

Биоорганическая химия, 2022, T. 48, № 2, стр. 194-206
Пептидные гормоны в медицине: 100-летняя история
В. П. Мартинович 1, К. В. Бородина 1, *
1 ГНУ “Институт биоорганической химии НАН Беларуси”
220141 Минск, ул. Акад. В.Ф. Купревича, 5/2, Беларусь
* E-mail: borodina@iboch.by
Поступила в редакцию 29.04.2021
После доработки 24.06.2021
Принята к публикации 28.06.2021
- EDN: BOUMXE
- DOI: 10.31857/S0132342322020154
Аннотация
В обзоре приводятся данные об исследованиях пептидных гормонов и создании препаратов на их основе в исторической перспективе, за 100 лет, начиная с выделения и введения в медицинскую практику инсулина в 1921 г. Рассмотрены основные группы пептидных гормонов: нейрогипофизарные гормоны, гипоталамические рилизинг-гормоны, инкретины, а также инсулин, адренокортикотропный гормон (ACTH) и кальцитонин. Первые препараты пептидных гормонов созданы на основе традиционного подхода, включающего выделение пептидов из тканей животных, их очистку до индивидуальных соединений, установление первичной структуры, далее их химический синтез или глубокую очистку и создание фармацевтического препарата. Современный подход к созданию препаратов пептидных гормонов основан на рассмотрении их как лигандов соответствующих клеточных рецепторов и предполагает использование компьютерного моделирования, эффективных методов синтеза, высокопроизводительного скрининга, что дает возможность создания более активных, чем природные аналоги, соединений с заданным спектром активности, выходящим за рамки гормональной регуляции, устойчивых к биодеградации. Такие препараты созданы на основе аналогов соматостатина и люлиберина, агонистов и антагонистов, которые нашли широкое применение в качестве гормональных регуляторов и при лечении злокачественных новообразований. Два последних десятилетия проводились интенсивные исследования и разработка препаратов на основе глюкагон-подобного пептида (GLP-1), в обзоре показано, какие модификации привели к созданию наиболее “успешных” препаратов – высокоактивных, с длительным периодом полураспада. На фармацевтическом рынке широко представлены как препараты гормонов с природной структурой, так и их аналоги.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ.......................................................194
КЛАССИЧЕСКИЕ ГОРМОНАЛЬНЫЕ ПЕПТИДНЫЕ ПРЕПАРАТЫ..........................195
ГИПОТАЛАМИЧЕСКИЕ РИЛИЗИНГ- ГОРМОНЫ И ИХ АНАЛОГИ...........................198
АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА-1......................................................201
ЗАКЛЮЧЕНИЕ................................................203
СПИСОК ЛИТЕРАТУРЫ................................204
ВВЕДЕНИЕ
Пептидные препараты занимают очень важную и объемную нишу в современной фармацевтической индустрии. Их разнообразие очень велико: от модифицированного дипептида лизиноприла (известного гипотензивного средства) до инсулина (препарата, незаменимого для больных диабетом первого типа). Пептиды – важнейшие регуляторы, обеспечивающие функционирование практически всех живых организмов, начиная с простейших. Эти соединения – сигнальные молекулы для многих физиологических функций, поэтому их использование дает возможность терапевтической коррекции широкого спектра биологических процессов, протекающих в организме. Ранее пептиды использовали в основном в заместительной гормональной терапии, которая восстанавливает их уровень в тех случаях, когда естественная выработка понижена или прекращена; сейчас сфера их применения очень широка. Начало применения пептидных гормонов в медицине относится к первой половине прошлого века, базовая веха – выделение и очистка инсулина в 1921 г.
Использование пептидов в качестве терапевтических средств эволюционировало во времени и продолжает развиваться вместе с прогрессом в области биохимии, молекулярной биологии и медицины. Анализ истории разработки пептидных гормональных препаратов позволяет выделить два основных периода [1–3]. Первый связан с традиционными подходами, включающими выделение пептидов из тканей животных, их очистку до индивидуальных соединений, установление первичной структуры, далее их химический синтез или глубокую очистку и создание фармацевтического препарата. Распространение пептидной терапии сдерживали такие свойства природных пептидов, как короткий период полувыведения из плазмы и незначительная пероральная биодоступность. Короткий период полураспада многих пептидных гормонов объясняется наличием многочисленных пептидаз и экскреторных механизмов, инактивирующих и выводящих пептиды. Эта лабильность позволяет организму быстро модулировать уровень гормонов для поддержания гомеостаза, но представляет серьезную проблему при разработке терапевтических препаратов.
Современный подход к созданию пептидных препаратов связан с развитием молекулярной биологии, позволившей привлечь к разработкам информацию о геноме, клонировать гены, кодирующие терапевтически важные биологические мишени, и экспрессировать их белковые продукты. Он основан на рассмотрении лиганд-рецепторного взаимодействия как универсального метода передачи информации клетками и тканями организма. Структурное соответствие, необходимое для связывания с белком-мишенью, служит основным критерием при дизайне пептидов – потенциальных кандидатов в лекарственные препараты. Этот подход включает также модификации структуры лигандов с целью повышения устойчивости к биодеградации и создания более активных соединений, чем природные аналоги. Кроме того, наличие массивных комбинаторных библиотек органических соединений и технологии высокопроизводительного скрининга позволяют конструировать маленькие молекулы непептидной природы, которые нацелены на пептидные рецепторы [3, 4]. Эти соединения обычно больше подходят для пероральной доставки и легче в производстве, чем пептиды. Однако такой подход никак не вытеснил пептидные соединения, их использование в медицине только расширяется. Несомненно, важнейший фактор прогресса в сфере лекарственных средств пептидной природы – совершенствование возможностей производства пептидов, которое связано с внедрением автоматизированного синтеза на твердофазных носителях и генно-инженерных технологий синтеза.
КЛАССИЧЕСКИЕ ГОРМОНАЛЬНЫЕ ПЕПТИДНЫЕ ПРЕПАРАТЫ
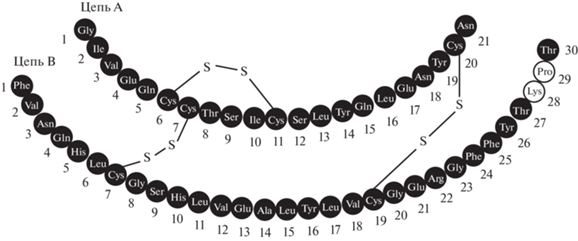
Первый пептид, на основе которого создан важнейший медицинский препарат, – инсулин, пептидный гормон, состоящий из 51 а.о. Инсулин продуцируется бета-клетками островков Лангерганса (образованиями, находящимися в поджелудочной железе) и регулирует биосинтез и потребление глюкозы клетками организма. Этот гормон был впервые выделен из поджелудочной железы свиньи 100 лет назад Banting et al. [5]. Очень скоро после подтверждения способности выделенного из животных инсулина регулировать потребление глюкозы был создан препарат на основе очищенного животного инсулина, хотя его структура оставалось неизвестной еще 30 лет. Только в 1954 г. первичную структуру инсулина (рис. 1) установили Sanger et al. [6, 7]. Инсулин состоит из двух полипептидных цепей, соединенных двумя дисульфидными мостиками, третий мостик находится в А-цепи, между Cys6 и Cys11. Вообще инсулин выступает хорошим маркером развития биохимии, не случайно многие вехи его исследований отмечены нобелевскими премиями. В 1923 г. за выделение инсулина премию в области физиологии и медицины получили F.G. Banting и J.J.R. Macleod, за установление первичной структуры инсулина F. Sanger получил премию в 1958 г., D. Hodgkin в 1964 г. получила премию за развитие кристаллографических исследований и установление пространственной структуры инсулина [8].
По оценкам аналитической компании MarketsandMarkets, мировой рынок препаратов человеческого инсулина в 2018 г. составил ~25 млрд долларов США с тенденцией постоянного роста [9]. Сегодня три крупнейших производителя инсулина – Eli Lilly, Novo Nordisk и Sanofi – контролируют 96% объема мирового рынка. В настоящее время большинство препаратов инсулина производится на основе генно-инженерных технологий, с использованием рекомбинантных плазмид, несущих синтетический ген проинсулина, которые внедряют в бактериальные или дрожжевые штаммы-продуценты. Экспрессированный бактериями или дрожжами проинсулин путем ферментативного гидролиза превращают в инсулин. На основе гормона, выделенного из поджелудочных желез животных, производятся препараты так называемого полусинтетического инсулина, аминокислотная последовательность которых изменена и соответствует инсулину человека. Наряду с инсулином природной последовательности созданы его структурные аналоги с различной длительностью действия и препараты на их основе, которые широко представлены на фармацевтическом рынке [10]. Значение исследований, связанных с инсулином, трудно переоценить. Полагают, что именно инсулин открыл новый этап целенаправленной заместительной терапии, введение инсулина ознаменовало конец так называемой “преинсулиновой эры”, открыв дорогу новым клиническим подходам [11].
В табл. 1 приведены важнейшие гормональные пептидные препараты, созданные в основном в рамках “классических” подходов [2].
Таблица 1.
Источник или химическая природа первых пептидных препаратов [2]
| Пептид | Источник получения | Год введения в клинику |
Природа структуры |
|---|---|---|---|
| Инсулин | Получен из поджелудочных желез крупного рогатого скота | 1920-е | Нативный гормон |
| Адренокортикотропный гормон (ACTH) |
Получен из гипофизов крупного рогатого скота и свиней |
1950-е | Нативный гормон |
| Кальцитонин | Получен из щитовидных желез лососей | 1971 | Нативный гормон |
| Окситоцин | Синтетический | 1962 | Нативный гормон |
| Вазопрессин | Синтетический | 1962 | Нативный гормон |
| Октреотид | Синтетический аналог соматостатина | 1988 | Циклический октапептидный аналог соматостатина-14 |
| Лейпрорелин | Синтетический аналог гонадорелина | 1984 | Нонапептидный аналог декапептида гонадорелина |
В результате изучения взаимосвязи гормональной и нервной систем, регуляции центральной нервной системой синтеза и выделения гормонов аденогипофиза были выделены и введены в клиническую практику несколько препаратов. Первым из них был адренокортикотропный гормон (ACTH) – 39-членный пептид с аминокислотной последовательностью Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-Gly-Lys-Lys-Arg-Arg-Pro-Val-Lys-Val-Tyr-Pro-Asn-Gly-Ala-Glu-Asp-Glu-Ser-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe, стимулирующий секрецию глюкокортикоидных гормонов из коры надпочечников. Он выделен из гипофиза крупного рогатого скота в 1942 г. [12], препарат на его основе создан в 1950-е гг. [2, 13]. В 1960 гг. был синтезирован его биологически активный 24-членный фрагмент ACTH1–24. И природный, и синтетический гормоны применяют при гормональных нарушениях, связанных с недостаточной выработкой ACTH и дисфункцией коры надпочечников (болезнь Аддисона, синдром Кушинга), при лечении ревматизма, полиартритов, подагры, бронхиальной астмы, экземы и других заболеваний [14], а также как компонент диагностических наборов при определении содержания ACTH методами иммуноферментного анализа. Уровень ACTH – маркер не только гормональных нарушений, но и гормон-зависимых онкологических заболеваний. На основе структуры ACTH созданы короткие пептиды со спектром действия, кардинально отличающимся от исходного гормона. В первую очередь следует отметить Семакс (Met-Glu-His-Phe-Pro-Leu-Pro) – российский препарат, внедренный в клиническую практику в 1997 г. [15]. Он создан на основе фрагмента ACTH4–10 и содержит в С-концевой области фрагмент с двумя остатками пролина, который делает пептид устойчивым к энзиматическому расщеплению и в десятки раз повышает его ноотропную активность по сравнению с природным фрагментом ACTH4–10. Препарат полностью лишен гормональной активности, но обладает широким спектром ноотропных и адаптивных свойств: применяется при лечении инсульта, транзиторной ишемической атаки, нарушений памяти и когнитивных функций, язвы желудка, заболеваний зрительного нерва, а также для укрепления иммунной системы [16, 17].
В начале 1960-х гг. был выделен в чистом виде 32-членный пептидный гормон кальцитонин, секретируемый парафолликулярными клетками щитовидной железы человека и других животных [18]. Он понижает содержание кальция в крови, оказывая эффект, противоположный действию паратиреоидного гормона. Первичная структура гормона – Cys-Gly-Asn-Leu-Ser-Thr-Cys-Met-Leu-Gly-Thr-Tyr-Thr-Gln-Asp-Phe-Asn-Lys-Phe-His-Thr-Phe-Pro-Gln-Thr-Ala-Ile-Gly-Val-Gly-Ala-Pro(S–S1–7), он содержит 7-членный цикл, образованный за счет дисульфидной связи между Cys1 и Cys7. Впервые препарат кальцитонина был создан на основе гормона из щитовидных желез лосося Cys-Ser-Asn-Leu-Ser-Thr-Cys-Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Thr-Gly-Ser-Gly-Thr-Pro(S–S1–7). Несмотря на существенную разницу в структуре (отличаются 16 а.о. из 32), кальцитонин лосося более активен в регуляции кальциевого обмена у людей, чем кальцитонин человека.
В настоящее время в качестве субстанции препарата используется только синтетический кальцитонин, который получают с помощью технологии рекомбинантной ДНК или путем твердофазного синтеза. К препаратам кальцитонинового ряда относится элькатонин – аналог кальцитонина угря, который содержит в 7-членном цикле стабильную связь C–N вместо S–S [19]. Препараты кальцитонина используются при гиперкальциемии и остеопорозе. Они подавляют абсорбцию и автолиз костей и, таким образом, приводят к снижению содержания кальция в крови, препятствуют растворению и переносу солей костей и способствуют выведению кальция и фосфора с мочой, подавляют реабсорбцию кальция, фосфора и натрия в почечных канальцах и поддерживают нормальный уровень кальция в крови.
Традиционный подход к разработке пептидных препаратов был продолжен исследованиями окситоцина и вазопрессина – нейрогипофизарными пептидными гормонами, продуцируемыми в гипоталамусе и секретируемыми в нейрогипофиз и далее в кровоток. Du Vigneaud et al. впервые выделили эти соединения из задней доли гипофиза и установили структуру обоих гормонов [20, 21]. Окситоцин Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly-NH2(S–S1–6) и вазопрессин Cys-Tyr-Phe-Gln-Asn-Cys-Pro-Arg-Gly-NH2(S–S1–6) – 9‑членные пептиды, содержащие 6-членный цикл, образованный дисульфидной связью между Cys1 и Cys6, содержание их в тканях мозга незначительно, поэтому препаративное выделение совершенно нерентабельно. Эти обстоятельства обусловили тот факт, что окситоцин стал первым синтетически полученным пептидным гормоном, первой синтетической пептидной субстанцией лекарственного препарата. Химический синтез окситоцина и вазопрессина осуществили Du Vigneaud et al. [22, 23] в 1953 г., медицинский препарат окситоцина введен в клиническую практику в 1962 г. С тех пор и по настоящее время окситоцин как стимулятор сокращения гладкой мускулатуры – незаменимый препарат при родовспоможении в гинекологии и ветеринарии. Вазопрессин известен как гормон, регулирующий водно-солевой обмен и кровяное давление. На основе субстанции вазопрессина также были созданы медицинские препараты, они используются в основном при лечении несахарного диабета.
В наши дни установлено, что роль вазопрессина и окситоцина не ограничивается гормональной регуляцией, эти пептиды – эволюционировавшие компоненты интегрированной адаптивной системы “окситоцин–вазопрессин”. Более древний пептид вазопрессин поддерживает индивидуальное выживание и играет роль в защитном поведении, включая мобилизацию и агрессию. Окситоцин ассоциируется с позитивным социальным поведением и может выступать в качестве биологической метафоры социальной привязанности или любви [24]. Также обнаружено влияние вазопрессина на биологические ритмы человека – он принимает непосредственное участие в регуляции циркадианных часов супрахиазматического ядра гипоталамуса [25].
На новый уровень исследования вазопрессина, окситоцина, их агонистов и антагонистов вышли после обнаружения и установления структуры их рецепторов. Как практически все пептидные гормоны, окситоцин и вазопрессин опосредуют свои биологические эффекты, как гормональные, так и адаптивные, воздействуя на специфические рецепторы. Известны три типа рецепторов вазопрессина (V1aR, V1bR, V2R) и один тип для окситоцина (OTR) [26, 27]. Они относятся к семейству рецепторов, сопряженных с G-белками, и обнаружены во многих органах и тканях, их широкая распространенность и вариабельность объясняет мультифункциональность нейрогипофизарных гормонов [28, 29]. Позднее были выявлены еще два типа атипичных рецепторов, обладающих аффинностью к аргинин-вазопрессину и опосредующих внутриклеточные ответы на него: ангиотензин-вазопрессин (AngII/AVP) и рецептор мобилизации кальция 1 (VACM-1) [30, 31]. Интерес представляет тот факт, что оба указанных рецептора имеют последовательности, отличные от семейства рецепторов вазопрессина, но их расположение сходно с таковым для V1aR и V1bR [30]. Кроме того, установлено, что AngII/AVP имеет высокое сродство к антагонистам V1-рецепторов, а VACM-1 способен различать вазопрессин и окситоцин [30–32]. Обнаружение и установление структуры рецепторов предоставило возможность нового подхода к дизайну аналогов нейрогипофизарных гормонов и оценки ранее синтезированных аналогов. К настоящему времени синтезировано несколько тысяч аналогов вазопрессина и окситоцина – агонистов и антагонистов, пептидной и непептидной природы [33, 34], многих отличает высокая эффективность и селективность действия, однако, по данным лаборатории Маннига, лидера по исследованиям в этой области, разрешены к применению в США только семь пептидов и два непептидных агониста (данные на 2012 г., табл. 2) [34].
Таблица 2.
Разрешенные к применению в США препараты вазопрессинового и окситоцинового рядов (2012 г.) [34]
| Пептидные препараты | Препараты непептидной природы |
|---|---|
| Карбетоцин (Депотоцин, Пабал) | Кониваптана гидрохлорид (Валпризол) |
| Десмопрессин (Минирин, DDAVP) | Толваптан |
| Орнитин-вазопресcин (Орнипрессин) | |
| Липрессин | |
| Окситоцин | |
| Терлипрессин (Глипрессин) | |
| Вазопрессин |
В России разрешены к применению препараты вазопрессина природной структуры (используются в основном для лечения несахарного диабета) и препараты на основе десмопрессина – структурного аналога вазопрессина, у которого дезаминирован остаток Cys1 и остаток L-Arg в восьмом положении заменен на D-Arg. Такие модификации существенно повысили энзиматическую устойчивость пептида и изменили рецепторную специфичность – десмопрессин избирательно активирует V2-рецепторы и не влияет на V1-рецепторы гладкомышечных клеток сосудов и внутренних органов, вследствие чего отношение антидиуретической активности к прессорной у такого пептида составляет ~2000 : 1 против 1 : 1 у аргинин-вазопрессина [35]. Энзиматическая стабильность десмопрессина позволяет применять его как в виде таблетированной формы, так и в качестве интраназального спрея.
Инъекционные препараты окситоцина природной структуры и его дезаминированного аналога – дезаминоокситоцина – находят широкое применение в гинекологии, разрешен к применению в России и интраназальный спрей окситоцина [35]. В настоящее время исследования в области пептидов вазопрессинового и окситоцинового ряда направлены в основном на создание соединений, лишенных гормональной активности, но селективно действующих на различные функции ЦНС. Пептиды, структурно родственные вазопрессину и окситоцину, исследуются как потенциальные средства лечения аутизма, тревожных расстройств, как стимуляторы памяти и обучаемости, адаптивные препараты в хирургии [36–38]. Предполагается, что активация системы вазопрессина может играть важную роль для профилактики и лечения когнитивных расстройств при болезни Альцгеймера [39]. Также системы вазопрессина и окситоцина представляют интерес как потенциальные мишени для лечения алкоголизма [40].
ГИПОТАЛАМИЧЕСКИЕ РИЛИЗИНГ-ГОРМОНЫ И ИХ АНАЛОГИ
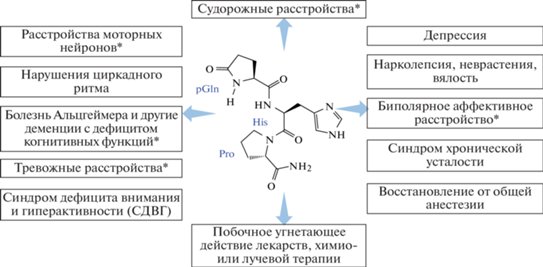
Новый этап возможностей медицинского применения пептидных гормонов начался в конце 1960-х – начале 1970-х гг. и связан выделением и установлением химической структуры гипоталамических регуляторов биосинтеза и секреции гормонов аденогипофиза. Эти соединения осуществляют связь между ЦНС и эндокринной системой и занимают ключевое положение в системе “гипоталамус–гипофиз–периферические эндокринные железы”. Структура первого из гипоталамических гормонов, тиролиберина или тиротропин-рилизинг-гормона (TRH) – трипептида pGlu-His-Pro-NH2, была установлена в 1969 г. двумя группами исследователей [41, 42]. Он стимулирует биосинтез и выделение аденотропных гормонов тиротропина и пролактина, играя ключевую роль в поддержании гомеостаза щитовидной железы; наряду с гормональным действием обладает ярко выраженным нейротропным эффектом, который проявляется в активации ряда функций ЦНС [43–47]. Тиролиберин оказывает противосудорожное, антикаталептическое и антигипотермическое действие, выступает антагонистом многих веществ с угнетающим действием на ЦНС. Он устраняет эффекты резерпина и хлорпромазина, галоперидола; обладает аналептическими свойствами, блокируя действие этанола, барбитуратов, некоторые реакции морфина и опиоидных пептидов. Анализ биологических исследований тиролиберина и его аналогов позволил выделить более десяти возможных направлений их использования в качестве активаторов функций ЦНС (рис. 2) [44].
Рис. 2.
Возможности клинического использования TRH и его аналогов [43]. * Могут быть особенно эффективны в качестве дополнительной терапии.
Препараты на основе нативной структуры тиролиберина были созданы и присутствуют на фармацевтическом рынке, но широкий спектр активности этого соединения, быстрая биодеградация и низкая биодоступность ограничивают его применение, в основном тиролиберин используется в диагностике тиреоидной функции. Введение TRH используют для оценки способности гипофиза выделять тиреотропный гормон (TSH), в этом тесте определяется уровень TSH до и после стимуляции тиролиберином. Тесты прямого определения TRH иммуноферментными методами используются реже. К настоящему времени синтезировано несколько сотен аналогов тиролиберина с различными профилями действия [43, 48–51], активные исследования новых синтетических аналогов TRH ведутся до сих пор [52, 53], однако успешно пройти все три фазы клинических испытаний мало кому из них удалось – в качестве лекарственного средства используется только талтирелин, разработанный в Японии [50]. Этот аналог тиролиберина с заменой Glp на остаток 2,4-диоксо-3-метилпиримидиновой кислоты используется при лечении различных нейродегенеративных расстройств [54], особенно перспективным представляется его способность повышать уровень дофамина при болезни Паркинсона [55].
В 1971 г. была установлена структура еще одного рилизинг-гормона – десятичленного пептида люлиберина, рилизинг-гормона гонадотропных гормонов (GnRH): pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2 [56, 57]. Этот гормон – регулятор репродуктивной сферы, стимулирует выделение и биосинтез лютеинизирующего и фолликулостимулирующего гормонов. В 1970-х гг. наблюдался расцвет пептидного синтеза – наряду с классическим пептидным синтезом в растворе, многостадийным, растянутым во времени, был введен в практику твердофазный пептидный синтез, который позволил автоматизировать и ускорить процесс получения пептидов, снять ограничения по длине пептидной цепи. С использованием и классического и твердофазного методов синтеза было синтезировано несколько тысяч аналогов люлиберина – агонистов и антагонистов – уже к середине 1980-х гг. [58, 59]. Были созданы суперактивные агонисты, превышающие по активности гормон-прародитель в сотни раз. В этих модифицированных пептидах остаток глицина в 6-м положении заменен на остаток D-аминокислоты, у некоторых соединений дополнительно С-концевой глициламид замещен на этиламид. На основе суперактивных агонистов были созданы препараты, стимулирующие овуляцию, позволяющие получать яйцеклетки, которые необходимы для процедур экстракорпорального оплодотворения. К ним относятся трипторелин (pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Pro-Gly-NH2), содержащий остаток D-триптофана в 6-м положении, лейпролид (H-Pyr-His-Trp-Ser-Tyr-D-Leu- Leu-Arg-Pro-NHEt), бусерилин (D-Ser(tBu)6EA10-LHRH) и другие. Лейпролид и другие суперагонисты GnRH используются также при лечении гормон-зависимых опухолей – рака простаты, молочных желез, миомы матки, эндометриоза. Лечение опухолей и гормональных нарушений основано на парадоксальном ингибирующем эффекте суперагонистов [60, 61]. Они вызывают начальную интенсивную стимуляцию гипофиза, но затем (при наличии постоянной концентрации препарата в крови) оказывают ингибирующее влияние. Ингибирование происходит в результате того, что суперагонисты GnRH устойчивы к деградации энзимами гипофиза, поэтому блокируют гонадотропные рецепторы в гипофизе и делают их невосприимчивыми после инициальной стимуляции, т.е. происходит десенсибилизация рецепторов. Это приводит к сильному снижению в крови уровня гонадотропинов и, соответственно, половых гормонов, происходит гормональная блокада. Ингибирующие эффекты агонистов полностью обратимы.
Структура антагонистов GnRH отличается от природной молекулы в большей степени, чем у агонистов, часто они содержат остатки неприродных аминокислот, у всех антагонистов изменена структура N-концевой области. Сохраняя способность связываться с рецепторами GnRH, они выступают в качестве конкурентов природного гормона, занимая места связывания, но генерации клеточного сигнала и выделения лютеинизирующего и фолликулостимулирующего гормонов при этом не происходит, действие природного GnRH блокируется. Антагонисты стабилизируют рецепторы в их неактивной конформации; в отличие от суперагонистов, фаза стимуляции отсутствует. С учетом их высокой резистентности к действию пептидаз, эффект отличается длительностью. На основе суперактивных антагонистов также были созданы эффективные противоопухолевые препараты и регуляторы гормональных нарушений, наиболее известны дегареликс, цетрореликс, абареликс, ганиреликс и озареликс [62, 63]. Эти пептиды содержат последовательность Ac-D-Nal-D-Cpa-D-Pal в аминоконцевой области, которая включает остатки 3-(2-нафтил)аланина (Nal), 3-(2-пиридил)аланина (Pal), β-циклопропилаланина (Cpa), различные производные D-аминокислот в 6-м положении и D-Ala в С‑концевой области. В табл. 3 приведены первичные структуры основных антагонистов GnRH. И суперагонисты, и антагонисты были введены в клиническую практику в середине 1980-х гг. и широко используются в настоящее время.
Таблица 3.
Структура антагонистов GnRH [61]
| Соединение | Номер аминокислотного остатка | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| GnRH | pGlu | His | Trp | Ser | Tyr | Gly | Leu | Arg | Pro | Gly-NH2 |
| Цетрореликс (Цетротид) | Ac-D-Nal | D-Cpa | D-Pal | Ser | Tyr | D-Cit | Leu | Arg | Pro | D-Ala-NH2 |
| Ганиреликс (Оргалутран) | Ac-D-Nal | D-Cpa | D-Pal | Ser | Tyr | D-hArg (Et2) | Leu | hArg (Et2) | Pro | D-Ala-NH2 |
| Абареликс | Ac-D-Nal | D-Cpa | D-Ala | Ser | Tyr | D-Asp | Leu | Lys (iPr) | Pro | D-Ala-NH2 |
| Дегареликс (Фирмагон) | Ac-D-Nal | D-Cpa | D-Pal | Ser | Aph (Hor) | D-Aph (Cba) | Leu | Lys (iPr) | Pro | D-Ala-NH2 |
В 1970-е гг. был выделен рилизинг-ингибитор гормона роста – циклический 14-членный пептид соматостатин Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys(S–S3–14) [64]. Исследования показали, что это соединение представляет собой мультифункциональный регулятор и вырабатывается не только в гипоталамусе, но и в желудке, кишечнике, поджелудочной железе. Соматостатин ингибирует секрецию гипофизарных соматотропного и тиреотропного гормонов, кроме того, представляет собой ингибитор различных пептидов, продуцируемых в желудке, кишечнике, печени и поджелудочной железе, а также серотонина. Соматостатин тормозит секрецию инсулина, глюкагона, гастрина, холецистокинина, вазоактивного интестинального полипептида, инсулиноподобного фактора роста-1 и других. Показаниями к применению соматостатина считаются гормональные нарушения, связанные с гиперпроизводством гормона роста соматотропина, кроме того, различные заболевания желудочно-кишечного тракта (ЖКТ) и поджелудочной железы [65]. В конце 1980-х гг. были разработаны и введены в практику препараты гормона природной структуры – модустатин, стиламин.
Структурно-функциональные исследования соматостатина привели к созданию высокоактивных аналогов-агонистов – октреотида и ланреотида [65–68]. Октреотид, циклический октапептид D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-Ol(S–S2–7), имитирует природный соматостатин фармакологически, но выступает более мощным ингибитором гормона роста, глюкагона и инсулина, чем естественный гормон. Важное его преимущество – высокая устойчивость к энзиматической деградации, которая служит следствием таких структурных преобразований, как введение остатков D‑аминокислот, изменение N- и С-концевой областей. Октреотид находит широкое применение при лечении гормон-зависимых опухолей. Ланреотид Naphtyl-D-Ala-Cys-Tyr-D-Trp-Lys-Val-Cys-Thr-NH2(S–S2–7) также устойчив к деградации, но спектру действия он отличается и от соматостатина, и от октреотида. Ланреотид обладает более выраженной тропностью к периферическим соматостатиновым рецепторам (гипофизарным и панкреатическим), чем к центральным [68]. Этим обусловлена его селективность в отношении секреции соматотропина и экзокринной секреции поджелудочной железы и желез кишечника.
На основе октреотида и ланреотида создано несколько препаратов, которые используются при внушительном перечне заболеваний. В России четыре фирмы наладили производство октреотида: Эллара, Фарм-Синтез, Фармкомпания и ФармФирма Сотекс, крупнейшие мировые производители октреотида: Novartis Pharma (Швейцария), Fresenius KABI Deutschland (Германия) и Sun Pharmaceutical Industries (Индия) [69]. Объемы производства ланреотида меньше, препарат выпускается под названиями соматулин и соматулин аутожель, основной производитель – Ipsen Pharma Biotech (Франция) [70].
АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА-1
В последней четверти XX века стало очевидным, что пептидные гормоны – мультифункциональные биорегуляторы, рецепторы которых представлены в организме очень широко. Было установлено, что пептидные гормоны продуцируются не только в системе “гипоталамус–гипофиз–железы внутренней секреции”, но и в других органах и тканях. Среди этих “неклассических” гормонов важное место занимают инкретины – пептидные гормоны, секретируемые ЖКТ [71]. Эти соединения, регулирующие углеводный обмен и ряд функций ЖКТ, стали исходным материалом для разработки новых препаратов. Среди инкретинов особое внимание исследователей привлек глюкагон-подобный пептид-1 (GLP-1) – 36-членный пептид, рецепторы которого широко распространены во многих органах и тканях. Спектр активности этого универсального биорегулятора широк: наряду со стимулированием биосинтеза и экскреции инсулина, его действие распространяются на важнейшие органы и ткани (табл. 4) [72]. Укороченные пептиды GLP-1(7–37) и GLP-1(7–36)-NH2 также обладают биологической активностью.
Таблица 4.
Воздействие GLP-1 на органы и системы организма [69]
| Орган или система организма | Эффект |
|---|---|
| Желудок | Тормозит секрецию соляной кислоты |
| Ослабляет моторную активность | |
| Поджелудочная железа | Усиливает секрецию инсулина |
| Стимулирует транскрипцию гена инсулина | |
| Ингибирует секрецию соматостатина и глюкагона | |
| Регулирует экспрессию АТФ-зависимых калиевых каналов бета-клеток | |
| Вызывает пролиферацию и неогенез бета-клеток | |
| Усиливает реакцию бета-клеток на глюкозу | |
| Сердечно-сосудистая система | Увеличивает частоту сердечных сокращений |
| Щитовидная железа | Стимулирует выведение тиреокальцитонина из железы |
| Легкие | Усиливает релаксацию легочных мышц |
| Усиливает мукозную секрецию | |
| Почки | Способствует диурезу и натрийурезу |
| Центральная нервная система | Стимулирует секрецию лютеинизирующего рилизинг-гормона из гипоталамуса |
| Стимулирует секрецию тиреотропного гормона, лютеинизирующего гормона, кортикостероидов | |
| Подавляет поступление пищи и воды |
Короткое время полураспада GLP-1, которое составляет менее 2 мин – главное препятствие для использования пептида в качестве субстанции лекарственного препарата. К настоящему времени этот недостаток устранен, разработано несколько препаратов на основе стабильных аналогов GLP-1 [73–76]. Стратегия создания этих препаратов представляет собой хороший пример использования современных подходов к созданию пептидных препаратов с целевыми свойствами и включает несколько этапов. Целью модификаций было сделать молекулу гормона устойчивой, повысить ее время полураспада, с сохранением высокого уровня активности. Был выбран путь создания аналогов, которые имеют высокое структурное соответствие клеточному рецептору GLP-1, устойчивы к действию дипептидилпептидазы-4 (DPP4) и способны обратимо связываться с альбумином, что предотвращало бы их быструю биодеградацию. С использованием такого подхода были созданы лираглутид и семаглутид [73].
Учитывая высокое сродство альбумина к гидрофобным соединениям, было принято решение прикрепить к молекуле гормона длинноцепочечную жирную кислоту. Для получения производных с жирными кислотами были определены положения, модификация которых мало влияет на конформационные особенности пептида, обеспечивающие высокое сродство к рецептору, в итоге был выбран Lys26. Для скрининга аналогов с различной структурой цепи жирной кислоты были получены производные с длиной цепи С12–С18 с карбоксилом и без него на свободном конце. В целом для выбора положения модификации, наиболее подходящих структур линкера и жирной кислоты были синтезированы и исследованы 36 соединений. В результате тестирования их связывания с рецептором GLP 1(7–37) выбрано одно наиболее активное и долгоживущее соединение – лираглутид, структура которого приведена на рис. 3.
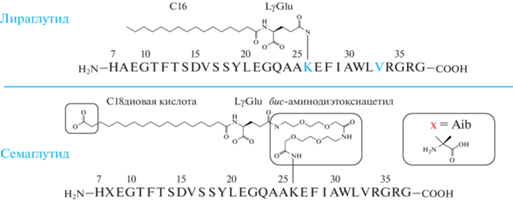
Дальнейшие изменения структуры GLP-1(7–37) состояли во введении во 2-е положение остатка аминоизомасляной кислоты, что значительно повышает устойчивость пептида к действию DPP4, не вызывая заметного снижения активности. Модификация положения Lys26 состояла в использовании бис-аминодиэтоксиацетильного линкера и введении остатка жирной диацилкислоты длиной С18 [74]. Семаглутид, полученный в результате этих модификаций, представляет собой высокоактивный и устойчивый аналог GLP-1, который вводится 1 раз в неделю. Он используется для лечения больных с диабетом второго типа, кроме того, он улучшает функционирование сердечно-сосудистой системы. Наибольший интерес вызывает способность семаглутида снижать массу тела у людей, страдающих ожирением, при этом никаких побочных последствий не отмечалось. Очевидно, из-за широкой распространенности болезней современности – диабета и ожирения – препараты-аналоги GLP-1 коммерчески очень успешны, темпы их разработки и внедрения очень высоки.
В табл. 5 представлены препараты пептидных гормонов, имеющие наибольшие объемы продаж.
Таблица 5.
Препараты пептидных гормонов, имеющие наибольшие объемы продаж в 2019 г. [1]
| Наименование пептида | Коммерческие названия самых распространенных препаратов | Производитель | Целевые заболевания для применения | Год одобрения и введения в практику | Продажи в 2019 г. (млн долларов) |
|---|---|---|---|---|---|
| Инсулин и аналоги | Ademelog, Apidra, Humulin, Humalog, Insuman, NovoMix, NovoRapid, Lantus, Levemir, Ryzodeg, Tresiba, Toujeo | Novo Nordisk, Eli Lilly, Sanofi | Диабет | 1982 | 25 000 |
| Дулаглутид | Trulicity | Eli Lilly, Dainippon Sumitomo | Диабет | 2014 | 4394 |
| Лираглутид | Victoza, Saxenda | Novo Nordisk | Диабет, ожирение | 2010 | 4142 |
| Лейпролид | Lupron, Eligard | AbbVie, Astellas, Takeda | Онкологические заболевания | 1985 | 2022 |
| Семаглутид | Ozempic, Rybelsus | Novo Nordisk | Диабет, ожирение | 2017, 2019 | 1694 |
| Октреотид | Sandostatin | Novartis | Онкологические заболевания | 1988 | 1585 |
| Терипаратид | Forteo | Eli Lilly | Остеопороз | 2002 | 1405 |
| Ланреотид | Somatuline | Ipsen | Акромегалия | 2007 | 1102 |
| ACTH | Acthar | Mallinckrodt | Множественный склероз, инфантильные спазмы | 1950 | 953 |
| Линаклотид | Linzess, Constella | Allergan, Astellas Pharma | Синдром раздраженного кишечника | 2012 | 877 |
| Госерилин | Zoladex | AstraZeneca | Онкологические заболевания | 1989 | 813 |
| Эксенатид | Buetta, Bydureon | AstraZeneca | Диабет | 2005 | 659 |
| Тедуглутид | Gattex, Revestive | Takeda | Синдром короткого кишечника |
2012 | 555 |
| Вазопрессин | Vasostrict | Endo Pharma | Несахарный диабет | 2014 | 532 |
ЗАКЛЮЧЕНИЕ
За 100 лет, прошедших со времени выделения и внедрения в медицинскую практику инсулина, пептидные гормоны, их аналоги и миметики стали важным элементом фармацевтической индустрии. Для нужд медицины созданы десятки препаратов, первое место стабильно занимают препараты инсулина, объем промышленного выпуска которых ведущими фирмами США и европейских стран составлял в 2019 г. 25 млрд долларов, около полутора десятков других пептидных лекарственных средств также имеют высокие стоимостные показатели (табл. 5) [1]. Лидирующие позиции имеют аналоги GLP-1 и GLP-2 (дулаглутид, лираглутид, семаглутид, эксенатид, тедуглутид), представлены два препарата-аналога GnRH (лейпролид, госерилин), высокие объемы продаж у терипаратида – препарата на основе рекомбинантного паратиреоидного гормона человека. Удерживают позиции и “классические” препараты ACTH и вазопрессин.
Разработка препаратов на основе пептидных гормонов – динамично развивающаяся отрасль с ясно очерченными целями, имеющая ряд направлений. По-прежнему интенсивно ведутся работы по созданию аналогов “классических” гормонов, высокоактивных, лишенных мультифункциональности, активно внедряются в медицинскую практику препараты на основе гормонов, структура которых установлена сравнительно недавно; например, в 2010 г. одобрен для медицинской практики тезаморелин – аналог рилизинг-гормона соматотропина. Установление структуры рецепторов пептидных гормонов позволяет проводить поиск их лигандов на основе структурного соответствия с использованием современных компьютерных технологий и баз данных, что приводит к созданию новых агонистов и антагонистов как пептидной, так и непептидной природы. Ведутся работы по повышению устойчивости и биодоступности пептидных препаратов, по усовершенствованию старых и разработке новых способов доставки гормонов в организм, актуальны исследования в области технологий получения пептидных гормонов и их аналогов и разработки новых “прорывных” методов синтеза. Нет сомнений, что в будущем наши знания о пептидных гормонах будут расширены и, как следствие, будут разработаны новые более совершенные препараты, которые найдут применение не только в гормональной терапии, но и в других областях медицины: онкологии, неврологии, лечении сердечно-сосудистых заболеваний, нарушений обмена веществ.
Список литературы
Muttenthaler M., King G.F, Adams D.J., Alewood P.F. // Nat. Rev. Drug Discov. 2021. V. 20. P. 309–325. https://doi.org/10.1038/s41573-020-00135-8
Lau J.L., Dunn M.K. // Bioorg. Med. Chem. 2018. V. 26. P. 2700–2707. https://doi.org/10.1016/j.bmc.2017.06.052
Fosgerau K., Hoffmann T. // Drug Discov. Today. 2014. V. 20. P. 122–128. https://doi.org/10.1016/j.drudis.2014.10.003
Filimonov D.A., Druzhilovskiy D.S., Lagunin A.A., Gloriozova T.A., Rudik A.V., Dmitriev A.V., Poroikov V.V. // Biomedical Chemistry: Research and Methods. 2018. V. 1. P. 1–21. https://doi.org/10.18097/bmcrm00004
Banting F.G., Best C.H., Collip J.B., Campbell W.R., Fletcher A.A. // Can. Med. Assoc. J. 1922. V. 12. P. 141–146.
Sanger F., Tuppy H. // Biochem. J. 1951. V. 49. P. 463–481. https://doi.org/10.1042/bj0490463
Sanger F., Thompson E.O. // Biochem. J. 1953. V. 53. P. 353–366. https://doi.org/10.1042/bj0530366
Candido R., Wyne K., Romoli E. // Diabetes Ther. 2018. V. 9. P. 927–949. https://doi.org/10.1007/s13300-018-0422-4
Infoholic Research LLP. Global Human Insulin Market 2018–2024. Research and Markets, ID: 4470733 2018. https://www.researchandmarkets.com/reports/4470733/ global-human-insulin-market-2018-2024
Hirsch I.B. // N. Engl. J. Med. 2005. V. 352. P. 174–183. https://doi.org/10.1056/NEJMra040832
Vecchio I., Tornali C., Bragazzi N.L., Martini M. // Front. Endocrinol. 2018. V. 9. P. 613. https://doi.org/10.3389/fendo.2018.00613
Li C.H., Simpson M.E., Evans H.M. // Science. 1942. V. 96. P. 450. https://doi.org/10.1126/science.96.2498.450
Elkinton J.R., Hunt A.D., Godfrey L., McCrory W.W., Rogerson A.G., Stokes J. // J. Am. Med. Assoc. 1949. V. 141. P. 1273–1279. https://doi.org/10.1001/jama.1949.02910180001001
Gallo-Payet N., Martinez A., Lacroix D. // Front. Endocrinol. 2017. V. 8. P. 101. https://doi.org/10.3389/fendo.2017.00101
Ашмарин И.П., Незавибатько В.Н., Мясоедов Н.Ф., Каменский А.А., Гривенников И.А., Пономарева-Степная М.А., Рясина Т.В. // Журнал высшей нервной деятельности. 1997. Т. 47. С. 419–425.
Левицкая Н.Г., Глазова Н.Ю., Себенцова Е.А., Манченко Д.М., Виленский Д.А., Андреева Л.А., Каменский А.А., Мясоедов Н.Ф. // Нейрохимия. 2008. Т. 25. С. 111–118.
Манченко Д.М., Глазова Н.Ю., Левицкая Н.Г., Андреева Л.А., Каменский А.А., Мясоедов Н.Ф. // Российский физиологический журнал. 2010. Т. 96. С. 1014–1023.
Copp D.H., Cheney B. // Nature. 1962. V. 193. P. 381–382. https://doi.org/10.1038/193381a0
Inzerillo A.M., Zaidi M., Huang C.L.H. // J. Pediatr. Endocrinol. Metab. 2004. V. 17. P. 931–940. https://doi.org/10.1515/JPEM.2004.17.7.931
Du Vigneaud V., Ressler C., Trippett S. // J. Biol. Chem. 1953. V. 205. P. 949–957. https://doi.org/10.1016/S0021-9258(18)49238-1
Du Vigneaud V., Lawler H.C., Popenoe E.A. // J. Am. Chem. Soc. 1953. V. 75. P. 4880–4881. https://doi.org/10.1021/ja01115a554
Du Vigneaud V., Ressler C., Swan J.M., Roberts C.W., Katsoyannis P.G. // J. Am. Chem. Soc. 1954. V. 76. P. 3115–3121. https://doi.org/10.1021/ja01641a004
Du Vigneaud V., Gish D.T., Katsoyannis P.G. // J. Am. Chem. Soc. 1954. V. 76. P. 4751–4752. https://doi.org/10.1021/ja01647a089
Carter C.S. // Front. Endocrinol. 2017. V. 22. P. 356. https://doi.org/10.3389/fendo.2017.00356
Ткачева М.А., Инюшкина Е.М., Карян С.Д., Инюшкин А.Н. // Ульян. мед.-биол. журнал. 2018. № 1. С. 145–158.
Gimpl G., Fahrenholz F. // Physiol. Rev. 2001. V. 81. P. 629–683. https://doi.org/10.1152/physrev.2001.81.2.629
Hoyle C.H. // Brain Res. 1999. V. 848. P. 1–25. https://doi.org/10.1016/S0006-8993(99)01975-7
Neumann I.D., Landgraf R. // Trends Neurosci. 2012. V. 35. P. 649–659. https://doi.org/10.1016/j.tins.2012.08.004
Manning M., Stoev S., Chini B., Durroux T., Mouillac B., Guillon G. // Prog. Brain Res. 2008.V. 170. P. 473–512. https://doi.org/10.1016/S0079-6123(08)00437-8
Hurbin A., Orcel H., Ferraz C., Moos F.C., Rabie A. // J. Neuroendocrinol. V. 12. P. 677–684. https://doi.org/10.1046/j.1365-2826.2000.00499.x
Herrera V.L.M., Bagamasbad P., Decano J.L., Ruiz-Opazo N. // Physiol. Genomics. 2011. V. 43. P. 32–42. https://doi.org/10.1152/physiolgenomics.00154.2010
Burnatowska-Hledin M., Lazdins I.B., Listenberger L., Zhao P., Sharangpani A., Folta V. // Am. J. Physiol. Renal. Physiol. 1999. V. 276. P. 199–209. https://doi.org/10.1152/ajprenal.1999.276.2.F199
Manning M., Stoev S., Bankowski K. // J. Peptide Sci. 2010. V. 16. P. 366–367.
Manning M., Misicka A., Olma A., Bankowski K., Stoev S., Chini B., Durroux T., Mouillac B., Corbani M., Guillon G. // J. Neuroendocrinol. 2012. V. 24. P. 609–628. https://doi.org/10.1111/j.1365-2826.2012.02303.x
Бирюкова Е.В. // Ожирение и метаболизм. 2017. Т. 14. № 4. С. 23–30. https://doi.org/10.14341/OMET2017423-30
Hammock E.A. // Neuropsychopharmacol. 2015. V. 40. P. 24–42. https://doi.org/10.1038/npp.2014.120
Belyakova F.S., Sinjushin A.A., Voskresenskaya O.G., Kamensky A.A., Golubovich V.P. // Neurochem. J. 2015. V. 9. P. 201–205. https://doi.org/10.1134/S1819712415030034
Altura B.M., Gebrewold A., Carella A., Altura B.T. // Int. J. Surg. Res. 2017. V. 4. P. 1–3. https://doi.org/10.19070/2379-156X-170002e
Pan Y.F., Jia X.T., Wang X.H., Chen X.R., Li Q.S., Gao X.P., Qi J.S. // Regul. Pept. 2013. V. 183. P. 7–12. https://doi.org/10.1016/j.regpep.2013.03.003
Harper K.M., Knapp D.J., Criswell H.E., Breese G.R. // Psychopharmacology (Berl). 2018. V. 235. P. 3363–3379. https://doi.org/10.1007/s00213-018-5099-x
Burgus R., Dunn T.F., Desidero D., Guillemin R. // C.R. Hebd. Seances Acad. Sci., Ser. D, Sci. Nat. (in French). 1969. V. 269. P. 1870–1873.
Boler J., Enzmann F., Folkers K., Bowser C.Y., Schally A.V. // Biochem. Biophys. Res. Commun. 1969. V. 37. P. 705–710. https://doi.org/10.1016/0006-291X(69)90868-7
Monga V., Meena C.L., Kaur N., Jain R. // Curr. Med. Chem. 2008. V. 15. P. 2718–2733.
Jantas D. // Adv. Cell Biol. 2010. V. 2. P. 139–154.
Pierpaoli W. // Curr. Aging Sci. 2013. V. 6. P. 92–98.
Yarbrough G.G. // Trends Pharmacol. Sci. 2003. V. 24. P. 617–618. https://doi.org/10.1016/j.tips.2003.10.001
Prokai L. // Prog. Drug Res. 2002. V. 59. P. 134–169. https://doi.org/10.1021/jm1012984
Faden A.I., Saksen I., Noble L.J. // Brain Res. 1988. V. 448. P. 287–293. https://doi.org/10.1016/0006-8993(88)91265-6
Kaur N., Monga V., Lu X., Gershengorn M.C., Jain R. // Bioorg. Med. Chem. 2007. V. 15. P. 433–443. https://doi.org/10.1016/j.bmc.2006.09.045
Yamamura M., Kinoshita K., Nakagawa H., Tanaka T., Maeda K., Ishida R. // Jpn. J. Pharmacol. 1990. V. 53. P. 451–461. https://doi.org/10.1254/jjp.53.451
Khomane K.S., Meena C.L., Jain R., Bansal A.K. // Expert Opin. Ther. Pat. 2011. V. 21. P. 1673–1691. https://doi.org/10.1517/13543776.2011.623127
Meena C.L., Thakur A., Nandekar P.P., Sangamwar A.T., Sharma S.S., Jain R. // Bioorg. Med. Chem. 2015. V. 23. P. 5641–5653. https://doi.org/10.1016/j.bmc.2015.07.022
Kobayashi K., Abe Y., Kawai A., Furihata T., Endo T., Takeda H. // Clin. Pharmacol. 2020. V. 60. P. 1314–1323. https://doi.org/10.1002/jcph.1628
Kinoshita K., Yamamura M., Sugihara J., Suzuki M., Matsuoka Y. // CNS Drug Rev. 1998. V. 4. P. 25–41. https://doi.org/10.1111/j.1527-3458.1998.tb00039.x
Zheng C., Chen G., Tan Y., Zeng W., Peng Q., Wang J., Cheng C., Yang X., Nie S., Xu Y., Zhang Z., Papa S.M., Ye K., Cao X. // Front. Cell. Neurosci. 2018. V. 12. P. 1–16. https://doi.org/10.3389/fncel.2018.00417
Matsuo H., Baba Y., Nair R.M.G., Arimura A., Schally A.V. // Biochem. Biophys. Res. Commun. 1971. V. 43. P. 1334–1341. https://doi.org/10.1016/S0006-291X(71)80019-0
Burgus R., Butcher M., Amoss M., Ling N., Monahan M.W., Rivier J., Fellows R., Blackwell R., Vale W., Guillemin R. // Proc. Natl. Acad. Sci. USA. 1972. V. 69. P. 278–284. https://doi.org/10.1073/pnas.69.1.278
Karter M.J., Rivier J.E. // Endocrine Rev. 1986. V. 7. P. 44–66. https://doi.org/10.1210/edrv-7-1-44
Schally A.V. // Gynecol. Endocrinol. 1999. V. 13. P. 401–409. https://doi.org/10.3109/09513599909167587
Walker L.M., Tran S., Robinson J.W. // Clin. Genitourin. Cancer. 2013. V. 11. P. 375–384. https://doi.org/10.1016/j.clgc.2013.05.004
Schultze-Mosgau A., Griesinger G., Altgassen C., von Otte S., Hornug D., Diedrich K. // Expert Opin. Investig. Drugs. 2005. V. 14. P. 1085–1097. https://doi.org/10.1517/13543784.14.9.1085
Van Poppel H., Klotz L. // Int. J. Urol. 2012. V. 19. P. 594–601. https://doi.org/10.1111/j.1442-2042.2012.02997.x
Kumar P., Sharma A. // J. Hum. Reprod. Sci. 2014. V. 7. P. 170–174. https://doi.org/10.4103/0974-1208.142476
Brazeau P., Vale W., Burgus R., Ling N., Butcher M., Rivier J., Guillemin R. // Science. 1973. V. 179. P. 77–79. https://doi.org/10.1126/science.179.4068.77
Rai U., Thrimawithana T.R., Valery C., Young S.A. // Pharmacol. Ther. 2015. V. 152. P. 98–110. https://doi.org/10.1016/j.pharmthera.2015.05.007
Grozinsky-Glasberg S., Shimon I., Korbonits M., Grossman A.B. // Endocr. Relat. Cancer. 2008. V. 15. P. 701–720. https://doi.org/10.1677/ERC-07-0288
Gomes-Porras M., Jersy Cárdenas-Salas J., Álvarez-Escolá C. // Int. J. Mol. Sci. 2020. V. 21. P. 1682–1689. https://doi.org/10.3390/ijms21051682
Günther T., Tulipano G., Dournaud P., Bousquet C., Csaba Z., Kreienkamp H.J., Lupp A., Korbonits M., Castaño J.P., Wester H.J., Culler M., Melmed S., Schulz S. // Pharmacol. Rev. 2018. V. 70. P. 763–835. https://doi.org/10.1124/pr.117.015388
Debnath D., Cheriyath P. // StatPearls. 2021. https://www.ncbi.nlm.nih.gov/books/NBK544333/
Lanreotide. https://go.drugbank.com/drugs/DB06791
Nauck M.A., Meier J.J. // Diabetes Obes. Metab. 2018. V. 20. P. 5–21. https://doi.org/10.1111/dom.13129
Аметов А.С. // Русский медицинский журнал. 2006. Т. 14. № 26. С. 11–16. https://doi.org/10.14341/2072-0351-5860
Knudsen L.B., Lau J. // Front. Endocrinol. 2019. V. 10. P. 155. https://doi.org/10.3389/fendo.2019.00155
Lau J., Bloch P., Schäffer L., Pettersson I., Spetzler J., Kofoed J., Madsen K., Knudsen L.B., McGuire J., Steensgaard B.D., Strauss H.M., Gram D., Knudsen S.M., Nielsen F.S., Thygesen P., Reedtz-Runge S., Kruse T. // J. Med. Chem. 2015. V. 58. P. 7370–7380. https://doi.org/10.1021/acs.jmedchem.5b00726
St Onge E.L., Miller S.A. // Expert. Opin. Biol. Ther. 2010. V. 10. P. 801–806. https://doi.org/10.1517/14712598.2010.481281
Rosenstock J., Balas B., Charbonnel B., Bolli G.B., Boldrin M., Ratner R., Balena R. // Diabetes Care. 2013. V. 36. P. 498–504. https://doi.org/0.2337/dc12-0709
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия