

Физикохимия поверхности и защита материалов, 2021, T. 57, № 5, стр. 494-521
Молекулярные машины в 3D и 2D системах: движение, механическая работа, переключения. Обзор
Д. С. Куцыбала 1, А. В. Шокуров 1, С. Л. Селектор 1, *
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский пр., 31, корп. 4, Россия
* E-mail: sofs@list.ru
Поступила в редакцию 27.04.2021
После доработки 28.04.2021
Принята к публикации 29.04.2021
ОГЛАВЛЕНИЕ
1. Введение
2. Обзор литературы
2.1. Молекулярные машины
2.1.1. Биологические молекулярные машины
2.1.2. Синтетические молекулярные машины
2.1.3. Внутримолекулярный перенос электрона – редокс-изомерия
2.1.4. Другие переключения под действием света
2.1.5. Электрохромизм
2.1.6. Ионофорные переключатели
2.2. Методы формирования 2D наноразмерных материалов
2.3. Методы исследования наноразмерных систем
2.3.1. Электрохимические методы
2.3.2. Электронная спектроскопия
2.3.3. Рентгеновские методы исследования молекулярных переключений
3. Заключение
1. ВВЕДЕНИЕ
Возможность использования молекулярных материалов и отдельных молекул как активных элементов электроники уже давно привлекает внимание исследователей различных областей науки. Примеры сверхкомпактной микроскопической записи информации в биологических системах просто поразительны. При этом разные виды информации не просто записываются, они видоизменяются и используются. Биологические клетки, несмотря на свои малые размеры, выполняют очень разнообразные и весьма активные действия: изменение своей собственной формы, выработка различных веществ, транспортировка, самосборка, а также целый ряд других достаточно сложных операций. До недавнего времени трудно было даже представить возможности человечества в случае изготовления микроскопических объектов, которые способны осуществлять подобные действия.
Учитывая успех машин человечества в макроскопическом мире, от колеса каменного века до современного смартфона, перспектива того, что однажды нашей целью станет достижение максимального уровня миниатюризации, была неизбежна. Однако для того, чтобы эта область начала активно развиваться, потребовалось некоторое время для овладения достаточным мастерством в сфере необходимой синтетической и супрамолекулярной химии и связанной с ней физики.
Как известно, в 2016 г. Нобелевскую премию по химии “за проектирование и синтез молекулярных машин” получили Жан-Пьер Соваж (Jean-Pierre Sauvage), Бернард Феринга (Bernard Lucas Feringa) и Фрезер Стоддарт (James Fraser Stoddart).
Жан-Пьер Соваж (Jean-Pierre Sauvage) – французский химик, специализирующийся на супрамолекулярной химии. Не стоит забывать о том, что он является первым химиком, которому удалось синтезировать ряд соединений из класса катенанов, молекулярная структура которых состоит из двух колец, механически сцепленных друг с другом. Данный вид связи называется топологическим.
Фрезер Стоддарт (James Fraser Stoddart) – шотландский ученый, работающий сейчас в США, расширил список соединений с подобными “нехимическими” связями, синтезировав ротаксан. Молекулы класса ротаксанов состоят из длинной цепочки, по которой свободно перемещается кольцо. Благодаря двум крупным конструкциям на концах синтетической цепочки кольцо не может с нее “свалиться”.
Бернард Феринга (Bernard Lucas Feringa) – специалист в области молекулярной нанотехнологии и гомогенного катализа – возглавляет группу химиков, которые первыми разработали и синтезировали молекулярный мотор — молекулу, которая под действием света претерпевала структурные изменения и начинала вращаться подобно лопасти ветряка в строго заданном направлении. В 1999 г. с помощью молекулярных моторов ученому удалось заставить вращаться стеклянный цилиндр в 10 тыс. раз превышавший размер самих моторов.
В настоящее время огромное число ученых исследователей под влиянием идеи миниатюризации активно изучает и разрабатывает новые виды молекулярных устройств. Но мало кто знает, что расцвет нанотехнологии был предсказан Ричардом Фейнманом (Richard Phillips Feynman) еще в 1960 г. на Рождественском обеде Американского физического общества. Речь Фейнмана состоялась еще до того, как химикам стали доступны синтетические методы и аналитические инструменты, которые могли бы дать возможность задуматься о создании “молекулярной машины”. Он считал, что в период 2010–2020 гг. ученые начнут синтезировать подобные биологическим системам соединения, состоящие из одной или двух молекул и способные под воздействием внешних факторов выполнять работу на молекулярном уровне. И, как мы можем видеть, его предсказание оказалось пророческим.
2. ОБЗОР ЛИТЕРАТУРЫ
2.1. Молекулярные машины
С развитием компьютерной техники человечество все чаще задумывается о создании идеальной по своим параметрам и функционалу вычислительной машины. Если только сократить диаметр существующих соединительных проводов до, например, ста атомов, то размер необходимой схемы не будет превышать нескольких тысяч ангстрем. Такие изменения значительно увеличат предел возможностей вычислительных машин. А рост числа элементов приведет к важным качественным изменениям характеристик электронных устройств.
Рассмотрим следующий пример. Мозг человека без особого труда воспринимает многоцветное изображение или лицо другого человека. Следовательно, встает вопрос о создании такого компьютера, который смог бы достаточно быстро считывать изображение и распознавать на нем человеческие лица на разных расстояниях или при разном освещении, взяв за основу микрокомпьютер, заложенный природой в наш мозг. Главное преимущество “человеческого процессора” в том, что число активных элементов в нашей маленькой черепной коробке значительно превышает число элементов в самых высококлассных компьютерах, когда-либо созданных человеком. А все потому, что элементы нашего мозга имеют микроскопические размеры. И если представить, что человек на основе сегодняшней микроэлектроники создал такой компьютер, который обладал бы всеми этими изумительными способностями, он бы, возможно, занимал площадь свыше нескольких десятков гектар, что приведет к ряду существенных проблем. Во-первых, это потребует слишком много материала, поставит проблему выработки тепла и энергопотребления. Во-вторых, такой компьютер будет ограничен определенной скоростью. В конечном итоге, когда наши компьютеры будут становиться более быстрыми, более мощными и все более и более сложными, придется делать их все меньше и меньше.
Возможность осуществления различных операций на микроуровне привлекает все больше специалистов в области физической химии, биологии и инженерии для дальнейшего изучения и реализации таких процессов. Ведь при помощи систем с электрическим или химическим управлением, которые представляют собой изготовленные обычным образом уменьшенные в разы копии “рук” оператора, можно будет легко выполнять разнообразные манипуляции в уменьшенном масштабе. Область применения таких микророботов, а также микромашин может быть довольно широкой – от хирургических операций до транспортировки и переработки опасных материалов. Существенное уменьшение масштабов потребует дальнейших конструкционных изменений и модификаций, но позволит изготовить новые, значительно более совершенные устройства.
Однако, по мере уменьшения размеров мы будем постоянно сталкиваться с очень необычными для макромира, но вполне естественными для микромира физическими явлениями. Все, с чем приходится встречаться в жизни, зависит от масштабных факторов. Кроме того, существует еще и проблема “слипания” материалов под действием сил межмолекулярного взаимодействия, или сил Ван-дер-Ваальса, которые могут приводить к эффектам, необычным для макроскопических масштабов. Например, нано-гайка не будет отделяться от нано-болта после откручивания, а в некоторых случаях будет плотно “приклеиваться” к поверхности. При проектировании и создании микроскопических устройств следует помнить о физических проблемах такого типа.
На сегодняшний день уже существует работающая нанотехнология, базирующаяся на молекулярных машинах, которая выполняет множество необходимых для жизни задач: она называется Биология. Природа богата многообразием различающихся по своим физико-химическим свойствам молекулярных машин, принцип действия и функционирование которых должны служить прототипом для создания схожих по свойствам синтетических наносистем.
2.1.1. Биологические молекулярные машины
Природа имеет свой собственный набор молекулярных машин, который работает веками и с течением времени в ходе естественного отбора подвергался изменениям для оптимизации работы и дизайна. Известные на сегодняшний день молекулярные машины подразделяются на три большие категории – системы на основе белков, на основе ДНК и химические молекулярные двигатели (моторы). Большинство природных молекулярных машин основаны на белках, тогда как молекулярные машины на основе ДНК в основном синтетические. Природа использует белки для выполнения различных задач на клеточном уровне: от перемещения грузов до каталитических реакций, ДНК при этом остается только носителем информации.
Молекулярные моторы на основе соединения, известного как аденозинтрифосфат (АТФ), которое представляет собой нуклеотид с тремя молекулами остатками фосфорной кислоты, играют ключевую роль во всех живых организмах и являются незаменимыми источниками энергии. Суперсемейства белковых молекулярных машин на основе кинезина, миозина и динеина прямо или косвенно зависят от АТФ в качестве движущей силы. Эти машины, которые на протяжении миллионов лет выполняли жизненно важные для организма функции как внутри, так и снаружи клеток, могут быть изолированы от своей естественной среды и рассматриваться как устройства преобразования энергии для генерации сил крутящего момента и движения.
Ведущая роль АТФ в процессе преобразования энергии в живых организмах была доказана Ф. Липпманом еще в 1941 г. [1]. Формируется АТФ в живых клетках с помощью известной молекулярной машины. Аденозиндифосфат (АДФ) и неорганический фосфат-ион, рекомбинация которых с использованием сверхэффективного ферментного моторного узла, так называемой АТФ-синтазы, приводит к образованию АТФ [2]. АТФ-синтаза присутствует в митохондриях клеток животных, в хлоропластах растений, в бактериях и в некоторых других организмах.
На самом деле АТФ-синтаза является комбинацией двух взаимодействующих друг с другом моторов F0 и F1 [3]. Оба мотора имеют отдельные структуры и функции. Мотор F0 в митохондриях животных встроен во внутреннюю мембрану и использует для своей работы ионно-движущую силу. Мотор F1, приводимый в действие гидролизом АТФ, состоит из центральной белковой основы, называемой γ-субъединицей, окруженной тремя копиями, каждая из которых состоит из α- и β-субъединиц. Есть δ-субъединицы, прикрепленные к периферии α-β-цилиндра, а ε-субъединицы присутствуют в основании γ-субъединиц [4], как показано на рис. 1. Субъединицы a, b, и c, показанные на рис. 1, являются частью мотора F0.
Рис. 1.
F0F1-АТФ мотор. Здесь α, β, γ и ε – субъединицы мотора F1; a, b, и c – субъединицы мотора F0 [4].

Главным образом принцип действия мотора F1 базируется на изменении состояний каждой из β‑субъединиц. β-цилиндры принимают три формы: так называемые O, L и T сайты связывания. Когда субъединица находится в форме O, она каталитически неактивна и имеет очень низкое сродство к связыванию субстратов. В L-форме субъединица слабо связывает субстраты (АДФ и фосфат-ион), хотя каталитически неактивна. В Т-форме АДФ и фосфат ион превращаются в прочно связанный АТФ до тех пор, пока конформационное изменение не преобразует Т-сайт в О-сайт, тем самым обеспечивая высвобождение вновь образованного АТФ [5]. Механизм показан на рис. 2.

Конформационное изменение β-субъединиц запускается вращением γ-субъединицы, которая действует как звено, соединяющее F1 с F0. Это было доказано экспериментально в 1997 [6]. В этом эксперименте F1 была прикреплена к покрытой никелем стеклянной поверхности. К другому концу γ-субъединицы присоединяли флуоресцентно меченую актиновую нить. За вращением можно было наблюдать при помощи флуоресцентного микроскопа. При условии, что сам двигатель имеет диаметр около 10 нм, он мог поддерживать и вращать структуру примерно в сто раз больше. Схема эксперимента показана на рис. 3.

Структура мотора F0 не так хорошо известна, как структура мотора F1, которая была полностью определена и подтверждена экспериментально.
С помощью современных микроскопических инструментов мы рассматриваем клетку не статической средой, а как набор различных движущихся компонентов, приводимых в действие молекулярными машинами. Молекулярные моторы, которые обеспечивают движение молекулярных фрагментов в одном направлении вдоль белковых полимеров (актина или микротрубочек), управляют движениями мышц, а также гораздо меньшими внутриклеточными “грузоперевозками”. В дополнение к двигателям АТФ внутри клетки присутствуют линейные транспортные двигатели, крошечные транспортные средства, известные как моторные белки, которые транспортируют молекулярные грузы [7]. Эти мельчайшие клеточные машины существуют в трех разновидностях: кинезины, миозины и динеины [8]. Грузы могут быть органеллами, липидами, белками и т.д. Они играют важную роль в делении и подвижности клеток.
Существует более 250 кинезиноподобных белков, и они участвуют в столь же разнообразных процессах, таких как движение хромосом и динамика клеточных мембран. Единственная их общая часть – это каталитический фрагмент. У них есть существенные различия в расположении внутри клеток, в их структурной организации и в перемещении, которое они производят [9]. Молекула кинезина была обнаружена только в 1985 году с использованием тестов подвижности [10]. Обычный кинезин – это высокопроизводительный мотор, который может совершать несколько сотен шагов по микротрубочке без отсоединения [11], тогда как мышечный миозин выполняет один шаг, а затем диссоциирует [12]. Подробный анализ и моделирование этих двигателей приведены в [13].
Мышечный миозин, начало исследования которого датируется 1864 г., десятилетиями служил модельной системой для изучения подобной моторики. Молекулярные машины на основе миозина транспортируют грузы по актиновым нитям. Они делают это путем гидролиза АТФ и использования высвобождаемой энергии [14]. Помимо транспорта, они также участвуют в процессе генерации силы во время мышечного сокращения, при котором тонкие актиновые нити и толстые миозиновые нити скользят друг мимо друга. Не все члены суперсемейства миозинов охарактеризованы. Однако об их структуре и функциях известно многое. Например, известно, что АТФ играет большую роль в связанном с миозином мышечном движении вдоль актина [15].
В отличие от миозина, который транспортируется только по актину, семейства белков кинезина и динеина участвуют в транспорте клеточного груза по микротрубочкам [16]. Микротрубочки – это трубочки диаметром 25 нм, состоящие из димеров тубулина, которые в организованном виде присутствуют в клетках живых организмов. Микротрубочки обладают полярностью: один конец – положительный (быстрорастущий), а другой – отрицательный (медленнорастущий) [17]. Кинезины перемещаются от отрицательного конца к положительному концу микротрубочки, тогда как динеины движутся от положительного к отрицательному. Расположение микротрубочек варьируется в разных клеточных системах. В нервных аксонах они расположены продольно так, что их положительные концы направлены от тела клетки в аксон. В эпителиальных клетках их положительные концы указывают на базальную мембрану. В фибробластах и макрофагах они выходят радиально из центра клетки, причем положительный конец выступает наружу [18]. Подобно миозину, кинезин также является двигателем, управляемым АТФ. Одной из уникальных характеристик белков семейства кинезина является их процессивность: они связываются с микротрубочками и буквально проходят по ним многие ферментативные циклы, прежде чем отсоединиться [19]. Кроме того, каждая из глобулярных головок/моторных доменов кинезина состоит из одного полипептида, в отличие от миозина.
Хотя кинезин также является двуглавым линейным мотором, его образ действия отличается от миозина в том смысле, что обе его головы выполняют совместную, хорошо скоординированную работу. Каждая из головок находится рядом с микротрубочкой в исходном состоянии и несет одну молекулу АДФ (рис. 4). Как только сила связывания моторных головок с микротрубочкой ослабевает, она теряет свою молекулу АДФ. Другая молекула АТФ заменяет молекулу АДФ, что приводит к конформационному изменению молекулярного мотора и способствует его дальнейшему продвижению по микротрубочке. Следовательно, движущей силой кинезина является последовательный гидролиз АТФ в двухмоторных головках.

Динеин был открыт в 1965 г. [21] и существует в двух формах: цитоплазматической и аксонемной. Поскольку динеин представляет собой более крупную и сложную структуру, чем другие моторные белки, принцип его действия не так хорошо известен. Однако механизм его работы схож с процессами, протекающими в молекуле кинезина. Движение головки динеина сопровождается разложением молекулы АТФ и высвобождением молекул АДФ и фосфат-иона.
Изучение биологических молекулярных машин, их дизайна и принципа действия спровоцировало здоровый интерес к методам воссоздания подобных наноразмерных соединений – синтетических молекулярных машин с похожими физико-химическими свойствами для осуществления механической работы на молекулярном уровне.
2.1.2. Синтетические молекулярные машины
Взяв за основу внешний вид и принцип работы макроскопических машин, ученым удалось воссоздать различные типы молекулярных механизмов. Сборка таких машин и устройств понятна с точки зрения их макроскопических аналогов, однако недостаток этого подхода заключается в том, что многие механические принципы, на которых основаны сложные макроскопические машины, не подходят для молекулярного мира.
Альтернативным путем развития синтетических молекулярных машин является изучение уже существующих нанотехнологий в биологии. Потенциальное преимущество этого биомиметического подхода связано с тем, что такие конструкции явно хорошо подходят для функциональных машин, работающих на наноуровне, даже когда они ограничены, как природа, использованием только около 20 различных строительных блоков (аминокислот). Однако, главная проблема в реализации этой второй стратегии заключается в том, что единственный “учебник”, которому мы должны следовать, неясен. Природные биологические машины настолько сложны, что зачастую трудно разобраться в причинах динамики отдельных деталей машин. Как и почему каждый пептидный остаток движется так, как он движется, чтобы кинезин шел по микротрубочкам; какие конформационные переходы, водородные связи и сольватационные изменения необходимы для осуществления транспорта? Тем не менее, подражание биологическим системам, безусловно, не единственный способ достижения сложной функциональности: компьютерные микросхемы, не смотря на свое сходство с нашим мозгом, совсем не влажные и не основаны на углероде, а изготовлены из кремниевых пластин. Возможно, наиболее продуктивные решения будут найдены, если мы не будем слепо подражать ни одному из этих “природных” подходов.
Во многом наука о искусственных молекулярных механизмах началась с изобретения Дж. Фрейзером Стоддартом (J.Fraser Stoddart) “молекулярного челнока” в 1991 г. (рис. 5) [22]. В этом ротаксане кольцо, механически заблокированное на оси стерически большими “пробками”, перемещается между двумя предпочтительными точками связывания (двумя гидрохиноновыми звеньями) за счет случайного теплового движением.
Рис. 5.
Первый молекулярный челнок, который перемещается вперед–назад между двумя идентичными станциями связывания, а именно гидрохиноновыми звеньями, симметрично расположенными в цепочке полиэфирной нити, на концах которой зафиксированы триизопропилсилильные группы в качестве объемных молекулярных стопперов [22].
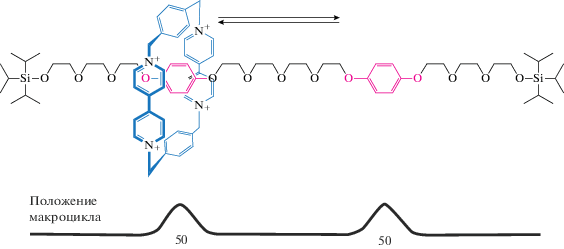
Используя две разных потенциальных точки связывания, или “станции”, относительное сродство к кольцу которых можно переключать, группы Стоддарта и Кайфера (Kaifer), возможно, изобрели первый искусственный молекулярный механизм движения (рис. 6) [23]. В примере, приведенном на рис. 6, катионное кольцо предпочитает находиться над бензидиновой группой, а не над бифенольным центром только до момента протонирования, или электрохимического окисления бензидиновой “станции”, что делает бифенольную группу более предпочтительной точкой связывания для катионного кольца, вызывая тем самым полное смещение кольца вдоль дорожки. Эта система представляет собой первый пример контролируемого переключения положения кольца вдоль молекулярной дорожки.
Рис. 6.
Первый переключаемый молекулярный челнок. В состоянии равновесия катионное кольцо занимает преимущественно бензидиновую станцию. Протонирование или электрохимическое окисление приводит к молекулярному переключению, в результате которого катионное кольцо располагается преимущественно над бифенольной станцией [23].

Группа Стоддарта внесла свой вклад в разработку и многих других процессов и стратегий для синтеза и сборки ротаксанов и катенанов, в период 1992–2007 гг., а также изобрела множество различных способов переключения между положениями компонентов в архитектурах ротаксана и катенана с различными внешними стимулами такими, как свет, электрохимическое воздействие, изменение рН или полярности среды, связывание катионов, связывание анионов, изменение температуры, обратимое образование ковалентных связей и т.д. Следующим ключевым шагом был поиск условий, в которых изменения положения компонентов молекулярной машины приводили бы к выполнению полезной работы, и методов регистрации таких изменений.
В 1999 г. появились две статьи, посвященные контролю направленного вращательного движения [24, 25]. Группа Келли (Kelly) использовала химические реакции образования уретана с последующим гидролизом, чтобы сместить вращение триптиценового остатка на 120° в одном направлении [24] (рис. 7). К сожалению, им так и не удалось наложить этот метод на систему, в которой происходило бы направленное вращение на 360°.

Однако в статье, которая последовала за этим, описана стерически затрудненная молекула алкена, в которой компоненты при облучении светом вращаются вокруг алкеновой оси. Эта молекула, полученная группой Б. Феринги (B. Feringa), была первым примером синтетического вращательного молекулярного мотора (рис. 8). Мало того, что этот элегантный дизайн обеспечивает полное вращение одной половины молекулы на 360° относительно другой, такой мотор непрерывно действует до тех пор, пока соединение облучается фотонами и находится выше критической температуры.

В течение следующего десятилетия было сделано много структурных улучшений для этого типа молекул-двигателей, которые позволили резко увеличить скорость вращения [26] и использовать их для выполнения таких задач, как вращение макроскопического объекта на поверхности жидкокристаллической среды (рис. 9) [27].
Рис. 9.
Вращение макроскопического объекта с помощью молекулярной машины. На фото стеклянный стержень, вращающийся на жидком кристалле во время облучения ультрафиолетовым светом. Кадры 1–4, сняты с интервалом 15 с, они показывают повороты стержня по часовой стрелке на 28° (кадр 2), 141° (кадр 3) и 226° (кадр 4) относительно положения на кадре 1. Масштабная линейка, 50 мкм [27].

Задачи, для выполнения которых необходимы молекулярные машины, также требуют детального рассмотрения и систематизации. Все существующие синтетические молекулярные машины можно разделить на три группы:
1) Молекулярные машины, способные выполнять механическую работу – “молекулярные мышцы”
Использование контролируемого движения на молекулярном уровне для создания силы в макроскопическом мире является привлекательной задачей для молекулярных машин потому, что именно так работают мышцы. В 2005 г. группы Ли (Leigh) и Стоддарта (Stoddart) продемонстрировали принцип действия искусственных молекулярных машин, способных выполнять механическую работу. Группа Ли использовала светочувствительный челнок поверхностно-связанного ротаксана, который позволил управляемо “открывать” и “скрывать” (прятать) фторуглеродную часть молекулярного ансамбля и тем самым управлять характеристиками смачиваемости поверхности, покрытой этим соединением. Изменение смачиваемости используется для продвижения капли вдоль поверхности и даже вверх по склону против силы тяжести (рис. 10) [28].
Рис. 10.
Светочувствительная поверхность на основе переключаемых фторированных молекулярных челноков. После облучения светом с длиной волны 240–400 нм ротаксаны изомеризуются, вызывая смещение молекулярного кольца, которое инкапсулирует фторалкановое звено, делая поверхность более полярофильной [28].

В другой работе группа Стоддарта использовала сжатие ротаксана в качестве молекулярного привода для изгиба золотого микрокантилевера (рис. 11) [29].
Рис. 11.
Вызванное окислением сокращение межкольцевого расстояния создает растягивающее напряжение, которое передается на нижележащий субстрат через “тросы”. В результате совокупное воздействие произвольно ориентированного ансамбля молекулярных мышц на субстрат приодит к его механическому изгибу. Данное механическое воздействие является обратимым [29].

2) Молекулярные машины, способные к молекулярному синтезу
Одно из ключевых применений молекулярных машин в биологии – это конструирование других молекул. В 2013 г. был изобретен искусственный низкомолекулярный аппарат, который собирал трипептиды определенной последовательности, путешествуя по дорожке, загруженной аминокислотными строительными блоками (рис. 12) [30].
Рис. 12.
Создание молекул с помощью молекулярных машин: искусственный синтезатор молекулярных пептидов [30].
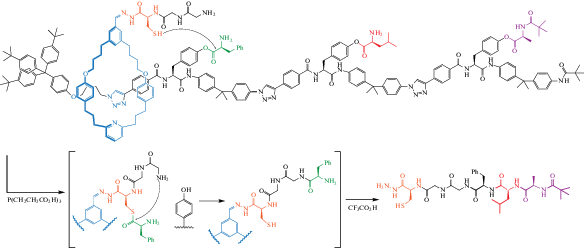
Это очень примитивный аналог задачи, выполняемой рибосомой в клетках, но, возможно, один из самых сложных, выполняемых искусственной молекулярной машиной на сегодняшний день. Для синтетической молекулярной машины она обладает действительно сложным механизмом работы, требующим интегрированного взаимодействия нескольких функциональных компонентов: “плеча” с регенерируемым каталитическим центром и сайтом удлинения пептида, кольца, которое проходит по дорожке без остаточных взаимодействий между молекулярными фрагментами, а также дорожки с аминокислотными строительными блоками, расположенными в заданной последовательности и разделенными жесткими “прокладками”. Можно ожидать, что в будущем, системы с интегрированным механизмом операций смогут привести к еще более амбициозным и потенциально полезным применениям синтетических молекулярных машин.
3) Молекулярные переключатели для молекулярной электроники
В серии экспериментов группы Стоддарта и Джима Хита (Heath) связали переключаемые ротаксаны и катенаны с электроникой на основе кремния, пытаясь использовать молекулярные челноки в твердотельной молекулярной электронике вычислительного устройства (рис. 13). Казалось бы, нелогично таким гибридным системам конкурировать с кремнием и другими полупроводниками, в которых движение электронов, по сути, происходит в миллиарды раз быстрее, чем изменения положения компонентов в ротаксане. Однако, десятилетие усилий и прогресса завершилось изготовлением и тестированием памяти объемом 160 кбит с плотностью носителей информации в 1011 бит см–2 на основе монослоя переключаемых ротаксанов в качестве элементов хранения информации [31, 32]. Вопрос о том, будут ли когда-либо эффективно использоваться ротаксаны в электронике, остается открытым, но важным наследием этой новаторской исследовательской программы является огромный объем полученных знаний.
Рис. 13.
Ротаксановые гибридные элементы на основе молекулярных переключателей. Схема молекулярной электронной памяти, изготовленная с плотностью 1011 бит на см–2. Элементом хранения данных служит монослой бистабильных молекул ротаксана. В центральной верхней части показана конформация системы в основном состоянии (потенциал +0.2 V), которое соответствует конформации с низкой проводимостью или “0”. Механизм переключения включает окисление сайта тетратиафульвалена (TTF) (зеленый) при приложении положительного напряжения (2 V) до состояния окисления TTF+1 или TTF+2 (фиолетовый) с последующим перемещением синего кольца с сайта TTF+ на сайт диоксинапталина (красный). Затем при подаче исходного потенциала 0.2 V TTF+ восстанавливается обратно до состояния окисления TTF0 с образованием метастабильного соконформера, который является состоянием с высокой проводимостью или состоянием “1”. Это состояние может релаксировать в основное с периодом полураспада около часа или подвергаться электрохимической принудительной релаксации [31] (рисунок адаптирован из [32]).

Другие типы молекулярных переключателей основаны на использовании процессов, связанных с переносом заряда и лежащих в основе таких явлений как внутримолекулярный перенос электрона, электро-, фото- и ионохромизм. Все эти явления, различающиеся по механизмам индуцирования процесса переноса заряда и по типу самого заряда, требуют более подробного рассмотрения.
Необходимо отметить, что “переключение” молекулярной системы в некоторых случаях приводит к таким изменениям ее свойств (например, линейных размеров), которые могут быть использованы для совершения механической работы.
2.1.3. Внутримолекулярный перенос электрона – редокс-изомерия
Молекулярные системы, демонстрирующие внутримолекулярный перенос электронов, индуцированный различными внешними стимулами, занимают особое место среди систем, поведение которых определяется процессами переноса заряда. Такие соединения, действительно, перспективны для приложений в области разработки наноразмерных устройств хранения и передачи информации. Можно предположить, что в системах, которые характеризуются сосуществованием двух различных электронных конфигураций одного и того же соединения (или/и возможностью переключения между ними), не только движущие силы, перечисленные выше, но и любое физическое воздействие, которое даже незначительно влияет на внутреннюю энергию системы, может сдвинуть динамическое равновесие к преобладанию одного из изомеров.
Рассмотрим общие представления об этом явлении.
Электромерия – это вид изомерии, заключающейся в существовании химических соединений, которые одинаковы по составу, но различаются способом распределения электронов между атомами и называются электромерами [33]. Одна из групп электромеров носит окислительно-восстановительный характер. Образование таких электромеров, или редокс-изомеров, происходит в результате внутримолекулярного переноса электрона с металлоцентра соответствующего комплекса на лиганд или с одного лиганда на другой.
Толчком для развития исследований в области редокс-изомерных превращений стала работа Пьерпонта (Pierpont) в 1980 г. над изменением редокс-состояния кобальта в семихиноновом комплексе (рис. 14) [34].

Явление редокс-изомерии набирает популярность среди исследователей в области физической, органической и супрамолекулярной химии. Ранее описанный и изученный ряд электроактивных хиноновых лигандов, способных к координации металлов, в особенности кобальта и рутения [34–36], пополняют краун-эфиры [37], ферроцены [38], тетратиафульвалены [39] и некоторые тетрапиррольные лиганды [40, 41].
Наряду с редкоземельными металлами переменной валентности, такие переходные металлы как рутений [42] и кобальт [34, 43, 44], обладают достаточной редокс-активностью и широко применяются для образования координационных связей в различных комплексах. С экспериментальной точки зрения, наибольший интерес вызывают редкоземельные металлы, которые обладают развитой электронной системой и способностью управляемо изменять валентное состояние в достаточно разнообразном по своим физико-химическим свойствам ряде комплексов [45, 46].
Несмотря на широкий список соединений, способных к редокс-изомеризации, исследовательских работ, посвященных изучению механизмов и движущих сил процессов внутримолекулярного переноса электрона на сегодняшний день крайне мало. Возможно, сложность состоит в подборе оптимального соединения, которое может выступать в качестве редокс-активного лиганда с различными металлами, так как для редокс-изомеризации должны одновременно соблюдаться несколько условий. Во-первых, оба лиганда или один из лигандов и ион металла должны быть редокс-активны; во-вторых, степень ковалентности взаимодействия между ионом металла и электроактивным лигандом должна быть довольно мала; и, в-третьих, энергии их граничных орбиталей должны быть соизмеримы (рис. 15) [47]. Другими словами, для возникновения редокс-изомерии необходимо, чтобы соответствующие редокс-изомеры имели минимальные энергетические различия. Следует отметить, что подобным требованиям соответствуют не только металлокомплексы, в которых перенос электрона осуществляется с мультивалентного иона металла на лиганд и обратно, но и некоторые соединения, в которых редокс-изомеризация осуществляется за счет внутримолекулярного переноса лиганд-лиганд [43] или металл-металл [42].
Рис. 15.
Кривые потенциальной энергии для систем со смешанной валентностью. Кривые причисляются к трем основным видам в соответствии с классификацией Робина и Дэя [47]. В случае I взаимодействие между редокс-центрами незначительно. В случае III электронное взаимодействие настолько сильно, что рассматриваемый электрон делокализован по всей молекуле. Случай II соответствует системе, где взаимодействие между редокс-центрами недостаточно интенсивно для полной делокализации электрона, но и не настолько слабо, чтобы его можно было исключить; именно в таких системах наблюдается внутримолекулярный перенос электрона [47].

В настоящее время наиболее изученными факторами, способными индуцировать редокс-изомеризацию химических соединений, являются изменение окружающих условий в растворе (состав растворителя) [34, 35, 48], термическое [36] и фотофизическое воздействия [41], а также рентгеновское облучение [44]. Кроме того, явление редокс-изомерии было обнаружено для монокристаллов некоторых соединений при приложении внешнего давления [49–51]. Тем не менее, если для объемных образцов влияние давления на процессы редокс-изомеризации упоминаются в литературе [49–51], то для планарных систем этот вопрос остается мало изученным. Следует отметить, что, хотя среди потока публикаций можно найти несколько работ, посвященных процессам изомеризации, происходящим на межфазной поверхности [41, 52], в них рассматривается только внутримолекулярный перенос электрона, индуцированный световым облучением в видимом диапазоне. Возможность реализации редокс-изомерных превращений в двумерных системах на межфазной границе под действием латерального сжатия была впервые продемонстрирована в работах нашей лаборатории [46, 53–56].
Другим уникальным способом индуцирования редокс-изомерных превращений является переход химического соединения от объемной фазы раствора к поверхности раздела. Так в исследовательских работах, посвященных изучению этого процесса на примере сэндвичевого краун-замещенного бисфталоцианината церия [53–56], было выявлено, что внутримолекулярный перенос электрона с лиганда на металлоцентр и обратно, то есть редокс-изомеризация комплекса с участием мультивалентного катиона Ce3+/4+, происходит при нанесении раствора на поверхность воды и последующем сжатии полученного таким образом монослоя. Позднее подобный переход был обнаружен и доказан для аналогичного комплекса европия, валентное состояние которого изменялось от Eu3+ в растворе к Eu2+ в монослое на поверхности воды [46].
Необходимо подчеркнуть, что описанные в работах [46, 53–56] двухпалубные тетрапиррольные комплексы металлов переменной валентности представляют особый интерес, с точки зрения расширения доступного количества обратимых электрохимических превращений, так как за счет возможности изменения валентного состояния металлоцентра они имеют большее число стабильных редокс-состояний и обратимых переходов между ними [56]. Кроме того, само существование стабильных редокс-изомеров, способных к управляемым взаимопревращениям, связанным с обратимым внутримолекулярным переносом электрона между ионом металлоцентром и лигандом, может представлять значительный интерес для молекулярной электроники, поскольку такие обратимые редокс превращения тетрапиррольных соединений могут сопровождаться изменением не только оптических, но и других, например, электрических и/или магнитных характеристик материала на их основе [56], а также изменением линейных размеров комплексов. Последнее может найти применение при разработке молекулярного мускула [46, 53–56].
Подчеркнем, что супрамолекулярные системы, обладающие способностью к редокс-изомеризации, которая может быть индуцирована различными внешними факторами, привлекают пристальное внимание мирового научного сообщества. В последние два десятилетия исследования в этом направлении охватывают все больший диапазон различных проявлений данных эффектов, а также все более широкий спектр воздействий, способных их индуцировать [57–60]. Однако, несмотря на широкие перспективы для применения соединений, способных к управляемой редокс-изомеризации, в качестве молекулярных переключателей, эта область остается практически не изученной. Разработка устройств молекулярной электроники на основе редокс-изомерных систем малой размерности, таких как монослои с высокой упорядоченностью активных элементов на границе раздела фаз, остается на стадии подбора перспективных соединений и поиска новых принципов индуцирования изомеризации в планарных системах и способов ее регистрации.
2.1.4. Другие переключения под действием света
Как уже отмечалось выше, воздействие света является важнейшим индуцирующим фактором не только для редокс-изомерных превращений, но и для целого ряда других управляемых переключений на молекулярном уровне. Свет – это универсальный источник энергии, который широко используется в биологических системах, таких как человеческое зрение и фотосинтез у растений. Воздействие излучения вызывает в светочувствительном веществе атомарные перестройки, изменение заселенности электронных уровней, а также другие изменения физико-химических свойств.
Фотохромизм является наиболее изученным и универсальным методом оптических манипуляций. Причем этот термин первоначально относился к явлению индуцированного светом обратимого изменения цвета. В настоящее время термин фотохромизм имеет более обобщенное значение и используется для описания обратимого химического превращения между двумя формами вещества, различающимися соответствующими спектральными характеристиками. При облучении светом наиболее распространенными процессами фотохромизма в таких соединениях являются транс-цис-изомеризация и перициклические реакции. Существуют различные классы органических соединений, которые обладают способностью к фотохромизму, такие как некоторые гидразоны [61, 62], сульфоксиды [63], индиго [64], дигидропирены [65], спиропираны [66] и т.д.
Однако не менее интересным приложением стимуляции светом является область молекулярных машин и переключателей. Внешнее оптическое излучение может действовать не только как кванты энергии в фотохимических процессах, но и как информационный элемент для мониторинга динамических движений наномашин [67, 68]. Более того, свет может применяться к наномашинам как стимул, исключающий физическое (механическое) воздействие, а его энергию можно контролировать, настраивая длину волны, интенсивность и поляризацию падающего излучения.
Группа Джузеппоне (Giuseppone) описала использование молекулярных роторных моторов с легким приводом для макроскопического сокращения геля под действием света (рис. 16) [69]. При УФ-облучении полимерные цепи свертываются, уменьшая таким образом габаритные размеры гелей миллиметрового масштаба. Приведение такой системы в действие является результатом работы, выполняемой двигателем по скручиванию полимерных цепей и преобразованию нановращения в сжатие в более высоких масштабах.
Рис. 16.
Преобразование вращательного движения в макроскопическое сжатие полимерного геля под действием света [69].

Воздействие света для реализации молекулярных переключений остается одним из наиболее изученных и популярных стимулов в химии новейших наноматериалов, и ему посвящен ряд современных обзоров [70–72].
2.1.5. Электрохромизм
Не менее перспективным типом молекулярных переключателей для 2D систем является электрохромное переключение, на основе которого разрабатываются электрохромные материалы – материалы, изменяющие свои оптические свойства при приложении к ним электрического напряжения. Актуальность таких систем заключается в том, что электрохромные материалы все более широко используются в оптоэлектронных устройствах различного типа: элементах отображения информации, умных окнах, зеркалах, системах маскировки и т.д.
Рис. 17.
Типичный пример электрохромного устройства на основе производного виологена, структура которого показана на части рисунка (а, I). (II) фотографии устройства в двухэлектродном исполнении в нейтральном состоянии (1) и при приложении разности потенциалов (2) –1.4 В, (3) –1.8 В. На рисунке (б, I) показана принципиальная схема устройства в трехэлектродном исполнении с гелевым электролитом. (II) Типичные цикловольтамперограммы устройства, (III) спектры пропускания ячейки в нейтральном состоянии и при приложении потенциалов, соответствующих разной окраске, (IV) параметры устройства в координатах цветности CIE l*a*b* (Comission Internationale de I’Eclairage) [рисунок адаптирован из [83] и [79]].

Электрохромизм в широком смысле определяется как обратимое оптическое изменение в материале, вызванное внешним напряжением, при этом многие неорганические и органические соединения проявляют электрохромизм во всем электромагнитном спектре [73]. В 1961 году Дж.Р. Платтом (J.R. Platt) были продемонстрированы первые примеры электрохромных материалов и устройств [74]. Среди электрохромных материалов оксиды переходных металлов, особенно полупроводниковый оксид вольфрама WO3, привлекли к себе наиболее пристальное внимание. Тонкие пленки аморфного или поликристаллического WO3 были получены вакуумным испарением, реактивным распылением и золь-гель процессом. Если в исходном состоянии пленка полностью прозрачна, то после восстановления катионов WO3 до MxWO3 (где M может быть водородом или щелочным металлом) происходит возникновение интенсивных полос поглощения в видимой области, что делает ее катодно-окрашенным материалом. После открытия такого эффекта для тонких пленок оксида вольфрама возможность проявления электрохромных свойств была изучена и для других неорганических материалов, таких как берлинская лазурь, оксиды V, Mo, Nb и Ti (катодное окрашивание) и оксиды Ni, Co и Ir (анодное окрашивание) [75].
Новый толчок к исследованиям электрохромных переключений дало открытие электрохромных соединений, включающих органические молекулы, такие как виологены. Материалы на их основе в нейтральном состоянии прозрачны, а при одноэлектронном восстановлении виологена образуется ярко окрашенный и исключительно стабильный катион-радикал. Тонкопленочный электрохромизм наблюдается для поливиологенов и N-замещенных виологенов [76–79]. Сопряженные полимеры – это третий класс электрохромных материалов, которые приобрели популярность благодаря простоте обработки, быстрому времени отклика, высоким оптическим контрастам и способности изменять свою структуру для создания многоцветных электрохромов. Из ряда сопряженных полимеров наиболее широко изучаются производные политиофена, полипиррола и полианилина [80]. Область применения электрохромов включает различные дисплеи, интеллектуальные окна, оптоэлектронные и зеркальные устройства. Не так давно на основе неорганических оксидов металлов были разработаны прототипы “умных окон” для зданий. Также очень многообещающим типом устройств являются сенсорные датчики для распознавания ионов на основе изменения характеристик хромогенных материалов, что и было продемонстрировано исследователями Национальной лаборатории возобновляемой энергии (National Renewable Energy Laboratory NREL) с использованием WO3 в качестве датчика молекулярного водорода [81, 82].
Использование явления электрохромизма в качестве инструмента для молекулярных переключений под действием электрического тока, которые могут быть легко зарегистрированы с помощью спектроскопии видимого диапазона, является еще одной перспективной задачей современной химии. С этой точки зрения, макрогетероциклические соединения и их комплексы, для которых характерна яркая окраска и большое число доступных редокс-состояний, представляются особо актуальными.
Отдельного внимания в этой области заслужили порфирины и фталоцианины [84, 85]. Как известно, порфирины и их металлокомплексы демонстрируют наиболее интенсивное поглощение в области от 400 до 500 нм, которое связано с так называемой полосой Соре. Порфирины обладают превосходными электронными и оптическими свойствами благодаря их большой π-электронной системе [86]. В 2004 г. группа Яхая (Yahaya) разработала электрохромный датчик на основе двух производных металлопорфиринов для обнаружения хлорид-иона [87]. Тонкая пленка на основе металлопорфиринового комплекса продемонстрировала хорошую регенерирующую способность, стабильность после двух циклов электрохимического процесса и время отклика на хлорид-ион в водном растворе около 10 с.
В частности, электрохромные фталоцианины широко используются в качестве тонкопленочных сенсоров для обнаружения, оксидов азота [84, 86], нуклеотидов [88] и ряда других аналитов [89].
Как известно, характеристики сенсорного устройства существенно зависят от характеристик самого чувствительного слоя: его размера и морфологии. Сенсоры на основе макроциклических электрохромных соединений в основном отличаются малым временем отклика и большим набором паров органических соединений в качестве аналитов, что указывает на широкие возможности применения таких сенсоров благодаря их структурным особенностям.
В то же время, поиск новых комбинаций на основе известных структурных фрагментов молекулярных рецепторов также является важной задачей супрамолекулярной химии, поскольку, изменяя некоторые фрагменты, которые связывают определяемые ионы или молекулы, можно контролировать способность рецептора к образованию специфических комплексов. Это, в свою очередь, позволяет создавать композиты, обладающие уникальными свойствами. Наиболее изученные примеры таких соединений – это краун- и бутокси-замещенные порфиринаты и фталоцианинаты, в которых заместители являются неотъемлемой частью фталоцианинового макроцикла [90–92].
Причем во многих случаях изменение оптических свойств при взаимодействии с определяемыми ионами или молекулами можно зарегистрировать и без наложения внешнего электрического поля, т. е. в таких системах мы имеем дело с ионохромизмом.
2.1.6. Ионофорные переключатели
Соединения, молекулярные переключения в которых индуцируются взаимодействием с какими-либо ионами и приводят к изменению оптических характеристик системы, называют хромо- и/или флуороионофорами. Например, процесс комплексообразования с металлами в краунированных фрагментах молекул фталоцианина или порфирина легко можно проследить на электронных спектрах и/или спектрах флуоресценции по явным откликам тетрапиррольных составляющих, благодаря которым возможно применение таких соединений в качестве молекулярных оптических датчиков.
В частности, производное фталоцианина, замещенное аза-краун-эфирным блоком, было предложено в качестве сенсора для щелочных и щелочноземельных металлов благодаря его переключаемому флуоресцентному поведению (рис. 18) [93]. В отсутствие катионов рецептор не является флуоресцентным, поскольку его возбужденное состояние релаксирует за счет внутримолекулярного переноса заряда от атома азота краун-эфирной группы к макроциклу фталоцианината цинка. Однако, когда ион щелочного металла связывается с краун-эфирным фрагментом, флуоресценция цинкового металлокомплекса разгорается из-за блокировки внутримолекулярного переноса заряда. В результате в присутствии ионов натрия и калия флуоресценция в растворе тетрагидрофурана усиливалась в 11.5 и 10.5 раз соответственно. Нужно отметить тот факт, что другие катионы щелочных, щелочноземельных и тяжелых металлов не способствовали таким большим значениям разгорания флуоресценции, что обусловлено специфическими характеристиками селективных супрамолекулярных взаимодействий катион – краун-эфир.
Рис. 18.
Аза-краун-фталоцианиновый флуоресцентный сенсор на катионы щелочных металлов и схематическое изображение связывания Na+ и K+, приводящего к усилению флуоресценции [93].

Аналогичные эффекты разгорания флуоресценции в присутствии ионов, комплементарных по отношению к краун-эфирному заместителю красителя на основе нафталимида, описаны в работах [94–97].
Возможен и широко используется и другой вариант участия ионофорного заместителя в цепи сопряжения, когда в координационных взаимодействиях с ионом участвуют атомы, входящие в состав сопряженной цепочки (например, атом азота в аза-краун-замещенных красителях при связывании комплементарных катионов металлов). В этом случае вовлечение электронов таких атомов в координационные взаимодействия может привести к обрыву сопряжения и соответствующему значительному гипсохромному сдвигу основной полосы поглощения [98]. Описанный принцип находит широкое применение в сенсорике с использованием в качестве ионофорных групп аза-краун-эфиров [96, 99–102]. Отметим, что такие эффекты могут регистрироваться как для объемных 3D систем, так и для ультратонких пленок, которые обладают целым рядом преимуществ, обусловленных использованием управляемой предорганизации таких наносистем [100–106].
Еще одна разновидность ион-индуцированных молекулярных переключателей основана на специфических взаимодействиях, связанных с кислотно-основным равновесием. Например, благодаря своим амфотерным свойствам тетрапиррольные макроциклы могут участвовать в кислотно-основном равновесии, что отражается на их физико-химических характеристиках, таких как электронные спектры поглощения, спектры флуоресценции и окислительно-восстановительные потенциалы [107]. В частности, мезо-N-атомы порфиразинов и фталоцианинов могут быть последовательно протонированы в сильнокислых условиях, допустим концентрированной серной кислотой. В отличии от β-замещенных фталоцианинов реакция α-замещения приводит к значительному батохромному сдвигу Q-полосы в ближнюю ИК-область на электронных спектрах поглощения. Дальнейшее последовательное протонирование атомов мезо-азота приводит к еще большему батохромному сдвигу Q-полосы [108].
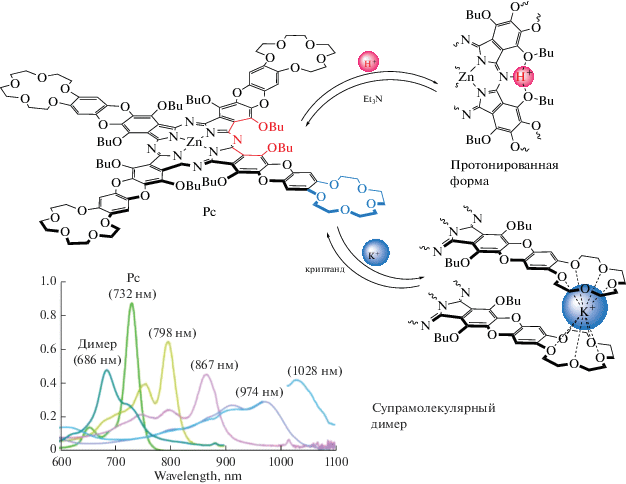
Этот подход был использован группой из лаборатории новых физико-химических проблем Института физической химии и электрохимии РАН для увеличения числа переключаемых состояний краун-замещенных оксантреноцианинов (рис. 19). Было показано, что непериферически замещенный комплекс демонстрирует два различных типа переключения [109]. Он может быть собран в супрамолекулярные димеры в присутствии ионов калия, что сопровождается гипсохромным сдвигом их Q-полос с 732 до 686 нм, или последовательно протонирован добавлением трифторуксусной кислоты. Второй процесс приводит к батохромному сдвигу Q-полосы комплекса с 732 до 1028 нм. Такой широкий охват спектра поглощения различными формами одного и того же соединения фталоцианина является уникальным, и данный комплекс был назван “молекулярным хамелеоном” из-за впечатляющих изменений его цвета при супрамолекулярной димеризации и протонировании.
Рис. 19.
Центры связывания протонов и катионов в окта-бутокси-тетра-15-краун-5-оксантреноцианинате цинка вместе с электронными спектрами поглощения различных форм, полученных супрамолекулярной димеризацией и протонированием [109].
Такая уникальная способность производных фталоцианинов к перемене цвета при различных воздействиях, как, например, описанное выше приложение потенциала, в сочетании с возможностью взаимодействия с малыми молекулами, находят применения и в сенсорике.
Тонкие пленки на основе фталоцианинов обладают высокой чувствительностью к воздействиям внешнего электрического поля, причем отклик системы на такие воздействия может существенно изменяться за счет различных физических эффектов, вызываемых в них взаимодействием с молекулами газа, например оксида азота [110]. Механизм такой реакции заключается в координировании молекулы аналита с металлоцентром фталоцианина. Поэтому многочисленные замещенные металлофталоцианины были исследованы на предмет их электрохромных свойств. У фталоцианинатов металлов есть два возможных центра адсорбции аналита: один – это центральный ион металла, а другой – π-электронная система. Причем на окислительно-восстановительные свойства таких соединений может влиять как координация металла и определяемого иона, так и периферийное присоединение дополнительных атомов или групп, которые увеличивают или уменьшают потенциал ионизации.
Рис. 20.
Схема оптоэлектронной газовой сенсорной системы на основе фталоцианинового металлокомплекса. Датчик цвета определяет интенсивность и изменение цветовых координат RGB (Red-Green-Blue) оптического сигнала в результате воздействия на чувствительную структуру газа определенного типа (в данном случае NH3). Оптический канал восприятия состоит из трех частей: входной порт, который включает в себя широкополосный источник света, многослойная чувствительная наноструктура, выходной порт, который включает кремниевый датчик цвета, обнаруживающий RGB сигнал. Из-за того, что на чувствительной наноструктуре измерительного канала происходит физико-химический процесс связывания, изменятся интерференционный цвет чувствительного элемента и цветовые координаты измеряемого оптического сигнала. По изменению интенсивности оптического сигнала RGB можно измерять концентрацию определенного газа в газовой смеси [111].

Необходимо подчеркнуть, что во многих случаях свойства функциональных материалов на основе соединений, способных к молекулярным переключениям, в значительной степени определяются межмолекулярными взаимодействиями, которые, в свою очередь, зависят от взаимной ориентации молекулярных фрагментов как относительно друг друга, так и относительно материала-носителя [112, 113]. Очевидно, что возможность управления молекулярной ориентацией выгодно отличает планарные 2D системы от 3D материалов и объектов. Данный факт вызывает огромный интерес у специалистов по прикладной химии и стимулирует развитие современных методов формирования высокоорганизованных наноразмерных материалов с заданным взаимным расположением молекул. В первую очередь это относится к ультратонким пленкам – наноразмерным 2D материалам.
2.2. Методы формирования 2D наноразмерных материалов
Выбор того или иного метода формирования ультратонких пленок определяется необходимостью получения конкретных функциональных свойств или обеспечения возможности реализации определенного отклика наноразмерной системы на внешние воздействия [114]. Например, в случаях, когда исследуются электрохромные свойства соединений на поверхности электродов, в основном используются довольно простые способы нанесения, такие как метод полива или метод центрифугирования. Такая технология не требует дополнительной функционализации исследуемых молекул, однако образующиеся пленки не обладают упорядоченностью, что в свою очередь приводит к замедлению полной электрохимической конверсии молекул на поверхности электрода из одного состояния в другое. С этой точки зрения важно развитие методов, позволяющих добиться высокой степени упорядоченности молекул на границе раздела электрод/электролит, что приводит к формированию систем с более коротким временем отклика и большим числом рабочих циклов.
Одним из подходов к созданию упорядоченных систем на основе переключаемых соединений является метод Ленгмюра-Блоджетт [115–117], который позволяет создавать упорядоченные монослои практически на любых поверхностях за счет контролируемого переноса монослоев, образованных дифильными соединениями на поверхности раздела вода/воздух. Данная технология широко применяется для создания сенсорных 2D-материалов на основе самых разнообразных молекулярных переключателей, используемых для детектирования различных аналитов как в водной, так и в газовой фазах [118, 119], а также для получения электроактивных материалов [120–122].
В настоящее время технология формирования наноразмерных материалов на основе тонких пленок толщиной в одну молекулу, полученных из монослоев Ленгмюра, вызывает огромный интерес у исследователей, работающих в области супрамолекулярной химии и не только. Были изучены характеристики монослоев и ультратонких пленок на основе самых разнообразных функциональных соединений и разработаны подходы, позволяющие направленно изменять характеристики формируемых 2D материалов. Показано, что в качестве управляющих факторов для этого можно использовать не только концентрацию формирующего раствора, температуру и поверхностное давление в монослое, но и состав субфазы, на которой он сформирован [100, 101, 112, 123–125].
Интересно отметить, что сжатие монослоя на поверхности субфазы во многих случаях сопровождается не только сближением молекул в монослое, но и существенными изменениями их расположения относительно подложки, а, следовательно, и друг относительно друга. Например, результаты, представленные в работах [126–129], показали, что молекулы в монослоях, полученных из растворов комплексов двухпалубных фталоцианинов РЗЭ в летучих растворителях, лабильны и под действием подвижных барьеров меняют ориентацию плоскостей дисков относительно плоскости межфазной поверхности с последующими стекинг-взаимоействиями. Полученные методом Ленгмюра–Блоджетт (ЛБ) ультратонкие пленки на основе данных монослоев имеют упорядоченную структуру и в нормальных условиях продолжительное время сохраняют исходную архитектуру.
На основании анализа целого ряда публикаций [100, 101, 112, 125, 130–133] можно достоверно утверждать, что техника ЛБ позволяет получать на твердых подложках монослойные пленки, структура и физические свойства которых соответствуют характеристикам монослоев прекурсоров на водной субфазе.
Другим подходом к созданию интеллектуальных наноматериалов является получение самоорганизованных монослоев на основе тиолированных переключаемых соединений на поверхности золота. Несмотря на сложности, связанные с наличием дополнительных стадий синтеза для введения тиольных групп, данный метод имеет важное преимущество, связанное с возможностью управления ориентацией молекулярных фрагментов относительно поверхности электрода за счет направленного получения соединений с заданным расположением якорных групп, обеспечивающих необходимую архитектуру наноматериала (рис. 21) [134–137]. Особый интерес, с этой точки зрения, представляют работы, направленные на поиск новых функциональных групп, обеспечивающих закрепление макроциклических соединений на поверхности раздела [138–141].
Рис. 21.
Архитектура слоев, полученных методом самосборки на поверхности раздела, на основе сэндвичевых комплексов тетрапирролов [134].

Еще один метод получения 2D пленок на различных твердых поверхностях – метод послойной сборки подробно описан в разделе 3.2 обзора [114].
2.3. Методы исследования наноразмерных систем
Развитие методов получения организованных ультратонких пленок и все более широкое их применение, в свою очередь, выносят на передний план задачи по развитию методов исследования таких планарных систем. Для изучения наноразмерных структур могут быть применимы практически такие же методы, как и для изучения кристаллических материалов. Однако для этих методов предъявляются повышенные требования к разрешающей способности, которая должна обеспечивать возможность исследовать участки поверхности образцов толщиной в одну или несколько молекул. Таким образом, можно выделить ряд методов структурного и химического анализа, применение которых позволяет учесть специфику наноразмерных материалов.
2.3.1. Электрохимические методы
В общем случае электрохимические методы анализа основаны на зависимости между составом анализируемой системы и ее электрохимическими характеристиками: стационарным потенциалом, потенциалами протекания редокс переходов, электрической проводимостью и др., проявляющимися в процессах, протекающих на поверхности электрода или в ближнем приэлектродном слое.
Аналитическим сигналом электрохимических методов являются: напряжение или разность потенциала, ток или его интегральная величина во времени – количество электричества, зависимость величины тока от приложенной разности потенциалов или сопротивление. В общем случае при классификации электрохимических методов по типу аналитического сигнала обычно выделяют: потенциометрические, амперометрические, кулонометрические, вольтамперометрические и электрогравиметрические [142].
С точки зрения исследования и эксплуатации молекулярных переключателей (как одиночных молекул, так и их ансамблей различной природы), наиболее распространенным и информативным электрохимическим методом остается вольтамперометрия. Регистрация электрического тока, протекающего в трехэлектродной ячейке при приложении закономерно изменяющегося потенциала, позволяет одновременно получить информацию о качественном и количественном составе пробы, компоненты которой восстанавливаются и/или окисляются на электроде, а также о характере электродного процесса [142]. Данный метод широко используется не только для управления молекулярными переключениями в исследуемых объектах, но и для фиксации происходящих при этих переключениях изменений. Регистрируемые при этом пиковые величины силы тока и соответствующие им значения напряжения могут служить характеристиками того или иного состояния молекулярного переключателя [143–148].
В настоящее время существует достаточно большое количество различных вольтамперометрических методов анализа, применимых к исследованию как растворов, так и тонких пленок, нанесенных на поверхность рабочего электрода. Их, в свою очередь, можно классифицировать по нескольким параметрам: по типу индикаторного электрода, по направлению развертки, по способу наложения потенциала, по способу представления вольтамперограмм: прямая и инверсионная вольтамперометрия [142].
Один из наиболее сложных электрохимических методов – это циклическая вольтамперометрия. На сегодняшний день этот метод используется довольно часто, поскольку позволяет получить богатую экспериментальную информацию о кинетике и термодинамике многих химических систем, таких как, например, аннелированный тетра-тиафульвален фталоцианин [149] или изучить редокс-переходы краун-замещенного фталоцианината цинка (рис. 22а) [150] в том числе в тонких пленках толщиной в одну или несколько молекул (рис. 22б) [151]. Главная особенность этого метода заключается в том, что потенциал изменяется циклически, сначала происходит окисление исследуемого вещества, а затем его восстановление на поверхности твердого электрода (или наоборот) согласно приведенным уравнениям реакций:

Red – ne = Ox+n,
Ox+n + ne = Red.
В результате в процессе измерений происходит циклическое изменение состояния исследуемого вещества. Графическим отображением происходящих на рабочем электроде электрохимических реакций является циклическая вольтамперограмма, которая состоит из двух ветвей: анодной и катодной. Таким образом, циклическая вольтамперометрия – это совокупность анодного (окисление) и катодного (восстановление) процессов, последовательно проходящих на электроде. Вид вольтамперограммы зависит от многих факторов: особенностей строения двойного электрического слоя исследуемого вещества, геометрии электрода, параметров ячейки и т.д.
Используя электрохимические методы анализа, можно узнать о протекании внутримолекулярных редокс-процессов, об электроактивности наноразмерных систем, степени покрытия электродов и т.д.
Так, например, метод ЦВА активно применяется в исследовании и разработке выше описанных электрохромных наноматериалов, в частности ультратонких пленок фталоцианинатов металлов [152, 153], материалов для модуляции поглощения в ИК-диапазоне [154], и т.д. Для электроактивных САМ, сформированных посредством самосборки, методы электрохимического анализа являются основным инструментом для получения информации о данных системах [141, 155]. Также, вольтамперометрические методы очевидно необходимы при исследовании новых наноматериалов – потенциальных электроактивных соединений [156–159].
С точки зрения молекулярных переключений, методы электрохимического анализа позволяют, во-первых, определить возможность реализации переключений за счет приложения электрического тока, а во-вторых, определить их скорость и обратимость [155, 160–162].
2.3.2. Электронная спектроскопия
Как известно, электронная спектроскопия охватывает оптические спектры в ультрафиолетовой, видимой и ближней инфракрасной областях электромагнитного излучения, связанные с переходами между различными электронными состояниями атомов и молекул. Молекулярные электронные спектры исследуются обычно как спектры поглощения или спектры флуоресценции. Электронные спектры поглощения молекул и ионов широко используются для качественного и количественного анализа в химии веществ.
Отметим, что в электронной спектроскопии молекул наряду с электронными переходами между орбиталями одного атома выделяют также переходы с переносом заряда [163, 164], которые представляют собой переходы электрона с орбитали, локализованной в одной части молекулы, на орбиталь, локализованную в другой части молекулы. Такие электронные переходы можно разделить на переходы с внутримолекулярным переносом заряда и переходы в комплексах с переносом заряда.
Изменение структурных характеристик молекулы при введении различного рода заместителей или изменении внешнего молекулярного окружения, взаимодействие с компонентами окружающей среды (аналитами), редокс процессы под действием внешнего электрического тока, изменение температуры и т.д. приводят к сдвигу полос поглощения хромофора и позволяют зарегистрировать и охарактеризовать происходящие изменения.
В работе [165] описан процесс структурного изменения порфиринового димера, вызванный изомеризацией, который, в свою очередь, отражается на спектральных характеристиках самого димера (рис. 23). Аналогичный димер был изучен и в ультратонких 2D системах, где разворот порфириновых макроколец относительно друг друга, также был зарегистрирован с помощью электронной спектроскопии [166].

Очевидно, что широкоизвестный метод электронной спектроскопии поглощения повсеместно применяется для характеризации окрашенных соединений – элементов молекулярных переключателей – в случаях, когда переключения приводят к изменению спектра поглощения. Например, вышеописанное явление электрохромизма, в котором окраска системы меняется при приложении потенциала, очевидно может быть успешно охарактеризовано одновременным применением электрохимических методов и электронной спектроскопии поглощения (так называемые спектро-электрохимические измерения) [84, 152–154].
Следует также отметить, что ряд неортодоксальных модификаций этого классического метода также был разработан для исследования монослоев окрашенных соединений на поверхности раздела фаз [167], определения молекулярной анизотропии в наноразмерных упорядоченных системах [168], измерения поглощения при межмолекулярном переносе заряда в молекулярных системах [169] и так далее.
2.3.3. Рентгеновские методы исследования молекулярных переключений
Все многообразие рентгеновских методов исследования наноматериалов можно разделить на несколько основных групп: рентгеновская дифракция, рентгеноспектральные методы, методы прямой визуализации. Дифракция рентгеновских лучей активно и довольно давно используется для идентификации фаз (рентгенофазовый анализ) и определения кристаллической структуры веществ, в том числе и наноматериалов. Толчком для развития методов рентгеновской спектроскопии (в частности, EXAFS (Extended X-Ray Absorption Fine Structure – Рентгеновское Поглощение Тонкой Структуры в Расширенном Диапазоне) и XANES (X-Ray Absorption Near Edge Structure – Рентгеновское Поглощение Краевой Структуры)) послужило появление синхротронных источников, обеспечивающих пучки достаточной интенсивности. Данные методы позволяют исследовать степень окисления и локальное окружение атомов (в пределах нескольких координационных сфер) [170, 171].
При облучении материала рентгеновским излучением возможен ряд процессов: отражение и преломление первичного излучения, поглощение рентгеновских лучей веществом и сопутствующие этому вторичные эффекты, процессы упругого и неупругого рассеяния.
Процессы отражения и преломления не позволяют получить какую-либо информацию о материале, поскольку излучение не взаимодействует с веществом. Эти эффекты широко используются в рентгеновской оптике и монокристальных монохроматорах [172]. При упругом рассеянии меняется направление импульса падающего излучения, но сохраняется его абсолютная величина и энергия. Следовательно, рассеянное на различных атомах рентгеновское излучение оказывается когерентным, и возможно возникновение дифракции. При наличии в твердом теле трансляционной симметрии появляются дифракционные максимумы в определенных направлениях, что позволяет получать информацию о симметрии элементарной ячейки исследуемого материала. На этом эффекте основаны широко используемые в химии и материаловедении дифракционные методы [173]. Использование этих методов для исследования структуры и организации наноматериалов (в том числе 2D систем) подробно описано в ряде обзоров [174, 175]. В тех случаях, когда молекулярное переключение сопровождается значительными структурными изменениями упорядоченной системы, рентгеновскую дифрактометрию можно использовать для регистрации происходящих изменений и характеризации ее переключаемых состояний.
С другой стороны, в подавляющем большинстве случаев для характеризации состояния отдельных молекул и элементов супрамолекулярных ансамблей, способных к переключениям разной природы, более интересны/применимы методы рентгеновской спектроскопии.
Вещество, облучаемое рентгеновскими фотонами, энергия которых сравнима с энергией ионизации атомов, способно поглощать данное излучение. Как правило, процесс поглощения сопровождается фотоэффектом (выбиванием электронов с основных уровней), в результате чего на внутренних уровнях возникает дырка. Данное состояние крайне неустойчиво и в течение 10–15–10–16 с релаксирует за счет перехода электронов с внешних уровней на освободившееся место. При этом на вышележащих уровнях образуется вакансия, что, в свою очередь, вызывает аналогичные процессы. Излишки энергии при этом теряются за счет процессов рентгеновской флуоресценции и выбивания Оже-электронов.
Таким образом, при поглощении веществом рентгеновского излучения наблюдаются процессы фотоэффекта, выбивания Оже-электронов и рентгеновской флуоресценции. Регистрируемое распределение испускаемых фотонов/электронов по энергии характеристично и позволяет получить информацию об электронных уровнях в исследуемом твердом теле. На данных эффектах основаны такие методы как рентгеновская фотоэлектронная спектроскопия (РФЭС), рентгеновская флуоресцентная спектроскопия (РФС), Оже-электронная спектроскопия (ОЭС). Необходимо отметить, что данные методы позволяют получить информацию лишь о тонком приповерхностном слое (в несколько атомных слоев). Это обстоятельство снижает возможности их применения к исследованию 3D систем, но обеспечивает высокую информативность для изучения ультратонких пленок.
Рентгеновская фотоэлектронная спектроскопия (РФЭС), или по-английски XPS (X-ray photoelectron spectroscopy), благодаря высокой поверхностной и элементной чувствительности является одним из наиболее информативных физико-химических методов, позволяющих проводить комплексные исследования пленок наноразмерной толщины. Даже при условии, что толщина пленок не превышает и нескольких монослоев, данный метод позволяет исследовать границу раздела пленка-подложка и реакции, которые протекают там во время температурных и радиационных воздействий.
Метод РФЭС дает информацию как о внешней, так и о внутренней электронной оболочке исследуемых атомов и молекул, уровнях энергии электронов в твердом теле. Для молекул энергии связи электронов во внутренних оболочках образующих их атомов зависят от типа химической связи (химические сдвиги), поэтому РФЭС успешно применяется в аналитической химии для определения состава вещества и в физической химии для исследования химической связи.
В работе [176] был впервые определен фазовый состав наноразмерных толстых пленок диоксида титана на титановой подложке с помощью рентгеновской фотоэлектронной спектроскопии путем развертки двухпиковой структуры валентной зоны на пять пиков и анализа относительной площади этих пиков. Переключение валентного состояния металлоцентра Се3+/4+ при редокс-изомеризации соответствующего бисфталоцианината при переходе комплекса из раствора в хлороформе на поверхность раздела воздух/вода было также напрямую подтверждено авторами методом РФЭС [56].
Рис. 24.
Спектры РФЭС оксидного слоя, сформированного на титановом образце, с разверткой валентной зоны [176].

Рис. 25.
Сопоставление двух областей рентгеновского поглощения на примере модельного двухатомного или одноатомного вещества. Слева приведены две модели расположения атомов в ближайшей к основному атому координационной сфере.

Рис. 26.
Спектры XANES, полученные для LnIII(C5H4SiMe3)3 (черный) и ${\text{L}}{{{\text{n}}}^{{{\text{II}}}}}\left( {{{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{4}}}}{\text{SiM}}{{{\text{e}}}_{{\text{3}}}}} \right)_{{\text{3}}}^{{{\text{1}} - }}$ (розовый) для Ln = Yb, Sm, Tm, Dy [177].

Сам спектр поглощения рентгеновского излучения веществом также чувствителен к строению электронных оболочек атомов и их локальному окружению. На анализе спектра поглощения основаны такие методы, как XANES и EXAFS-спектроскопия.
Спектроскопия XANES (X-ray absorbtion near edge structure) – отвечает за низкоэнергетическую область, называемую ближней, или околопороговой, тонкой структурой, которой соответствует энергия фотоэлектронов до ~30 эВ, где существенным оказывается многократное рассеяние.
Cпектроскопия EXAFS (Extended X-ray absorbtion fine structure), в свою очередь, отвечает за высокоэнергетическую область, называемую протяженной, или дальней, тонкой структурой, где главный вклад в поглощение дает однократное рассеяние фотоэлектрона.
На рис. 25 видно, что изменение симметрии (углового распределения) фотонов, окружающих поглощающий атом в веществе, влияет на характер спектра в области XANES, тогда как спектр в области EXAFS остается неизменным.
В работе [177] описывается использование рентгеновской спектроскопии XANES для характеризации электронных конфигураций лантаноидных соединений с валентностью металла 2+, общая формула которых ${\text{L}}{{{\text{n}}}^{{{\text{II}}}}}\left( {{{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{4}}}}{\text{SiM}}{{{\text{e}}}_{{\text{3}}}}} \right)_{{\text{3}}}^{{{\text{1}} - }},$ где Ln = Pr, Nd, Sm, Gd, Tb, Dy, Y, Ho, Er, Tm, Yb и Lu. При этом для характеризации проводится сравнение с тщательно подобранной серией стандартов. Спектроскопия XANES была также применена для исследования валентного состояния цериевого металлоцентра гетеролептических фталоцианиново-порфириновых комплексов в зависимости от их строения [178].
Исследования, связанные с молекулярными переключениями, в которые вовлечены атомы металла, например, описанные ранее редокс-изомерные превращения, также хорошо дополняются методами XANES и EXAFS [46, 54, 179, 180], позволяющими, зачастую in situ, наблюдать изменения валентности и ближайшего координационного окружения металла в процессе молекулярного переключения. Так, в работе [46] авторами была продемонстрирована редокс-изомеризация комплекса европия, вызванная изменением молекулярной ориентации на границе раздела фаз воздух/вода, что является не только необычным стимулом для индуцирования данного явления, но и первым примером редокс-изомеризации комплекса европия. Применение время-разрешенной версии методики регистрации спектров XANES также позволило зарегистрировать редокс-изомеризацию комплекса кобальта in situ под действием импульса света [181]. Ряд других интересных примеров подобных исследований с использованием рентгеновского излучения приведен в обзоре [182].
3. ЗАКЛЮЧЕНИЕ
Цель данного обзора – показать многообразие возможностей молекулярных переключений: от ферментативных машин в живых организмах до синтетических супрамолекулярных систем, реализованных в самых различных архитектурах и работающих под действием всевозможных внешних факторов. Очевидно, переход к наноразмерным элементам электроники и мультифункциональным системам атомарного масштаба неизбежен с точки зрения дальнейшего развития целого ряда областей науки и техники.
Проведенный анализ литературы показывает, что для дальнейшего расширения потенциальной сферы практического применения принципов молекулярного переключения необходим поиск новых объектов, разработка новых методов характеризации и формирования таких систем, причем наибольший интерес представляют системы с пониженной размерностью. Как продемонстрировано в обзоре, усилия мирового научного сообщества шаг за шагом приближают нас к давней мечте Р. Фейнмана о принципиально новых устройствах, основанных на феноменах, которые имеют место лишь на нано-уровне.
Список литературы
Lipmann F. // A Source B. Chem. 1900–1950. 2014. P. 381–383. https://doi.org/10.4159/harvard.9780674366701.c141
Yasuda R., Nojii H., Kazuhiko Kinosita J. et al. // FEBS Lett. 1989. V. 253. № 1. P. 289.
Oster G., Wang H. // Structure. 1999. V. 7. № 4. P. 67–72. https://doi.org/10.1016/S0969-2126(99)80046-X
Knowles A.F., Penefsky H.S. // J. Biol. Chem. 1972. V. 247. № 20. P. 6624–6630. https://doi.org/10.1016/S0021-9258(19)44737-6
Walker J.E. // Rev. ATP Synth. by Rotary Catal. Nobel Lect. 1962.
Noji H., Yasudat R. // Nature. 1997. V. 386 120. P. 299–302.
Howard J. // Nature. 1997. V. 389. № 6651. P. 561–567. https://doi.org/10.1038/39247
Vale R.D. // J. Cell Biol. 1996. V. 135. № 2. P. 291–302. https://doi.org/10.1083/jcb.135.2.291
Farrell C.M., Mackey A.T., Klumpp L.M. et al. // J. Biol. Chem. 2002. V. 277. № 19. P. 17079–17087. https://doi.org/10.1074/jbc.M108793200
Vale R.D., Milligan R.A. // Science. 2000. V. 288. № 5463. P. 88. https://doi.org/10.1126/science.288.5463.88
Howard J., Hudspeth A.J., Vale R.D. // Nature. 1989. V. 342. № 6246. P. 154–158. https://doi.org/10.1038/342154a0
Finer J.T., Simmons R.M., Spudich J.A. // Nature. 1994. V. 368. № 6467. P. 113–119. https://doi.org/10.1038/368113a0
Hackney D.D. // Annu. Rev. Physiol. 1996.
Howard J. // Nature. 1994. V. 368. № 6467. P. 98–99. https://doi.org/10.1038/368098a0
Huxley H.E. // Am. Heart J. 1959. V. 58. № 5. P. 777–796. https://doi.org/10.1016/0002-8703(59)90238-8
Howard J. // Annu. Rev. Physiol. 1996. V. 58. P. 703–729. https://doi.org/10.1146/annurev.ph.58.030196.003415
Howard J., Hyman A.A. // Nature. 2003. V. 422. № 6933. P. 753–758. https://doi.org/10.1038/nature01600
Hirokawa N. // Science. 1998. V. 279. № 5350. P. 519–526. https://doi.org/10.1126/science.279.5350.519
Berliner E., Young E.C., Anderson K. et al. // Nature. 1995. V. 373. № 6516. P. 718–721. https://doi.org/10.1038/373718a0
https://www.popmech.ru/science/235122-nanoroboty-vnutri-nas-kak-rabotayut-kletki/ [Electronic resource].
Gibbons IR R.A. // Science. 1965. V. 149. № 3682. P. 424–426. https://doi.org/10.1126/science.149.3682.424
Anelli P.L., Spencer N., Stoddart J.F. // J. Am. Chem. Soc. 1991. V. 113. № 13. P. 5131–5133. https://doi.org/10.1021/ja00013a096
Bissell R.A., Córdova E., Kaifer A.E. et al. // Nature. 1994. V. 369. № 6476. P. 133–137. https://doi.org/10.1038/369133a0
Kelly T.R., De Silva H., Silva R.A. // Nature. 1999. V. 401. № 6749. P. 150–152. https://doi.org/10.1038/43639
Koumura N., Zijistra R.W.J., Van Delden R.A. et al. // Nature. 1999. V. 401. № 6749. P. 152–155. https://doi.org/10.1038/43646
Klok M., Boyle N., Pryce M.T. et al. // J. Am. Chem. Soc. 2008. V. 130. № 32. P. 10484–10485. https://doi.org/10.1021/ja8037245
Eelkema R., Pollard M.M., Vicario J. et al. // Nature. 2006. V. 440. № 7081. P. 163–163. https://doi.org/10.1038/440163a
Berná J., Leigh D.A., Lubomska M. et al. // Nat. Mater. 2005. V. 4. № 9. P. 704–710. https://doi.org/10.1038/nmat1455
Liu Y., Flood A.H., Bonvallet P.A. et al. // J. Am. Chem. Soc. 2005. V. 127. № 27. P. 9745–9759. https://doi.org/10.1021/ja051088p
Lewandowski B., De Bo G., Ward J.W. et al. // Science. 2013. V. 339. № 6116. P. 189–193. https://doi.org/10.1126/science.1229753
Green J.E., Wook Choi J., Boukai A. et al. // Nature. 2007. V. 445. № 7126. P. 414–417. https://doi.org/10.1038/nature05462
Kay E.R., Leigh D.A. // Angew. Chemie Int. Ed. 2015. V. 54. № 35. P. 10080–10088. https://doi.org/10.1002/anie.201503375
Bally T. // Nat. Chem. 2010. V. 2. № 3. P. 165–166. https://doi.org/10.1038/nchem.564
Buchanan R.M., Pierpont C.G. // J. Am. Chem. Soc. 1980. V. 102. № 15. P. 4951–4957. https://doi.org/10.1021/ja00535a021
Pierpont C.G. // Coord. Chem. Rev. 2001. V. 216–217. P. 99–125. https://doi.org/10.1016/S0010-8545(01)00309-5
Abakumov G.A., Cherkasov V.K., Nevodchikov V.I. et al. // Inorg. Chem. 2001. V. 40. № 10. P. 2434–2436. https://doi.org/10.1021/ic001449w
Lapierre H.S., Kameo H., Halter D.P. et al. // Angew. Chemie – Int. Ed. 2014. V. 53. № 28. P. 7154–7157. https://doi.org/10.1002/anie.201402048
Wei J., Diaconescu P.L. // Acc. Chem. Res. 2019. V. 52. № 2. P. 415–424. https://doi.org/10.1021/acs.accounts.8b00523
Rovira C. // Chem. Rev. 2004. V. 104. № 11. P. 5289–5318. https://doi.org/10.1021/cr030663+
Kaim W., Schwederski B. // Pure Appl. Chem. 2004. V. 76. № 2. P. 351–364. https://doi.org/10.1351/pac200476020351
Nitahara S., Akiyama T., Inoue S. et al. // J. Phys. Chem. B. 2005. V. 109. № 9. P. 3944–3948. https://doi.org/10.1021/jp046776u
Laye R.H., Couchman S.M., Ward M.D. // Inorg. Chem. 2001. V. 40. № 16. P. 4089–4092. https://doi.org/10.1021/ic010054p
Lange C.W., Conklin B.J., Pierpont C.G. // Inorg. Chem. 1994. V. 33. № 7. P. 1276–1283. https://doi.org/10.1021/ic00085a012
Poneti G., Mannini M., Sorace L. et al. // Angew. Chem. Int. Ed. Engl. 2010. V. 49. № 11. P. 1954–1957. https://doi.org/10.1002/anie.200906895
Fedushkin I.L., Maslova O.V., Morozov A.G. et al. // Angew. Chem. Int. Ed. Engl. 2012. V. 51. № 42. P. 10 584–10 587. https://doi.org/10.1002/anie.201204452
Shokurov A.V., Kutsybala D.S., Martynov A.G. et al. // Langmuir. 2020. V. 36. № 6. P. 1423–1429. https://doi.org/10.1021/acs.langmuir.9b03403
Robin M., Day P. // Adv. Inorg. Chem. Radiochem. 1968. V. 10. P. 247–422.
Creutz C., Taube H. // J. Am. Chem. Soc. 1969. V. 91. № 14. P. 3988–3989. https://doi.org/10.1021/ja01042a072
Gütlich P., Gaspar A.B., Ksenofontov V. et al. // J. Phys. Condens. Matter. 2004. V. 16. № 14. P. S1087–S1108. https://doi.org/10.1088/0953-8984/16/14/019
Roux C., Adams D.M., Itié J.P. et al. // Inorg. Chem. 1996. V. 35. № 10. P. 2846–2852. https://doi.org/10.1021/ic951080o
Caneschi A., Dei A., Fabrizi de Biani F. et al. // Chemistry. 2001. V. 7. № 18. P. 3926–3930. https://doi.org/10.1002/1521-3765(20010917)7:18<39-26::AID-CHEM3926>3.0.CO;2-6
Zhao W., Tong B., Pan Y. et al. // Langmuir. 2009. V. 25. № 19. P. 11796–11801. https://doi.org/10.1021/la901427j
Селектор С.Л., Шокуров А.В., Райтман О.А. и др. // Коллоидный журн. 2012. Т. 74. № 3. С. 359–370.
Shokurov A.V., Kutsybala D.S., Martynov A.G. et al. // Macroheterocycles. 2019. V. 12. № 3. P. 264–267. https://doi.org/10.6060/mhc191071s
Селектор С.Л., Шокуров А.В., Арсланов В.В. и др. // Электрохимия. 2012. V. 48. № 2. P. 240–256.
Selektor S.L., Shokurov A. V., Arslanov V. V. et al. // J. Phys. Chem. C. 2014. V. 118. № 8. P. 4250–4258. https://doi.org/10.1021/jp411936k
Hay M.A., Boskovic C. // Chem. – A Eur. J. 2021. V. 27. № 11. P. 3608–3637. https://doi.org/10.1002/chem.202003761
de Saja J.A., Rodríguez-Méndez M.L. // Adv. Colloid Interface Sci. 2005. V. 116. № 1–3. P. 1–11. https://doi.org/10.1016/j.cis.2005.03.004
Vesper B.J., Salaita K., Zong H. et al. // J. Am. Chem. Soc. 2004. V. 126. № 50. P. 16653–16658. https://doi.org/10.1021/ja045270m
Gottfried J.M. // Surf. Sci. 2015. V. 70. № 3. P. 259–379. https://doi.org/10.1016/j.surfrep.2015.04.001
Cvrtila I., Fanlo-Virgós H., Schaeffer G. et al. // J. Am. Chem. Soc. 2017. V. 139. № 36. P. 12459–12465. https://doi.org/10.1021/jacs.7b03724
Qian H., Pramanik S., Aprahamian I. // J. Am. Chem. Soc. 2017. V. 139. № 27. P. 9140–9143. https://doi.org/10.1021/jacs.7b04993
McClure B.A., Abrams E.R., Rack J.J. // J. Am. Chem. Soc. 2010. V. 132. № 15. P. 5428–5436. https://doi.org/10.1021/ja9099399
Huang C.-Y., Bonasera A., Hristov L. et al. // J. Am. Chem. Soc. 2017. V. 139. № 42. P. 15205–15211. https://doi.org/10.1021/jacs.7b08726
Mitchell R.H., Ward T.R., Chen Y. et al. // J. Am. Chem. Soc. 2003. V. 125. № 10. P. 2974–2988. https://doi.org/10.1021/ja0288136
Kortekaas L., Browne W.R. // Chem. Soc. Rev. 2019. V. 48. № 12. P. 3406–3424. https://doi.org/10.1039/C9CS00203K
Balzani V., Credi A., Venturi M. // Chem. Soc. Rev. 2009. V. 38. № 6. P. 1542. https://doi.org/10.1039/b806328c
Credi A., Silvi S., Venturi M. // Molecular Machines and Motors. 2014. P. 1–34.
Li Q., Fuks G., Moulin E. et al. // Nat. Nanotechnol. 2015. V. 10. № 2. P. 161–165. https://doi.org/10.1038/nnano.2014.315
GouletHanssens A., Eisenreich F., Hecht S. // Adv. Mater. 2020. V. 32. № 20. P. 1905966. https://doi.org/10.1002/adma.201905966
Andréasson J., Pischel U. // Coord. Chem. Rev. 2021. V. 429. P. 213695. https://doi.org/10.1016/j.ccr.2020.213695
Šípová-Jungová H., Andrén D., Jones S. et al. // Chem. Rev. 2020. V. 120. № 1. P. 269–287. https://doi.org/10.1021/acs.chemrev.9b00401
Paul M.S. Monk, Roger J. Mortimer, David R. Rosseinsky. Electrochromism: Fundamentals and Applications. 1995. 239 p.
Platt J.R. // J. Chem. Phys. 1961. V. 34. № 3. P. 862–863. https://doi.org/10.1063/1.1731686
Habib M.A. // Electrochem. 1992. P. 51–62.
Palenzuela J., Viñuales A., Odriozola I. et al. // ACS Appl. Mater. Interfaces. 2014. V. 6. № 16. P. 14 562–14 567. https://doi.org/10.1021/am503869b
Madasamy K., Velayutham D., Suryanarayanan V. et al. // J. Mater. Chem. C. 2019. V. 7. № 16. P. 4622–4637. https://doi.org/10.1039/C9TC00416E
Sun X.W., Wang J.X. // Nano Lett. 2008. V. 8. № 7. P. 1884–1889. https://doi.org/10.1021/nl0804856
Alesanco Y., Viñuales A., Rodriguez J. et al. // Materials (Basel). 2018. V. 11. № 3. P. 414. https://doi.org/10.3390/ma11030414
Mortimer R.J. // Electrochim. Acta. 1999. V. 44. № 18. P. 2971–2981. https://doi.org/10.1016/S0013-4686(99)00046-8
https://www.nrel.gov/research/index.html. [Electronic resource]
Argun A.A., Aubert P., Thompson B.C. et al. // Chem. Mater. 2004. V. 16. № 23. P. 4401–4412. https://doi.org/10.1021/cm049669l
Alesanco Y., Viñuales A., Palenzuela J. et al. // ACS Appl. Mater. Interfaces. 2016. V. 8. № 23. P. 14795–14801. https://doi.org/10.1021/acsami.6b01911
Martynov A.G., Safonova E.A., Tsivadze A.Y. et al. // Coord. Chem. Rev. 2019. V. 387. P. 325–347. https://doi.org/10.1016/j.ccr.2019.02.004
Kutsybala D.S., Shokurov A.V., Kroitor A.P., Martynov A.G., Selektor S.L., Tsivadze A.Yu. et.al. // Macroheterocycles. 2021. P. [рукопись принята в печать].
Biesaga M. // Talanta. 2000. V. 51. № 2. P. 209–224. https://doi.org/10.1016/S0039-9140(99)00291-X
Yahaya M.B., Mat Salleh M., Yusoff N.Y.N. et al. // Proc. SPIE 5276, Device and Process Technologies for MEMS, Microelectronics, and Photonics III, (2 April 2004) P. 422. https://doi.org/10.1117/12.522857
Basova T., Jushina I., Gürek A. G. et al. // J. R. Soc. Interface. 2008. V. 5. № 24. P. 801–806. https://doi.org/10.1098/rsif.2007.1241
Rodriguez-Mendez M.L., Antonio de Saja J. // J. Porphyr. Phthalocyanines. 2009. V. 13. P. 606–615. https://doi.org/10.1142/S1088424609000814
Jux N. 2012. Coordination Chemistry of Crown Ether-Porphyrin Conjugates // Handbook of Porphyrin Science. V. 24. Р. 181–269. doi:10.1142/9789814397605_0014
Lukyanets E.A., Nemykin V.N. // J. Porphyr. Phthalocyanines. 2010. V. 14. № 1. P. 1–40. https://doi.org/10.1142/S1088424610001799
Martynov A.G., Gorbunova Y.G., Tsivadze A.Y. // Russ. J. Inorg. Chem. 2014. V. 59. № 14. P. 1635–1664. https://doi.org/10.1134/S0036023614140046
Novakova V., Lochman L., Zajícová I. et al. // Chem. – A Eur. J. 2013. V. 19. № 16. P. 5025–5028. https://doi.org/10.1002/chem.201300079
Selektor S.L., Shokurov A.V., Revina A.A. et al. // Macroheterocycles. 2015. V. 8. № 2. P. 135–142. https://doi.org/10.6060/mhc150765s
Panchenko P.A., Fedorov Y.V., Fedorova O.A. et al. // Mendeleev Commun. 2011. V. 21. № 1. P. 12–14. https://doi.org/10.1016/j.mencom.2011.01.005
Panchenko P.A., Fedorova O.A., Fedorov Y.V. // Russ. Chem. Rev. 2014. V. 83. № 2. P. 155–182. https://doi.org/10.1070/RC2014v083n02ABEH004380
Panchenko P.A., Fedorov Y.V., Fedorova O.A. // J. Photochem. Photobiol. A Chem. 2018. V. 364. P. 124–129. https://doi.org/10.1016/j.jphotochem.2018.06.003
Ушаков Е.Н.Н., Алфимов М.В.В., Громов С.П.П. // Успехи Химии. 2008. V. 77. № 1. P. 42–59.
Mitewa M., Mateeva N., Antonov L. et al. // Dye. Pigment. 1995. V. 27. № 3. P. 219–225. https://doi.org/10.1016/0143-7208(94)00060-F
Shokurov A.V., Alexandrova A.V., Shcherbina M.A. et al. // Soft Matter. 2020. V. 16. № 43. P. 9857–9863. https://doi.org/10.1039/d0sm01078b
Shokurov A.V., Shcherbina M.A., Bakirov A.V. et al. // Langmuir. 2018. V. 34. № 26. P. 7690–7697. https://doi.org/10.1021/acs.langmuir.8b01369
Shokurov A.V., Alexandrova A.V., Lukovskaya E.V. et al. // Macroheterocycles. 2017. V. 10. № 4–5. P. 560–566. https://doi.org/10.6060/mhc171140s
Shokurov A.V., Alexandrova A.V., Shepeleva I.I. et al. // Mendeleev Commun. 2019. V. 29. № 1. P. 74–76. https://doi.org/10.1016/j.mencom.2019.01.025
Saleem M., Rafiq M., Hanif M. // J. Fluoresc. 2017. V. 27. № 1. P. 31–58. https://doi.org/10.1007/s10895-016-1933-x
Ermakova E.V., Koroleva E.O., Shokurov A.V. et al. // Dye. Pigment. 2021. V. 186. P. 108967. https://doi.org/10.1016/j.dyepig.2020.108967
Chang X., Zhou Z., Shang C. et al. // J. Am. Chem. Soc. 2019. V. 141. № 4. P. 1757–1765. https://doi.org/10.1021/jacs.8b12749
Stuzhin P.A. // J. Porphyr. Phthalocyanines. 1999. V. 3. № 6–7. P. 500–513. https://doi.org/10.1002/(sici)1099-1409(199908/10) 3:6/7<500::aid-jpp168>3.0.co;2-9
Donzello M.P., Ercolani C., Gaberkorn A.A. et al. // Chem. – A Eur. J. 2003. V. 9. № 17. P. 4009–4024. https://doi.org/10.1002/chem.200304929
Safonova E.A., Martynov A.G., Nefedov S.E. et al. // Inorg. Chem. 2016. V. 55. № 5. P. 2450–2459. https://doi.org/10.1021/acs.inorgchem.5b02831
Claessens C.G., Blau W.J., Cook M. et al. // Monatshefte fuer Chemie/Chemical Mon. 2001. V. 132. № 1. P. 3–11. https://doi.org/10.1007/s007060170140
Maciak E., Pustelny T., Opilski Z. // Procedia Eng. 2012. V. 47. P. 738–741. https://doi.org/10.1016/j.proeng.2012.09.253
Selektor S.L., Shokurov A.V. // Prot. Met. Phys. Chem. Surf. 2015. V. 51. № 2. P. 171–203. https://doi.org/10.1134/S2070205115020161
Okada Y., Chiba K. // Chem. Rev. 2018. V. 118. № 9. P. 4592–4630. https://doi.org/10.1021/acs.chemrev.7b00400
Antipin I.S., Alfimov M.V., Arslanov V.V. et al. // Russ. Chem. Rev. 2021. V. 90. [рукопись принята в печать].https://doi.org/10.1070/RCR5011
Zhavnerko G., Marletta G. // Mater. Sci. Eng. B. 2010. V. 169. № 1–3. P. 43–48. https://doi.org/10.1016/j.mseb.2009.12.005
Lee I.C., Frank C.W., Yamamoto T. et al. // Langmuir. 2004. V. 20. № 14. P. 5809–5828. https://doi.org/10.1021/la0361518
Kan J., Chen Y., Qi D. et al. // Adv. Mater. 2012. V. 24. № 13. P. 1755–1758. https://doi.org/10.1002/adma.201200006
Li X., Zhang G., Bai X. et al. // Nat. Nanotechnol. 2008. V. 3. № 9. P. 538–542. https://doi.org/10.1038/nnano.2008.210
Muramatsu M., Akatsuka K., Ebina Y. et al. // Langmuir. 2005. V. 21. № 14. P. 6590–6595. https://doi.org/10.1021/la050293f
Gütlich P., Garcia Y., Woike T. // Coord. Chem. Rev. 2001. V. 221. P. 839–879.
Barszcz B., Bogucki A., Biadasz A. et al. // J. Photochem. Photobiol. A Chem. Elsevier. 2011. V. 218. № 1. P. 48–57. https://doi.org/10.1016/j.jphotochem.2010.12.002
Obraztsov I., Noworyta K., Hart A. et al. // ACS Appl. Mater. Interfaces. 2014. V. 6. № 11. P. 8688–8701. https://doi.org/10.1021/am501446g
Sadullah M.S., Panter J.R., Kusumaatmaja H. // Soft Matter. 2020. V. 16. № 35. P. 8114–8121. https://doi.org/10.1039/D0SM00766H
Kalinina M.A., Golubev N.V., Raitman O.A. et al. // Sensors Actuators B Chem. 2006. V. 114. № 1. P. 19–27. https://doi.org/10.1016/j.snb.2005.04.021
Selektor S.L., Shcherbina M.A., Bakirov A.V. et al. // Langmuir. 2016. V. 32. № 2. P. 637–643. https://doi.org/10.1021/acs.langmuir.5b04075
Selector S.L., Arslanov V.V., Birin K.P. et al. // J. Porphyr. Phthalocyanines. 2008. V. 12. № 3–6. P. 713.
Selector S.L., Arslanov V.V., Gorbunova Y.G. et al. // J. Porphyr. Phthalocyanines. 2008. V. 12. № 11. P. 1154–1162. https://doi.org/10.1142/S1088424608000522
Selektor S.L., Sheinina L.S., Shokurov A.V. et al. // Prot. Met. Phys. Chem. Surf. 2011. V. 47. № 4. P. 447–456. https://doi.org/10.1134/S2070205111040162
Shokurov A.V., Selektor S.L., Arslanov V.V. et al. // Macroheterocycles. 2012. V. 5. № 4–5. P. 358–365. https://doi.org/10.6060/mhc2012.121106s
Ariga K., Yamauchi Y., Mori T. et al. // Adv. Mater. 2013. V. 25. № 45. P. 6477–6512. https://doi.org/10.1002/adma.201302283
Hussain S.A., Dey B., Bhattacharjee D. et al. // Heliyon. 2018. V. 4. № 12. P. e01038. https://doi.org/10.1016/j.heliyon.2018.e01038
Hussain S.A. // arXiv preprint arXiv:0908.1814, 2009. P. 1–9.
Ariga K. // Langmuir. 2020. V. 36. № 26. P. 7158–7180. https://doi.org/10.1021/acs.langmuir.0c01044
Schweikart K.-H., Malinovskii V.L., Diers J.R. et al. // J. Mater. Chem. 2002. V. 12. № 4. P. 808–828. https://doi.org/10.1039/b108520d
Lu X., Zhang L., Li M. et al. // ChemPhysChem. 2006. V. 7. № 4. P. 854–862. https://doi.org/10.1002/cphc.200500492
Yasseri A.A., Syomin D., Malinovskii V.L. et al. // J. Am. Chem. Soc. 2004. V. 126. № 38. P. 11944–11953. https://doi.org/10.1021/ja047723t
Gryko D.T., Clausen C., Roth K.M. et al. // J. Org. Chem. 2000. V. 65. № 22. P. 7345–7355. https://doi.org/10.1021/jo000487u
Martin C.A., Ding D., Sørensen J.K. et al. // J. Am. Chem. Soc. 2008. V. 130. № 40. P. 13198–13199. https://doi.org/10.1021/ja804699a
Wang L., Polyansky D.E., Concepcion J.J. // J. Am. Chem. Soc. 2019. P. jacs.9b01044. https://doi.org/10.1021/jacs.9b01044
Sosa-Vargas L., Kim E., Attias A.-J. // Mater. Horizons. 2017. V. 4. № 4. P. 570–583. https://doi.org/10.1039/C7MH00127D
Shokurov A.V., Yagodin A.V., Martynov A.G. et al. // ECS J. Solid State Sci. Technol. 2020. V. 9. № 5. P. 051 006. https://doi.org/10.1149/2162-8777/ab9a5e
Плэмбек Д. Электрохимические методы анализа: основы теории и применение. 1985. 504 p.
Guille-Collignon M., Delacotte J., Lemaître F. et al. // Chem. Rec. 2021. https://doi.org/10.1002/tcr.202100022
Amatore C., Bouret Y., Maisonhaute E. et al. // Comptes Rendus Chim. 2003. V. 6. № 1. P. 99–115. https://doi.org/10.1016/S1631-0748(03)00019-5
Kaifer A.E., Gómez-Kaifer M. Supramolecular electrochemistry. 2008. books.google.com.
Chen Q., Sun J., Li P. et al. // J. Am. Chem. Soc. 2016. V. 138. № 43. P. 14242–14245. https://doi.org/10.1021/jacs.6b09880
Barcena H.S., Liu B., Mirkin M.V. et al. // Inorg. Chem. 2005. V. 44. № 21. P. 7652–7660. https://doi.org/10.1021/ic051048m
Ashton P.R., Ballardini R., Balzani V. et al. // Chem. – A Eur. J. 1997. V. 3. № 1. P. 152–170. https://doi.org/10.1002/chem.19970030123
Loosli C., Jia C., Liu S.X. et al. // J. Org. Chem. 2005. V. 70. № 13. P. 4988–4992. https://doi.org/10.1021/jo0501801
Biyiklioǧlu Z., Çakir V., Çakir D. et al. // J. Organomet. Chem. 2014. V. 749. P. 18–25. https://doi.org/10.1016/j.jorganchem.2013.07.079
Crespilho F.N., Zucolotto V., Siqueira J.R. et al. // Int. J. Electrochem. Sci. 2006. V. 1. № 4. P. 151–159.
Selector S.L., Shokurov A.V., Arslanov V.V. et al. // Russ. J. Electrochem. 2012. V. 48. № 2. P. 218–233. https://doi.org/10.1134/S1023193512020164
Basova T., Jushina I., Gürek A.G. et al. // J. R. Soc. Interface. 2008. V. 5. № 24. P. 801–806. https://doi.org/10.1098/rsif.2007.1241
Cui B.-B., Tang J.-H., Yao J. et al. // Angew. Chemie Int. Ed. 2015. V. 54. № 32. P. 9192–9197. https://doi.org/10.1002/anie.201504584
Eckermann A.L., Feld D.J., Shaw J.A. et al. // Coord. Chem. Rev. 2010. V. 254. № 15–16. P. 1769–1802. https://doi.org/10.1016/j.ccr.2009.12.023
Morlanés N., Takanabe K., Rodionov V. // ACS Catal. 2016. V. 6. № 5. P. 3092–3095. https://doi.org/10.1021/acscatal.6b00543
Xiao Y.-H., Gu Z.-G., Zhang J. // Nanoscale. 2020. V. 12. № 24. P. 12712–12730. https://doi.org/10.1039/D0NR03115A
Lin S., Pineda-Galvan Y., Maza W.A. et al. // ChemSusChem. 2017. V. 10. № 3. P. 514–522. https://doi.org/10.1002/cssc.201601181
Shokurov A.V., Kutsybala D.S., Martynov A.G. et al. // Thin Solid Films. 2019. V. 692. P. 137591. https://doi.org/10.1016/j.tsf.2019.137591
Seo S., Kim Y., You J. et al. // Macromol. Rapid Commun. 2011. V. 32. № 8. P. 637–643. https://doi.org/10.1002/marc.201000791
Tang J.-H., He Y.-Q., Shao J.-Y. et al. // Sci. Rep. 2016. V. 6. № 1. https://doi.org/10.1038/srep35253
Liu Z., Ren S., Guo X. // Top. Curr. Chem. 2017. V. 375. № 3. P. 56. https://doi.org/10.1007/s41061-017-0144-5
Oquendo L.E., Ehamparam R., Armstrong N.R. et al. // J. Phys. Chem. C. 2019. V. 123. № 12. P. 6970–6980. https://doi.org/10.1021/acs.jpcc.8b10301
Lehtivuori H., Kumpulainen T., Efimov A. et al. // J. Phys. Chem. C. 2008. V. 112. № 26. P. 9896–9902. https://doi.org/10.1021/jp8026918
Kärnbratt J., Hammarson M., Li S. et al. // Angew. Chemie Int. Ed. 2010. V. 49. № 10. P. 1854–1857. https://doi.org/10.1002/anie.200906088
Shokurov A.V., Meshkov I.N., Bulach V. et al. // New J. Chem. 2019. V. 43. № 28. P. 11419–11425. https://doi.org/10.1039/C9NJ01807G
Stuchebryukov S.D., Selektor S.L., Silantieva D.A. et al. // Prot. Met. Phys. Chem. Surf. 2013. V. 49. № 2. P. 189–197. https://doi.org/10.1134/S2070205113020044
Flora W.H., Mendes S.B., Doherty W.J. et al. // Langmuir. 2005. V. 21. № 1. P. 360–368. https://doi.org/10.1021/la0480078
Kim H.-B., Kim J.-J. // Org. Electron. 2018. V. 62. P. 511–515. https://doi.org/10.1016/j.orgel.2018.06.022
Alagna L., Capobianchi A., Casaletto M.P. et al. // J. Mater. Chem. 2001. V. 11. № 7. P. 1928–1935. https://doi.org/10.1039/b100041l
Chang Y.H., Choi K.H., Ford W.T. et al. // J. Chem. Soc. Chem. Commun. 1994. № 6. P. 785–788. https://doi.org/10.1039/C39940000785
Ceglio N.M. // J. Xray. Sci. Technol. 1989. V. 1. № 1. P. 7–78. https://doi.org/10.3233/XST-1989-1103
Warren B.E. // J. Appl. Phys. 1941. V. 12. № 5. P. 375–383. https://doi.org/10.1063/1.1712915
Narayanan T., Konovalov O. // Materials (Basel). 2020. V. 13. № 3. P. 752. https://doi.org/10.3390/ma13030752
Sedigh Rahimabadi P., Khodaei M., Koswattage K.R. // X-Ray Spectrom. 2020. V. 49. № 3. P. 348–373. https://doi.org/10.1002/xrs.3141
Krishna D.N.G., George R.P., Philip J. // Thin Solid Films. 2019. V. 681. P. 58–68. https://doi.org/10.1016/j.tsf.2019.04.044
Fieser M.E., Ferrier M.G., Su J. et al. // Chem. Sci. 2017. V. 8. № 9. P. 6076–6091. https://doi.org/10.1039/C7SC00825B
Bian Y., Jiang J., Tao Y. et al. // J. Am. Chem. Soc. 2003. V. 125. № 40. P. 12257–12267. https://doi.org/10.1021/ja036017+
Evangelio E., Ruiz-Molina D., Ruiz-Molina D. // Eur. J. Inorg. Chem. 2005. V. 2005. № 15. P. 2957–2971. https://doi.org/10.1002/ejic.200500323
Kim S., Bae I.T., Sandifer M. et al. // J. Am. Chem. Soc. 1991. V. 113. № 24. P. 9063–9066. https://doi.org/10.1021/ja00024a006
Ash R., Zhang K., Vura-Weis J. // J. Chem. Phys. 2019. V. 151. № 10. P. 104201. https://doi.org/10.1063/1.5115227
Collet E., Guionneau P. // Comptes Rendus Chim. 2018. V. 21. № 12. P. 1133–1151. https://doi.org/10.1016/j.crci.2018.02.003
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов