напомним, что шкале ASP принадлежат две особые точки $E_{{\text{N}}}^{0}$ = –$G_{{\text{S}}}^{0}$/zF и $E_{{\text{S}}}^{0}$ = $G_{{\text{S}}}^{0}$/zF, где $\Delta G_{{\text{S}}}^{0}$ – cтандартная величина энергии образования поверхности грани металла. В точке ПНЗ грани, принимая ES = $E_{{\text{N}}}^{0}$ = 0, имеет место NV(S) = = exp (–$G_{{\text{S}}}^{0}$/RT) минимум адсорбции ОЗВ. Катодная поляризация, при которой электроду навязывается потенциал Ek, равный поверхностному, т.е. как ΔE = Ek – $E_{{\text{N}}}^{0},$ снижает ПЭ и повышает концентрацию ОЗВ. Целесообразно использовать шкалу ASP, оперируя величиной ES = Ek в (5.17) и парциальной катодной зависимостью NV(S) от потенциала в области от ES = $E_{{\text{N}}}^{0}$ = 0 до его предельного значения ES = –$E_{{\text{S}}}^{0}$ (см. рис. 4.1 и ур. (4.3), ч. 1). От ур. (5.17) легко перейти к уравнению для катодной реакции ES < 0
(5.18)
${{N}_{{V\left( {\text{S}} \right)}}} = {\text{exp}}\left[ { - \frac{{{\Delta }G_{{\text{S}}}^{0} + zF\left( {{{E}_{{\text{S}}}} - E_{{\text{N}}}^{0}} \right)}}{{RT}}} \right].$Далее, от термодинамического соотношения (5.18) для свободной энергии Гиббса мы переходим, используя правило Horiuty–Polanyi [13], к кинетическому соотношению с аррениусовской энергией активации замедленного разряда ионов H+ c коэффициентом переноса α. Кроме того, мы учитываем участие ОЗВ с зарядом z =1, получив, в итоге, для реакции (5.15) формулу концентрации “активных центров” – вакансий на поверхности металла для разряда ионов водорода:
(5.19)
${{N}_{{V\left( {\text{S}} \right)}}} = {\text{exp}}\left( { - \frac{{\Delta G_{{\text{S}}}^{0}}}{{RT}}} \right){\text{exp}}\left[ {\frac{{ - \alpha F\left( {{{E}_{{\text{S}}}} - E_{{\text{N}}}^{0}} \right)}}{{RT}}} \right].$Окончательно, делая подстановку (5.19) в (5.16) и логарифмируя полученное выражение, получим уравнение
(5.20)
$\begin{gathered} {{\eta }_{{{{{\text{H}}}_{2}}}}} = - \left( {{{E}_{{\text{S}}}} - E_{{\text{N}}}^{0}} \right) = - \frac{{RT}}{{\alpha F}}{\text{ln}}{{K}_{{{\text{ch}}}}} - \\ - \,\,\frac{{RT}}{{\alpha F}}{\text{ln}}{{C}_{{{{{\text{H}}}^{ + }}}}} + \frac{{\Delta G_{{\text{S}}}^{0}}}{{\alpha F}} + \frac{{RT}}{{\alpha F}}{\text{ln}}{{i}_{{{\text{H}} - {\text{ad}}}}}. \\ \end{gathered} $Это – теоретическая формула электрокаталитической реакции выделения атомарного водорода на металлах, обладающих различной ПЭ ΔGS (hkl) с минимумом ПЭ сингулярной грани (hkl). Скорость выделения Had определяется энергией адсорбции атомарного водорода $\Delta G_{{{\text{ad}}}}^{0}$ = $ - \Delta G_{{\text{S}}}^{0}$ и замедленностью разряда его ионов. Используя условия ${{C}_{{{{{\text{H}}}^{ + }}}}}$ = 1 и Т = 298 K, получим из ур. (5.20) величину перенапряжения водорода при токе обмена I0,H–ad
(5.21)
${{\eta }_{{{{{\text{H}}}_{2}}}}}\left( {{{i}_{{0,{\text{H}} - {\text{ad}}}}}} \right) = - {\text{const}} + \frac{{\Delta G_{{\text{S}}}^{0}}}{{\alpha F}}.$Сравнение формулы (5.21) с экспериментальным ур. (5.4) показывает, что они практически совпадают по тангенсу угла наклона прямой tgγ = = 0.0201 с теоретическим tgγ = ($ \propto $F)–1 = 0.0207, где $ \propto $ = 0.5. Кроме того, согласно (5.21) величина const равна перенапряжению выделения водорода при $\Delta G_{S}^{0}$ = 0, т.е для некоторого гипотетического металла, не обладающего ПЭ и потому не оказывающего никакого деполяризующего влияния на перенапряжение (ближе всех к нему подходит Pb или жидкая Hg). В этом случае энергия перенапряжения затрачивается на выделение водорода $\Delta G_{{{{{\text{H}}}_{{\text{2}}}}}}^{0} = - 2F\eta _{{{{{\text{H}}}_{{\text{2}}}}}}^{0} = - 2F{{\overline K }_{1}}$ = 381 кДж/моль, где K1 = –1.97 В – константа в ур. (5.4). Эта величина коррелирует с используемой в литературе величиной энергии диссоциации молекулы водорода ${{D}_{{{\text{H}} - {\text{H}}}}}$ = 420–435 кДж/моль. Отклонение опытной величины от ${{D}_{{{\text{H}} - {\text{H}}}}}$ может быть связано с учетом теплоты физической адсорбции газов на металлах порядка ${{q}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ ≈ –(30–40) кДж/моль. Тогда константу хемосорбции Kch из ур. (5.20) представляем с учетом ур. (5.5) в качестве константы диссоциативной хемосорбции молекул Н2, что позволяет использовать вместо ур. (5.20) нижеследующее уравнение с более определенным физико-химическим содержанием
(5.22)
${{\eta }_{{{{{\text{H}}}_{2}}}}} = - \frac{{{{K}_{{{\text{dis}}}}} - {{q}_{{{{{\text{H}}}_{2}}}}}}}{{2F}} + \frac{{\Delta G_{{\text{S}}}^{0}}}{{\alpha F}} + \frac{{RT}}{{\alpha F}}{\text{ln}}{{i}_{{{\text{H}} - {\text{ad}}}}}.$Заметим, что, используя шкалу ASP, мы получили термодинамическую формулу (5.11) зависимости перенапряжения водорода от величины ПНЗ грани металла, на которой выделяется водород. Вводя в эту формулу кинетический коэффициент $ \propto ,$ получим ранее неизвестное соотношение между перенапряжением выделения водорода на металле и величиной ПНЗ низкоиндексной грани металла:
(5.23)
${\eta }_{{{{{\text{H}}}_{2}}}}^{0} = - 1.97 - z{{E_{{\text{N}}}^{0}} \mathord{\left/ {\vphantom {{E_{{\text{N}}}^{0}} \alpha }} \right. \kern-0em} \alpha }\,\,\left( {{\text{SHE}}} \right).$Проверим это соотношение, используя теоретически вычисленные при Т = 298 K (с использованием известных величин $\Delta U_{S}^{0}$ [8]) значения EN (111) (табл. 4.2 ., ч. 1) для Ag с величиной ( минус) 0.52 В и Z = 1 и для Ni c (минус) 0.33 В и Z = 2. Расчет дает для ${{\eta }_{{{{{{{\text{H}}}_{2}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}_{2}}} {{\text{Ni}}}}} \right. \kern-0em} {{\text{Ni}}}}}}}$ = –0.65 В (по Трасатти [4] –0.62 В) и для ${{\eta }_{{{{{{{\text{H}}}_{2}}} \mathord{\left/ {\vphantom {{{{{\text{H}}}_{2}}} {{\text{Ag}}}}} \right. \kern-0em} {{\text{Ag}}}}}}}$= –0.93 В (здесь расчет совпадает с опытными данными).
Подводя итог проведенным расчетам, мы полагаем, что атомы водорода H0 могут стабильно существовать только в адсорбированном состоянии на поверхности металла, а их избыточное накопление в ходе электролиза приводит к их непрерывному переходу во внешний слой, где атомы H0 рекомбинируют в молекулы H2. Для сравнения, в водородной энергетике существует проблема получения, сохранения и использования атомарного водорода. Газообразный атомарный водород образуется при прохождении электрического разряда через разреженную атмосферу водорода (Wood, 1922). В газоразрядном аппарате, Bounhoffer (1924) на выходе получал “активный” водород ${{{\text{H}}}^{{\text{0}}}}$ (см. также [14]). В его потоке небольшие зерна Pt, Pd или W сильно нагреваются. Boungoffer объяснял этот эффект выделением большого количества тепла в реакции рекомбинации атомов ${{{\text{H}}}^{0}} + {{{\text{H}}}^{0}} = {{{\text{H}}}_{2}},$ протекающей вследствие каталитической активности этих металлов, а затем использовал это объяснение для обоснования ряда металлов (5.14). В энергетическом сравнении этот процесс противоположен первому – электролитическому выделению ${{H}_{2}}.$ Важно дать обоим процессам объяснение с позиций закона сохранения и превращения энергии и особенно – участия в нем поверхностной энергии металла. В первом процессе, согласно ур. (2.8), ПС металла аккумулирует энергию за счет поляризации электрода с уменьшением поверхностной энергии $\Delta {{G}_{{\text{S}}}},$ а затем отдает ее в виде энергии адсорбции атомов ${{{\text{H}}}^{{\text{0}}}}$ как $\Delta {{G}_{{\text{S}}}} = - \Delta {{G}_{{{\text{ads}},{{{\text{H}}}^{0}}{\;}}}}.$ Во втором – наоборот происходит адсорбция атомов ${{{\text{H}}}^{{\text{0}}}}$ на металле. В пределе, полное покрытие поверхности θ → 1 может снизить поверхностную энергию до нуля $\Delta {{G}_{{\text{S}}}}$ → 0, но для незаряженной поверхности металла, которой соответствует точка ПНЗ $E_{{\text{N}}}^{0}$ с величиной ${\;}\Delta G_{S}^{0}$ (рис. 5.1), это снижение является избыточным, так что происходит превращение энергии адсорбции – в поверхностную $\Delta {{G}_{{{\text{ads}},{{{\text{H}}}^{0}}{\;}}}} = - {\;}\Delta G_{{\text{S}}}^{0}.$ Этот избыток энергии передается атомам ${{{\text{H}}}^{0}}\quad$ в виде энергии возбуждения, которая будет тем больше, чем больше $\Delta {{G}_{{\text{S}}}}$ металла. Процесс заканчивается рекомбинацией возбужденных атомов ${{{\text{H}}}^{0}}\quad$ с выделением количества тепла, эквивалентного сумме энергии рекомбинации атомов $\Delta {{U}_{{{{{\text{H}}}^{0}} - {{{\text{H}}}^{0}}}}}$ и поверхностной энергии ${\;}\Delta G_{{\text{S}}}^{0}\left( T \right)$ при данной температуре (такой механизм хорошо объясняет максимальный разогрев в потоке атомарного водорода тех металлов (Pt и W), которые имеют максимальную поверхностную энергию). По существу, этот механизм действует и в процессе атомно-водородной сварки металлов с тем отличием, что диссоциация Н2 с целью образования Н0 происходит в электрической дуге.
6. ЭЛЕКТРОХИМИЧЕСКАЯ ПАССИВНОСТЬ МЕТАЛЛОВ И ТЕРМОДИНАМИЧЕСКИЙ РАСЧЕТ ФЛАДЕ-ПОТЕНЦИАЛОВ Ni И Cr
Другим примером совмещения шкалы обратимых потенциалов электродных реакций и шкалы поверхностных потенциалов металла является случай образования поверхностного оксида на металле. Неудачи в теории электрохимической пассивности металлов, по нашему мнению, были связаны долгое время с тем, что образование этого оксида на металле рассматривалось вне свойств поверхности металла как простая реакция:
(6.1)
$m{\text{М е }} + n{{{\text{H}}}_{{\text{2}}}}{\text{O}} = {\text{М }}{{{\text{e}}}_{m}}{{{\text{O}}}_{n}} + 2n{{{\text{H}}}^{ + }} + 2n{{{\text{e}}}^{ - }}.$Потенциал образования такого оксида по водородной шкале можно рассчитать как:
(6.2)
$E_{{{{{\text{М е }}} \mathord{\left/ {\vphantom {{{\text{М е }}} {{\text{М }}{{{\text{e}}}_{m}}}}} \right. \kern-0em} {{\text{М }}{{{\text{e}}}_{m}}}}{{{\text{O}}}_{n}}}}^{0} = \frac{{{\;}\Delta G_{{{\text{М }}{{{\text{e}}}_{m}}{{{\text{O}}}_{n}}}}^{0}}}{{2nF}} + E_{{{{{\text{O}}}_{2}}}}^{0}.$Именно с этой величиной в литературе сравнивали экспериментальное значение Фладе-потенциала ${{E}_{F}},$ являющегося важной характеристикой пассивности металла. Большие расхождения в величинах между расчетом по ур. (6.2) и экспериментальной величиной ${{E}_{F}}$ препятствовали развитию теории пассивности. Совмещение шкалы электродных потенциалов для реакции (6.1) и шкалы поверхностных потенциалов ${{E}_{{\text{S}}}}$ позволяет дать анализ ранее выведенной формулы Фладе-потенциала на основе хемосорбционной модели, учитывающей поверхностную энергию металла [15, 16]:
(6.3)
$E_{{F\left( {\text{S}} \right)}}^{0} = \frac{{{\;}\Delta G_{{{\text{М }}{{{\text{e}}}_{m}}{{{\text{O}}}_{n}}}}^{0}}}{{2nF}} + \frac{{{\;}m\Delta G_{{{\text{S}},{\text{Me}}}}^{0}}}{{2nF}} - \frac{{{\;}\Delta G_{{{{{\text{H}}}_{2}}{\text{O}}}}^{0}}}{{2F}}.$В свете излагаемой теории шкалы ASP, первый терм в правой части ур. (6.3) относится к абсолютному значению потенциала образования оксида металла, а второй – абсолютному значению поверхностной энергии металла. Так как $\Delta G_{{{{{\text{H}}}_{2}}{\text{O}}}}^{0} = - 2F\left( {E_{{{{{\text{O}}}_{2}}}}^{0} - E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{0}} \right),$ то формула (6.3) соответствует переходу от абсолютной шкалы к – водородной относительно величины $E_{{{{{{{\text{H}}}^{ + }}} \mathord{\left/ {\vphantom {{{{{\text{H}}}^{ + }}} {{{{\text{H}}}_{2}}}}} \right. \kern-0em} {{{{\text{H}}}_{2}}}}}}^{0} = {\text{O,}}$ так что в ур. (6.3) и ур. (6.2) величина ${{\Delta G_{{{{{\text{Н }}}_{2}}{\text{О }}}}^{0}} \mathord{\left/ {\vphantom {{\Delta G_{{{{{\text{Н }}}_{2}}{\text{О }}}}^{0}} {2F}}} \right. \kern-0em} {2F}}$ = 1.229 B равна также и стандартному потенциалу кислорода. Но самое существенное в применении щкалы ASP это – придание ей свойства шкалы адсорбционных потенциалов точечных дефектов – вакансий или адатомов. Отсюда формулу второго терма в (6.3) можно связать с формулой (5.17), устанавливающей соотношение между поверхностным потенциалом ЕS грани (hkl) и мольной долей адатомов Nad. Здесь величину $\quad{{N}_{{{\text{ad}}}}}$ рассматривать как концентрацию адатомов, участвующих в реакции их окисления водой, например, при пассивации Ni как:
(6.4)
${\text{Ni}} - {\text{ad}} + {{{\text{H}}}_{2}}{\text{O}} = {\text{NiO}} + 2{{{\text{H}}}^{ + }} + \quad2{{{\text{e}}}^{ - }}.$Действительно, в литературе [17] используют модельные представления о специфической адсорбции ионов на электроде, вызванной силами различной природы . В контексте данной работы определение “специфической адсорбции”относится исключительно к действию поверхностных сил. Cледуя ур. (5.17) применительно к анодной поляризации, с ростом потенциала EA = ${{E}_{{\text{S}}}}$ величина ${{N}_{{{\text{ad}}}}}$ может возрастать вплоть до максимума поверхностной концентрации адатомов ${{N}_{{{\text{ad}}}}}$ = 1 (см. также правую ветвь адсорбции адатомов на рис. 4.1), при котором достигается поверхностный потенциал для m молей окисляемого металла $E_{{\text{S}}}^{0} = m{\;}{{\Delta G_{{\text{S}}}^{0}} \mathord{\left/ {\vphantom {{\Delta G_{{\text{S}}}^{0}} {2nF}}} \right. \kern-0em} {2nF}},$ обеспечивающий образование хемосорбированного оксида по реакции (6.1). При этом, величина поверхностного потенциала $E_{{\text{S}}}^{0}$ = $\Delta G_{{\text{S}}}^{0}$/zF грани (hkl) берется относительно $E_{{\text{N}}}^{0}$ = 0 и термодинамического заряда zF для 1 моля атомов в ПС металла. Реакция хемосорбции образования оксида состоит в окислении адатома металла с переходом ze-электронов на атом кислорода. Условие электронейтральности в стехиометрическом оксиде для реакции (6.3) дает соотношение n = zm. Его использование, а также учет стехиометрии реакции Н2 – электрода приводит к формуле (6.3). Представляет интерес образование важного для практики нестехиометрического оксида [16], для которого термодинамический расчет дает баланс поверхностной энергии Гиббса
(6.5)
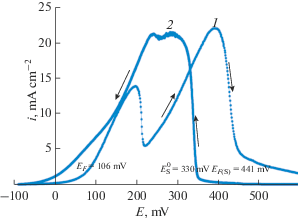
$\Delta {{G}_{{M{{e}_{{m - x}}}{{O}_{n}}}}} = \Delta {{G}_{{M{{e}_{m}}{{O}_{n}}}}} + \left( {m - x} \right)\Delta {{G}_{{S,Me}}}.$Термодинамический расчет Фладе потенциалов нестехиометрических оксидов с величиной $X \ll 1$ в ур. (6.5) показывает незначительную разницу в его величине относительно расчета для стехиометрического оксида. Отсюда численный расчет Фладе-потенциала электрода Ni/NiO по формуле (6.3) приводит к величине EF(S) = 0.46 В, что превосходно подтверждается результатами измерения Фладе-потенциала Ni-электрода методом хронопотенциометрии, полученными Sato и Okamoto [18] (см. обсуждение в [15, 16]). Кроме того, по даным РФЭС [19] пассивная пленка на Ni состоит из внутреннего слоя NiO и внешнего – Ni(OH)2, и рост пленки начинается при Е = 0.48 В, что также удовлетворительно согласуется с теоретическим расчетом Фладе-потенциала никеля. Из наших поляризационных измерений анодной пассивации никеля (рис. 6.1) следует [16], что потенциал перехода никеля из пассивного в активное состояние (так назывемый потенциал реактивации пассивного состояния) близок к термодинамической величине ЕF(S) = 0.46 В. За вычетом величины 0.106 В, отвечающей энергии Гиббса образованию NiO вне поверхности Ni, получается величина 0.35 В для поверхностного потенциала $E_{{\text{S}}}^{0}$ для Ni(111), близкого к величине $E_{{\text{N}}}^{0}$ = –0.33 В для Ni(111), взятого с обратным знаком (см. формулы (4.2) и (4.3), ч. 1 и рис. 6.1).
Рис. 6.1.
Анодные поляризационные кривые для никеля в растворе 0.5 М H2SO4 при развертке потенциала в прямом (1) и обратном (2) направлении. Кривая 2 отражает процесс катодного восстановления пассивной пленки, т.е. активации поверхности никеля. Видно, что потенциал начала активации Ea и теоретический Фладе – потенциал по ур.(6.3) практически совпадают. Ход кривой активации удовлетворяет протеканию реакции (6.4) с образованием адионов с ожидаемым максимумом их концентрации Nad = 1.
Принципиально важным в расчете EF(S) как характеристики реакции (6.1) является также учет стехиометрии поверхностного оксида – числа атомов металла m в оксиде. Для оксида хрома только при m = 2 расчет дает величину Фладе-потенциала EF(S) = –0.192 В близкую к опытным электрохимическим данным прошлых лет: E = = ‒0.25 В [20] или E = –0.2 В [21]. Методом РФЭС в растворе 0.5 М H2SO4 получена линейная зависимость толщины d пассивной пленки оксида хрома от потенциала, и экстраполяция этой зависимости на толщину d ≤ 1 нм привела к потенциалу пассивации Cr, равному Е = –0.2 В [22], практически совпадающему с теоретическим расчетным значением в нашей работе. Важно отметить, что в качестве исходных величин расчета Фладе-потенциалов сплавов Ni–Cr [23], были использованы табличные значения поверхностной энергии [8] низкоиндексных граней с ее минимумом соответственно для никеля U(S) (111) и хрома U(S) (110), пересчитанные на Т = 298 K по формуле (3.14). Если использовать величины U(S) (hkl) с более высокой энергией, то отмеченная выше удовлетворительная корреляция между опытными данными и расчетом пропадает. Точно такая же закономерность проявляется и при сравнении расчета и опыта в анализе зависимости перенапряжения водорода от величины поверхностной энергии граней монокристалла металлов (см. п. 5).
ЗАКЛЮЧЕНИЕ
1. Теория шкалы абсолютных поверхностных потенциалов (asp) базируется на основных положениях:
− знание численных величин поверхностной энергии ΔGS (hkl) низкоиндексных граней металлов;
− связь концентрации поверхностых вакансий (ПВ) и адатомов с величиной ΔGS металла;
− использование поверхностых потенциалов ES = ΔGS/zF атомов, относящихся к существованию точки ПНЗ с минимумом концентрации ПВ или адатомов и с их максимумом в точке ES= 0.
2. Основные результаты применения шкалы ASP
2.1. Определена общность энергии хемосорбционных процессов выделения водорода на металлах и пассивации металлов, которая выражается в том, что наряду с энергией Гиббса химического (или электрохимического) ΔGch < 0 взаимодействия всегда проявляется влияние поверхностной энергии металла в виде минимальной энергии Гиббса грани (hkl) для ГЦК, ГПУ или ОЦК металлов ΔGS (hkl) > 0. Исходя из теоретического расчета, подтвержденного экспериментальными данными, предлагается рассматривать энергию Гиббса хемосорбции как сумму их величин
(A)
$\Delta {{G}_{{{\text{ch - ads}}}}} = \Delta {{G}_{{{\text{ch}}}}} + \Delta {{G}_{S}}\left( {hkl} \right).$Формула (А) может быть заменена суммой обратимых электродных потенциалов химической реакции и поверхностного потенциала металла по шкале абсолютных потенциалов с последующим переходом к водородной шкале.
2.2. Электрокаталитические реакции катодного направления (выделение атомарного водорода на различных металлах) и анодного направления (электрохимическая пассивация никеля и хрома) рассчитываются с использованием единого уравнения (5.17), определяющего зависимость концентрации каталитически активных ПВ или адатомов (адионов) от электродного потенциала;
2.3. В процессах выделения водорода на металлах и пассивации металлов наблюдается переход от поликристаллической структуры поверхности ГЦК металлов к монокристаллической (111), который объясняется минимумом поверхностной энергии грани (111) по сравнению с энергией других граней поликристалла, а также быстрой диффузией атомов, связанной с высокой концентрацией атомных вакансий в поверхностном слое металла (см. часть 1).
Список литературы
Андреев Ю.Я. // Физикохимия поверхности и защита материалов. Часть 1. 2018. Т. 54. С. 523
Bard A.J., Inzelt G., Scholz F. Electrochemical Dictionary, 2nd Edition. Springer, 2012.
Bockris J.O’.M. // Trans. Faraday Soc. 1947. V. 43. P. 417.
Trassatti S. // J. Electroanal. Chem. 1972. V. 39. P. 163.
Trasatti S. // Inter. Union of Pure and Appl. Chem. 1986. V. 58. P. 955.
Trasatti S., Lust E. The Potential of Zero Charge, in: Modern Aspects of Electrochemistry. № 33 (edit. White R.E., Bockris J.O.M. and Conway B.E.). 1999.
Андреев Ю.Я. // Физикохимия поверхности и защита материалов. 2012. Т. 48. С. 242.
Vitos L., Ruban A.V., Skriver H.L., Kollar J. // Surf. Sci. 1998. V. 441. P. 186.
Антропов Л.И. Теоретическая электрохимия, М.: Высшая школа, 1975.
Roberts M.W., McKee C.S. Chemistry of the Metal-Gas Interface, Clarendon Г. Press, Oxford, 1978.
Фрумкин А.Н. Избранные труды. Электродные процессы. М.: Наука, 1987.
Халдеев Г.В. Структурная коррозия металлов. Пермь: ПГУ, 1994.
Horiuti J., Polanyi U. // Acta physicochim. URS. V. 2. № 4. P. 505.
Реми Г. Курс неорганической химии, т. 1. М.: Мир, 1966.
Андреев Ю.Я., Бобков Т.В. // Физикохимия поверхности и защита материалов. 2015. Т. 51. С. 456.
Андреев Ю.Я. Электрохимия металлов и сплавов. М.: Высшее образование и наука, 2016.
Дамаскин Б.Б., Петрий О.А., Цирлина Г.А. Электрохимия. – М.: Химия, КолосС, 2008.
Sato N., Okamoto G. // J. Soc. 1963. V. 110. P. 605.
Haupt S., Strehblow H.-H. // J. Electroanal. Chem. 1987. V. 228. P. 365.
Ulig H.H. Corrosion and Corrosion control. N.Y. Wiley Int., 1962.
Kolotyrkin Ya.M. // Z. Elektroch. 1958. Bd. 62. S. 664.
Hoppe H.-W., Strehblow H.-H. // Surf. Interf. Anal. 1989. V. 14. P. 121.
Андреев Ю.Я., Сафонов И.А., Дуб А.В. // Физикохимия поверхности и защита материалов. 2010. Т. 46. С. 435.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов