

Физикохимия поверхности и защита материалов, 2020, T. 56, № 1, стр. 53-57
Монолитная колонка с сорбентом на основе 1-винил-1,2,4-триазола для гидрофильной ВЭЖХ
Ю. В. Патрушев 1, 2, *, Ю. С. Сотникова 1, 2, В. Н. Сидельников 1
1 Институт катализа им. Г.К. Борескова Сибирского отделения
Российской академии наук
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: patrush@catalysis.ru
Поступила в редакцию 18.02.2019
После доработки 18.05.2019
Принята к публикации 22.05.2019
Аннотация
Впервые приготовлена монолитная хроматографическая колонка для ВЭЖХ с сорбентом на основе 1-винил-1,2,4-триазола. Показано, что в зависимости от состава подвижной фазы (ацетонитрил/вода) данная колонка может работать в режиме обращенно-фазовой хроматографии или в режиме гидрофильной хроматографии. Исследованы зависимости фактора удерживания от объемной доли ацетонитрила в подвижной фазе для веществ кислотной и основной природы. Приведен пример разделения компонентов лекарственного препарата “Аскофен-П” на приготовленной колонке.
ВВЕДЕНИЕ
В современной жидкостной хроматографии наибольшее число определений приходится на обращенно-фазовую ВЭЖХ (ОФ ВЭЖХ). Это связано с тем, что большинство коммерческих хроматографических колонок для ОФ ВЭЖХ заполнены силикагельными сорбентами, поверхность которых модифицирована алкильными группами с длиной цепи от С8 до С18. Селективность разделения на обращенно-фазовых сорбентах можно регулировать изменением состава подвижной фазы. Однако не все аналитические задачи удается решить методом ОФ ВЭЖХ.
Для разделения небольших по размеру и полярных молекул лучше всего подходят хроматографические колонки с неподвижными фазами, проявляющими способность к гидрофильным взаимодействиям [1]. В качестве таких неподвижных фаз выступают силикагельные сорбенты с полярными группами, такими, как NH2, CN, амиды, диолы, химически связанными с поверхностью силикагеля [2]. Селективность, характерную для гидрофильных взаимодействий, проявляет так же и сорбент с силикагельной основой, поверхность которого модифицирована 1,2,4-триазолом. Следствием этих взаимодействий является сильное удерживание колонкой с данным сорбентом кислот [3–5].
В случае гидрофильной хроматографии неподвижной фазой является тонкий слой воды, адсорбированный на поверхности гидрофильных фрагментов сорбента, а подвижной фазой является раствор ацетонитрила и воды, в котором высока доля ацетонитрила. Объектами разделения для гидрофильной хроматографии являются полярные или ионизированные в растворе молекулы [6]. В этом случае хорошо известная для обращенно-фазовой хроматографии зависимость фактора удерживания от содержания органической составляющей в элюенте принимает иной вид. При наличии гидрофильных взаимодействий удерживание полярных аналитов возрастает с увеличением доли органической составляющей в подвижной фазе.
В отличие от ОФ ВЭЖХ, селективность разделения в гидрофильной хроматографии зависит в большей степени не от состава элюента, а от свойств неподвижной фазы [7]. Поэтому поиск новых неподвижных фаз для гидрофильной ВЭЖХ расширяет возможности метода.
Следует отметить, что, помимо насадочных колонок для ВЭЖХ существуют и развиваются технологии приготовления колонок, в которых неподвижная фаза (НФ) является проницаемым монолитом. Интерес к монолитным материалам органической и неорганической природы в качестве НФ для высокоэффективной жидкостной хроматографии связан как с относительной простотой приготовления, так и с возможностью создания колонок, обладающих одновременно хорошей эффективностью и высокой проницаемостью [8]. В качестве материала для создания монолита внутри колонки наиболее распространенными являются либо силикагели, либо органические полимеры на основе акрилатов и ароматических мономеров. В настоящее время описаны монолитные колонки с пористыми полимерами на основе таких исходных мономеров, как дивинилбензол (ДВБ), стирол (СТ), винилбензилхлорид, алкилметакрилаты, N-винилкарбазол, этиленгликоль диметакрилат, глицидилметакрилат, пентаэритриол тетраакрилат, пентаэритриол триакрилат, этилвинилбензол, 2-гидроксиэтил метакрилат, 1,2-(п-вилинфенилэтан), 1-винилимидазол, 4-винилпиридин [9–18].
В данной работе описан синтез и некоторые свойства монолитной колонки с органическим сорбентом на основе 1-винил-1,2,4-триазола (ВТр), сополимеризованного с дивинилбензолом (ДВБ) и стиролом (Ст). Поскольку монолит ВТр-ДВБ-Ст состоит как из гидрофильных фрагментов (ВТр), так и из гидрофобных фрагментов (ДВБ, Ст), появляется возможность работы такой колонки как в режиме ОФ хроматографии, так и в режиме гидрофильной хроматографии в зависимости от состава подвижной фазы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали жидкостный хроматограф Милихром А-02 с автоматическим дозатором. Для приготовления элюента использовали деионизованную воду (0.05 мкСм/см), ацетонитрил для ВЭЖХ (НПО Экрос, Санкт-Петербург) и трифторуксусную кислоту (ТФК) 99% (Sigma-Aldrich Chemie GmbH, Steinheim, Germany). Для обработки внутренней поверхности стеклянных трубок использовали 3-(триметоксисилил)пропилметакрилат (ТМСПМ). Для синтеза монолитных сорбентов применяли: стирол (99%), дивинилбензол (80%, смесь изомеров), 1-винил-1,2,4-триазол (99%), 1-метил-2-пирролидон (99%), перекись бензоила (75%), Pluronic 10R5 (бифункциональный блок сополимер полипропиленгликоль-полиэтиленгликоль-полипропиленгликоль) в качестве порообразователя (все реактивы от Sigma-Aldrich Chemie GmbH, Steinheim, Germany).
Обработка стеклянных трубок
Для приготовления монолитных колонок использовали стеклянные трубки из свинцового стекла (содержание PbO около 40%) внутренним диаметром 2 мм и внешним диаметром 4 мм. Исходные стеклянные трубки предварительно обрабатывали уксусной кислотой. Для этого трубки заполняли 60% раствором уксусной кислоты и выдерживали в течение суток. Затем трубки промывали дистиллированной водой и сушили в токе воздуха. Далее трубки обрабатывали 3-(триметоксисилил)пропилметакрилатом (ТМСПМ). Это вещество используется при модифицировании поверхности стекла или силикагеля для образования на ней функциональных групп с двойной связью, способных вступать в реакцию радикальной полимеризации [19]. Для модифицирования поверхности стеклянных трубок готовили раствор: ТМСПМ, этанол, 95% раствор трифторуксусной кислоты в объемном соотношении 10 : 10 : 0.1 соответственно. Трубки заполняли свежеприготовленным раствором и выдерживали при комнатной температуре в течение суток. Затем трубки промывали этанолом и сушили в токе азота при комнатной температуре.
Приготовление монолита
Для приготовления полимеризационного раствора последовательно смешивали: 1-метил-2-пирролидон (0.4 мл), дивинилбензол (1 мл), стирол (0.2 мл) и 1-винил-1,2,4-триазол (0.8 мл). В раствор добавляли перекись бензоила в количестве 1 мас. % к суммарному объему мономеров. В качестве порообразователя использовали Pluronic 10R5 объемом 3 мл.
Предварительно обработанную стеклянную трубку заполняли полимеризационной смесью и запаивали оба конца. Далее трубку помещали в воздушный термостат на 4 ч при температуре 80°С. После этого трубку с монолитом охлаждали до комнатной температуры и обрезали по длине.
Колонку устанавливали в хроматограф при помощи стандартных фитингов. Для удаления порообразователя и остатков полимеризационной смеси колонку промывали ацетонитрилом со скоростью 50 мкл/мин [18]. После этого колонка была готова к работе.
На рис. 1 представлен внешний вид готовых колонок и микрофотография синтезированного монолитного сорбента. Видно, что сорбент представляет собой твердый полимерный каркас, сформированный из ассоциатов глобул диаметром около 2–3 мкм. Каркас пронизан полостями размером около 5 мкм. По этим полостям, которые являются транспортными порами, элюат движется вдоль колонки.

Зависимость фактора удерживания от состава подвижной фазы
Для исследования закономерности удерживания полярных молекул на приготовленной монолитной колонке с 1,2,4-триазольными группами в нее вводили аналиты с кислотными и основными свойствами. Растворы ацетилсалициловой кислоты, салициловой кислоты, бензойной кислоты, аскорбиновой кислоты, напроксена, кофеина, парацетамола, пропифеназона и пиридоксина готовили из аптечных препаратов в растворителе с объемным соотношением ацетонитрил/0.1% ТФК в воде = 98/2. Для хроматографирования использовали подвижную фазу, состоящую из ацетонитрила и воды с 0.1% ТФК. Соотношение ацетонитрил/вода в ПФ меняли от 40 до 99%. Объем вводимой пробы составлял 1 мкл. По временам удерживания тестовых веществ строили зависимость фактора удерживания (k) от объемной доли ацетонитрила в ПФ (рис. 2).
Рис. 2.
Зависимость фактора удерживания тестовых соединений от объемной доли ацетонитрила в подвижной фазе. Колонка ВТр длиной 78 мм. Объемная скорость подвижной фазы – 200 мкл/мин; (а) вещества с кислотными свойствами; (б) вещества с основными свойствами.
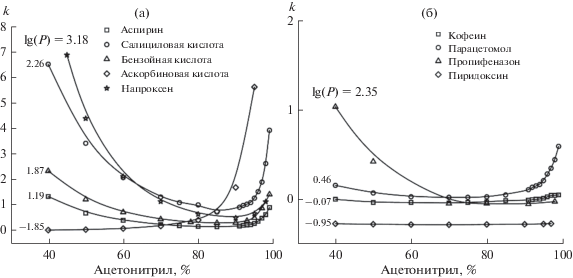
В табл. 1 представлены тестовые вещества, использованные в эксперименте. Для каждого вещества приведены константы кислотности pKa и значения коэффициентов гидрофобности lg (P). Коэффициент гидрофобности – это логарифм константы равновесия для данного вещества в системе 1-октанол–вода. Чем выше значение lg(P), тем более гидрофобным является аналит.
Таблица 1.
Тестовые вещества, использованные для исследования зависимости фактора удерживания от состава ПФ. Значения коэффициентов гидрофобности и констант кислотности взяты из базы данных PubChem (https://pubchem.ncbi.nlm.nih.gov)
| Аналит | lg (P) | pKa |
|---|---|---|
| Напроксен | 3.18 | 4.15 |
| Салициловая кислота | 2.26 | 2.97 |
| Бензойная кислота | 1.87 | 4.19 |
| Аспирин | 1.19 | 3.49 |
| Аскорбиновая кислота | –1.85 | 4.7 |
| Пропифеназон | 2.35* | 13.1* |
| Парацетамол | 0.46 | 9.38 |
| Кофеин | –0.07 | 10.4 |
| Пиридоксин | –0.95* | 9.4* |
Хроматографирование лекарственного препарата “Аскофен-П”.
Таблетки Аскофена измельчали и растворяли в смеси ацетонитрил/0.1% ТФК = 98/2 объемом 4 мл. Полученную суспензию обрабатывали в ультразвуковой ванне в течение 10 мин, после чего 1 мл суспензии центрифугировали. Полученный раствор разбавляли в 10 раз смесью ацетонитрил/0.1% ТФК = 98/2. В колонку вводили пробу объемом 1 мкл.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 2 представлены зависимости факторов удерживания исследованных соединений от содержания ацетонитрила в подвижной фазе. Как видно, для молекул с кислотными свойствами наблюдается снижение фактора удерживания в диапазоне содержания ацетонитрила в подвижной фазе от 40 до 85% (рис. 2а). Это говорит о том, что колонка работает в режиме обращенно-фазовой хроматографии при данных значениях состава ПФ. Чем больше значение коэффициента гидрофобности lg (P) аналита, тем выше его фактор удерживания при одном и том же составе элюента. Это не удивительно, т.к. при высоком содержании воды в ПФ равновесие сорбции-десорбции для гидрофобных веществ сдвигается в сторону органического сорбента, который является в данных условиях более гидрофобным, чем подвижная фаза. При повышении содержания ацетонитрила с 85 до 99% происходит увеличение фактора удерживания для молекул с кислотными свойствами. То есть колонка начинает работать в режиме гидрофильной хроматографии. Это можно объяснить тем, что в качестве неподвижной фазы выступает вода, адсорбированная на гидрофильных триазольных группах сорбента. Поэтому удерживание полярных молекул будет возрастать с повышением доли органического модификатора в подвижной фазе, поскольку равновесие сорбция-десорбция будет сдвигаться в сторону полярной неподвижной фазы. Исключением является аскорбиновая кислота, для которой во всем исследованном диапазоне состава ПФ фактор удерживания увеличивается с повышением содержания ацетонитрила. Такое поведение может быть связано с низким значением коэффициента гидрофобности (lg(P) = –1.85) аскорбиновой кислоты.
Для аналитов, проявляющих основные свойства в данном эксперименте, картина иная. Пропифеназон из всех основных аналитов, представленных в табл. 1, является наиболее гидрофобным (lg(P) = 2.35). Поэтому при большом содержании воды в ПФ он удерживается лучше остальных тестовых соединений. В диапазоне содержания ацетонитрила в ПФ от 80 до 40% фактор удерживания пропифеназона возрастает с уменьшением доли ацетонитрила, в то время как для остальных основных аналитов он почти не меняется. При высоком содержании ацетонитрила в ПФ (от 80 до 99%) увеличивается удерживание парацетамола, в то время как фактор удерживания остальных основных аналитов не меняется. Такое поведение парацетамола может быть связано с тем, что у него наименьшая величина pKa среди основных аналитов, и в условиях малого содержания воды в ПФ он может сильнее взаимодействовать с 1,2,4-триазольными группами, проявляющими основные свойства.
Теперь рассмотрим разделительные свойства колонки. На рис. 3 представлены хроматограммы препарата “Аскофен-П” в изократическом режиме при разных соотношениях ацетонитрил/вода в подвижной фазе. Данный препарат содержит несколько компонентов (кофеин, парацетамол, ацетилсалициловую кислоту и салициловую кислоту как примесь), которые проявляют как кислотные, так и основные свойства.
Рис. 3.
Разделение компонентов лекарственного препарата “Аскофен-П”. Колонка ВТр длина 78 мм. Подвижная фаза: ацетонитрил/0.1% трифторуксусная кислота: (а) 50/50; (б) 75/25; (в) 98/2. Детектор – УФ-спектрометр, λ = 220 нм; 1 – кофеин, 2 – парацетамол, 3 – ацетилсалициловая кислота, 4 – салициловая кислота.
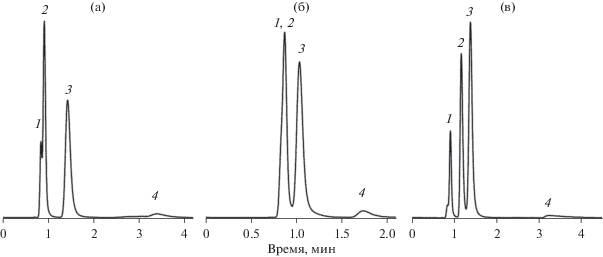
При составе подвижной фазы ацетонитрил/вода = 50/50 разделение происходит преимущественно по обращенно-фазовому механизму (рис. 3а). При этом кофеин и парацетамол делятся неудовлетворительно. При соотношении ацетонитрил/вода в подвижной фазе равном 75/25 разделение происходит быстрее, поскольку факторы удерживания для всех компонентов принимают минимальные значения (см. рис. 2). Но при этом разница между факторами удерживания также минимальная, и это приводит к тому, что разделение ухудшается. При дальнейшем увеличении содержания ацетонитрила в подвижной фазе происходит увеличение фактора удерживания для всех компонентов смеси, кроме кофеина (рис. 2). Это говорит о том, что повышается вклад гидрофильных взаимодействий в удерживание аналитов. На рис. 3в видно, что при составе элюента ацетонитрил/вода = 98/2 разделение всех компонентов смеси удовлетворительное, и хроматографирование проходит за 4 мин.
ЗАКЛЮЧЕНИЕ
Монолитная колонка с сорбентом на основе 1-винил-1,2,4-триазола в зависимости от состава подвижной фазы проявляет способность к различным типам механизмов удерживания. В зависимости от соотношения ацетонитрил/вода колонка может работать как в режиме обращенно-фазовой хроматографии, так и в режиме хроматографии гидрофильных взаимодействий (ХГВ). В варианте ОФ ЖХ фактор удерживания уменьшается с увеличением содержания ацетонитрила в ПФ, в то время как во втором режиме фактор удерживания увеличивается при повышении доли ацетонитрила в ПФ. Для исследованных тестовых аналитов кислой природы смена механизма удерживания происходит при содержании ацетонитрила от 80 до 90%. При этом для аскорбиновой кислоты гидрофильный режим элюирования наблюдается во всем диапазоне содержания ацетонитрила в элюенте: от 40 до 99%. Для тестовых веществ основной природы нет такой же явно выраженной зависимости фактора удерживания от состава подвижной фазы, как для кислотных компонентов. Вероятно, это связано с тем, что триазольные группы монолитного сорбента проявляют основные свойства, и сила взаимодействия основных аналитов с неподвижной фазой не столь существенна, как для кислотных аналитов. Помимо возможности работы в двух режимах (обращенно-фазовой и гидрофильной хроматографии), описанная в работе колонка благодаря органической матрице, обладает химической устойчивостью при работе с сильнокислыми и сильноосновными подвижными фазами и может быть использована для разделения компонентов с кислотными свойствами.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-33-90006.
Список литературы
Dejaegher B., Vander Heyden Y. // J. Sep. Sci. 2010. V. 33. № 6–7. P. 698.
Guo Y., Gaiki S. // J. Chromatogr. A. 2011. V. 1218. № 35. P. 5920.
Huang Z. et al. // J. Pharm. Biomed. Anal. 2014. V. 102. P. 17.
Iwasaki M. et al. // Biomed. Chromatogr. 2016. V. 30. № 3. P. 384.
Xiong X., Liu Y. // Talanta. 2016. V. 150. P. 493.
Alpert A.J. // J. Chromatogr. A. 1990. V. 499. P. 177.
Kumar A., Heaton J.C., McCalley D.V. // J. Chromatogr. A. 2013. V. 1276. P. 33.
Guiochon G. // J. Chromatogr. A. 2007. V. 1168. № 1–2. P. 101.
Urban J., Svec F., Fre J.M.J. // Anal. Chem. 2010. V. 82. № 5. P. 1621.
Koeck R. et al. // Anal. Bioanal. Chem. 2014. V. 406. № 24. P. 5897.
Merhar M. et al. // J. Sep. Sci. 2003. V. 26. № 3–4. P. 322.
Korolev A.A. et al. // Russ. J. Phys. Chem. A. 2014. V. 88. № 9. P. 1609.
Smirnov K.N. et al. // Moscow Univ. Chem. Bull. 2011. V. 66. № 6. P. 351.
Trojer L. et al. // J. Chromatogr. A. 2009. V. 1216. № 35. P. 6303.
Wang Q.C., Svec F., Fréchet J.M.J. // Anal. Chem. 1993. V. 65. № 17. P. 2243.
Svec F., Fréchet J. // Anal. Chem. 1992. V. 64. P. 820.
Smirnov K.N. et al. // J. Chromatogr. A. 2011. V. 1218. № 30. P. 5010.
Patrushev Y., Yudina Y., Sidelnikov V. // J. Liq. Chromatogr. Relat. Technol. 2018. V. 41. № 8. P. 458.
Brüggemann O. et al. // J. Chromatogr. A. 1997. V. 781. № 1–2. P. 43.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов