

Журнал аналитической химии, 2019, T. 74, № 6, стр. 455-463
Методика контроля остаточного содержания хлорамфеникола (левомицетина) в пищевой продукции животного происхождения
Е. И. Полянских 1, А. Г. Полоневич 1, *, Л. Л. Белышева 1, Е. М. Рахманько 2, С. М. Лещев 2
1 Республиканское унитарное предприятие “Научно-практический центр гигиены”
220012 Минск, ул. Академическая, 8, Республика Беларусь
2 Белорусский государственный университет, химический факультет
220050 Минск, ул. Ленинградская, 14, Республика Беларусь
* E-mail: gannapalanevich@gmail.com
Поступила в редакцию 29.12.2017
После доработки 19.06.2018
Принята к публикации 15.10.2018
Аннотация
Разработана унифицированная методика определения хлорамфеникола методом ВЭЖХ с тандемным масс-спектрометрическим детектированием в различных группах пищевой продукции животного происхождения с пределом определения 0.2 мкг/кг. Изучена экстракция хлорамфеникола из водных растворов органическими растворителями. Оптимизированные процедуры извлечения хлорамфеникола, очистки получаемых экстрактов и концентрирования аналита, а также применение дейтерированной формы хлорамфеникола позволили проводить количественное определение с помощью внешней градуировки методом внутреннего стандарта с максимальной расширенной неопределенностью получаемых результатов 18.4%.
Принадлежащий к классу ароматических соединений антибиотик хлорамфеникол (C11H12Cl2N2O5; 323.1 г/моль, схема) обладает широким спектром лечебного действия, однако его прием может также негативно влиять на здоровье человека. Особенностью хлорамфеникола является его способность вызывать апластическую анемию – редкое, но крайне тяжелое заболевание с возможным летальным исходом. Поскольку развитие апластической анемии не зависит от количества принятого препарата и может проявиться после прекращения курса лечения, в медицинской практике данный антибиотик, как правило, заменяют аналогами, не вызывающими таких тяжелых осложнений, а в ветеринарной практике многих стран его применение ограничено [1, 2]. Так, на территории стран Европейского союза использование хлорамфеникола для лечения животных не разрешено и установлен минимальный требуемый уровень представления результатов (MRPL) 0.3 мкг/кг для продовольственного сырья животного происхождения (мяса, яиц, молока, аквакультурных продуктов и меда) [3]. В Республике Беларусь данный антибиотик также запрещен к применению в ветеринарии и наличие его в сырье и в готовой продукции животного происхождения не допускается – не более 0.3 мкг/кг [4 ] . На территории Таможенного союза остаточное содержание хлорамфеникола в пищевых продуктах животного происхождения не должно превышать 10 мкг/кг [5], в молоке, мясе и в продуктах на их основе, предназначенных для детского питания, – 0.3 мкг/кг [6, 7].

Структурная формула хлорамфеникола.
В настоящее время имеется ряд официальных методик контроля содержания хлорамфеникола, в основе которых лежат микробиологические [8, 9], иммуноферментные методы [10], ВЭЖХ с различными типами детектирования [10–13]. Простые и дешевые микробиологические методы определения [8, 9] недостаточно специфичны, распространяются только на сырье (молоко и мясо) и имеют пределы определения выше установленных нормативов. Для скринингового контроля содержания хлорамфеникола в продовольственном сырье широко используют иммуноферментный метод анализа (ИФА), который позволяет за короткий срок проанализировать большое число образцов и характеризуется высокими специфичностью и точностью, низкими пределами определения. Так, предел определения тест-системы Ридаскрин Левомицетин в зависимости от пищевой матрицы (яйца, молоко, мясо) составляет 0.0375–0.15 мкг/кг [10]. Следует отметить, что полученные методом ИФА положительные результаты должны быть подтверждены арбитражными методами.
Методики [10–12] основаны на определении хлорамфеникола методом ВЭЖХ со спектрофотометрическим детектированием при заданной длине волны. Однако данный метод может считаться арбитражным только в случае одновременного использования двух разных хроматографических систем или второго, отличного от спектрофотометрического, способа детектирования [14]. Каждый из стандартов [10–12] распространяется лишь на определенную группу пищевых продуктов: на молоко, мясо и яйца [10], на все виды мяса и мясопродуктов [11], на молоко и молочную продукцию [12]. Пределы определения методик [10] и [11] не соответствуют действующему нормированию: 10 [10] и 6.5 [11] мкг/кг. Кроме того, описанная в данных стандартах процедура пробоподготовки длительна и трудоемка, что неприемлемо для рутинного контроля. В методике [12] заявлен предел определения 0.1 мкг/кг, свойственный методу ВЭЖХ с тандемной масс-спектрометрией (ВЭЖХ–МС/МС), однако границы относительной погрешности в диапазоне 0.1–1.0 мкг/кг составляют ±60%.
Методика одновременного определения остаточного содержания различных групп антибиотиков методом ВЭЖХ–МС/МС [13] относится к арбитражным методам контроля [14], распространяется на широкий круг пищевых матриц, имеет достаточный предел количественного определения хлорамфеникола (0.2 мкг/кг), но характеризуется высоким относительным значением расширенной неопределенности в диапазоне от 0.2 до 1.0 мкг/кг – 93%. Кроме того, предложенный в данном документе способ приготовления матричных градуировочных растворов путем добавления аликвот стандартных растворов хлорамфеникола и дейтерированного хлорамфеникола к сухому остатку “чистых” проб на заключительной стадии анализа не позволяет компенсировать потери аналита в процессе пробоподготовки.
Целью данной работы явилась разработка методики определения остаточного содержания хлорамфеникола методом ВЭЖХ–МС/МС во всех группах пищевых продуктов, производимых на основе животноводческого сырья, соответствующей требованиям [3–7 ] и лишенной недостатков, свойственных существующим официальным методикам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы, материалы, оборудование. В качестве стандартных образцов использовали хлорамфеникол (99.9%, Sigma-Aldrich, Германия) и хлорамфеникол-D5 (99.8%, Sigma-Aldrich, Швейцария). Использовали ацетонитрил и метанол для ВЭЖХ (≥99.9%, Sigma-Aldrich, Германия); н-гексан и ацетон для ВЭЖХ (≥99.9%, Panreac, Испания); этилацетат для ВЭЖХ (≥99.9%, Fisher Chemical, Германия); хлороформ х. ч. и н-бутанол ч. д. а. (ЗАО “База № 1 Химреактивов”, Россия); муравьиную кислоту (98%, Acros Organics, Бельгия); формиат аммония (99%, Acros Organics, Бельгия); ацетат аммония (≥97%, Carl Roth, Германия); аммиак водный ос. ч. (Сигма Тек, Россия); сульфат аммония х. ч. (ЗАО “ВЕКТОН”, Россия). Деионированную воду получали с помощью системы очистки воды Easy pure II RF/UV (Thermo Scientific, США). Условия хроматографирования оптимизировали на колонке длиной 150 мм, внутренним диаметром 2.1 мм и с зернением сорбента 3.5 мкм Zorbax SB C18 (Agilent Technologies, США). Твердофазную экстракцию (ТФЭ) проводили на патронах SampliQ OPT (60 мг, 3 мл, Agilent Technologies, США). Пробы перед анализом методом ВЭЖХ–МС/МС фильтровали с помощью мембранных шприцевых фильтров из регенерированной целлюлозы (13 мм, 0.2 мкм, Agilent Technologies, Китай). Использовали центрифугу охлаждаемую 3–18 K (Sigma, Германия), электровстряхиватели Multi Reax и Reax Control (Heidolph, Германия), испаритель аналитический ZipVap 20 (Glas-Col, США). Для количественного определения использовали жидкостный хроматограф Agilent 1200 с масс-спектрометрическим детектором Agilent 6410 (Agilent Technologies, Германия).
Экстракция хлорамфеникола. В градуированные пробирки емк. 15.0 мл вводили по 5 мл водного раствора хлорамфеникола с концентрацией 4.0 нг/мл и по 5 мл органического экстрагента. Содержимое пробирок взбалтывали в течение 4 мин, оставляли в покое на 10 мин до достижения межфазного равновесия и разделения фаз. Измеряли объемы равновесных фаз и рассчитывали их соотношение: соотношение объемов равновесных фаз могло изменяться относительно исходных вследствие взаимной растворимости. Затем органический слой переносили в полипропиленовые пробирки, помещали пробирки в нагревательный модуль и упаривали растворитель в токе воздуха: н-бутанол при 55°С, этилацетат и н-гексан при 45°С, хлороформ и смесь хлороформ–ацетон (2 : 1) при 35°С. Сухой остаток растворяли в деионированной воде в объеме, равном равновесному объему органической фазы.
Содержание хлорамфеникола в полученных растворах определяли методом ВЭЖХ–МС/МС. Значения коэффициентов распределения D и степени извлечения R антибиотика рассчитывали по формулам (n = 3):
Очистка экстрактов методом твердофазной экстракции. Очистку экстрактов проб и концентрирование аналита проводили на полимерных полиамидных патронах SampliQ OРT (60 мг, 3 мл), проявляющих и гидрофильные, и гидрофобные свойства. Патроны последовательно кондиционировали 3 мл метанола и 3 мл воды, затем пропускали 5 мл водного раствора хлорамфеникола с концентрацией 4.0 нг/мл. Патроны промывали 6 мл воды и высушивали под вакуумом в течение 10 мин. Хлорамфеникол элюировали 3 мл метанола, элюат упаривали в нагревательном модуле при 45°С досуха. Сухой остаток растворяли в 5 мл воды и анализировали методом ВЭЖХ–МС/МС.
Выполнение определения. Жидкие, пастообразные и твердые пищевые продукты. Тщательно гомогенизированные навески массой 1.00 г взвешивали в полипропиленовых пробирках емк. 50 мл, вводили по 0.1 мл раствора внутреннего стандарта хлорамфеникола-D5 с концентрацией 10 нг/мл, через 10 мин добавляли по 5 мл 4 М раствора сульфата аммония и тщательно перемешивали, затем добавляли по 15 мл этилацетата и помещали пробирки на 10 мин в электровстряхиватель. При анализе Сухие пищевые продукты (сухое молоко, сухие детские молочные адаптированные смеси, спортивное питание и т.п.). Навески продуктов массой 1.00 г взвешивали в полипропиленовых пробирках емк. 50 мл, вводили по 0.1 мл раствора хлорамфеникола-D5 с концентрацией 10 нг/мл, приливали по 5 мл воды и интенсивно встряхивали до растворения пробы. Затем к содержимому пробирок приливали по 5 мл 4 М раствора сульфата аммония, перемешивали, добавляли по 18 мл этилацетата и помещали пробирки на 10 мин в электровстряхиватель. Далее пробы центрифугировали при 10000 об./мин в течение 10 мин при 5 ± 2°С. После центрифугирования этилацетатный слой переносили в полипропиленовые пробирки емк. 15 мл. Экстракты упаривали в нагревательном модуле в токе воздуха при 50 ± ± 2°С до удаления органического растворителя. К полученным после упаривания остаткам приливали по 5 мл воды и 5 мл н-гексана, встряхивали в течение 5 мин в элетровстряхивателе. После расслоения фаз гексановые фракции отбирали и отбрасывали. Полученные водные экстракты пропускали через предварительно уравновешенные 3 мл метанола и 3 мл воды патроны SampliQ OPT, которые затем промывали 6 мл воды и высушивали под вакуумом в течение 10 мин. Аналит и внутренний стандарт элюировали 3 мл метанола. Элюат упаривали в нагревательном модуле при 45 ± 2°С. К сухим остаткам приливали по 1 мл смеси ацетонитрил–вода (3 : 7, по объему), тщательно перемешивали в электровстряхивателе, фильтровали через мембранные шприцевые фильтры из регенерированной целлюлозы в микрофлаконы, анализировали методом ВЭЖХ–МС/МС.
Условия хроматографического разделения: колонка Zorbax SB C18 (150 × 2.1 мм, 3.5 мкм); компоненты подвижной фазы вода (А) и ацетонитрил (Б); режим градиентного элюирования (0–5 мин – от 30 до 50, 5–6 мин – от 50 до 100, 6–9 мин – 100; 9–13 мин – 30 об. % Б); расход подвижной фазы 0.3 мл/мин; температура термостата колонки 40°С; объем вводимой пробы 20 мкл.
Параметры масс-спектрометрического детектирования: ионизация электрораспылением в режиме регистрации отрицательно заряженных ионов; расход газа для десольватации 600 дм3/ч; температура газа для десольватации 350°С; давление на распылителе 45 psi (310.3 кПа); напряжение на капилляре 4000 В; режим мониторинга заданных реакций для хлорамфеникола m/z 321.0 → 152.0 и m/z 326.0 → 157.0 для внутреннего стандарта.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
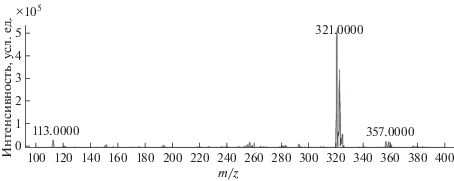
Выбор условий масс-спектрометрического детектирования. Ионизацию хлорамфеникола осуществляли методом электрораспыления в режиме регистрации отрицательно заряженных ионов. Для определения исходного иона непосредственно в масс-спектрометрический детектор вводили раствор хлорамфеникола в воде с концентрацией 1 мкг/мл и регистрировали полный масс-спектр (рис. 1). Видно, что доминирующими ионами являются депротонированные изотопологи хлорамфеникола (М–Н)– с разными комбинациями изотопов двух атомов хлора в своем составе: m/z 321, 35Cl35Cl; m/z 323, 35Cl37Cl; m/z 325, 37Cl37Cl. В качестве исходного иона выбрали наиболее интенсивный псевдомолекулярный ион C11H1135Cl2N2${\text{O}}_{5}^{ - }$ с m/z 321.0.
Рис. 1.
Масс-спектр водного раствора хлорамфеникола (1 мкг/мл) при напряжении на фрагментаторе 120 В.
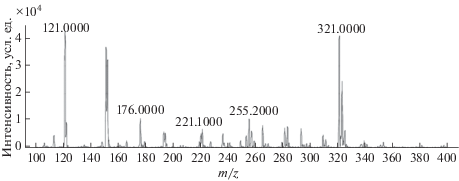
Большое влияние на величину отклика оказывают параметры настройки масс-детектора. Так, повышение напряжения на фрагментаторе (НФ) способствует лучшей передаче ионов в первый квадруполь, однако при чрезмерно высоком значении НФ исходный ион аналита может распадаться на фрагменты, не достигнув первого квадрупольного фильтра. Для установления оптимального значения НФ изучили влияние на отклик исходного иона m/z 321.0 величины напряжения в диапазоне значений от 60 до 180 В. Максимального отклика достигли при 120 В, в то время как высокое НФ приводило к образованию множества ионов с небольшими значениями m/z и, как следствие, к существенному снижению интенсивности сигнала выбранного исходного иона с m/z 321.0. Масс-спектр, полученный для водного раствора хлорамфеникола концентрацией 1 мкг/мл при НФ 180 В, представлен на рис. 2. Таким образом, оптимальное значение НФ для эффективного переноса исходного иона с m/z 321.0 составило 120 В.
Рис. 2.
Масс-спектр водного раствора хлорамфеникола (1 мкг/мл) при напряжении на фрагментаторе 180 В.
Изучено влияние величины энергии соударений (ЭС) на характер фрагментации исходного иона хлорамфеникола в ячейке соударений для выбора наилучших пар ионных реакций. Для иона-предшественника с m/z 321.0 выделили три доминирующих иона-продукта и проанализировали влияние ЭС на величину их отклика. Результаты представлены на рис 3. Видно, что максимальный отклик получен для иона-продукта ${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}}{\text{NO}}_{3}^{ - }$ с m/z 152.0 при ЭС 5 эВ. На втором месте по интенсивности находится сигнал фрагмента C10H1035ClN2${\text{O}}_{5}^{ - }$ с m/z 257.0 при ЭС 0 эВ. Отклик иона ${{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{3}}}}{\text{NO}}_{2}^{ - }$ с m/z 121.0 достиг своего наибольшего значения при ЭС 25 эВ, однако по интенсивности он значительно уступает двум предыдущим. Таким образом, для определения содержания хлорамфеникола использовали ион-продукт с m/z 152.0 (ЭС 5 эВ), для подтверждения достоверности определения – отношение откликов ионов m/z 152.0 (5 эВ)/m/z 257.0 (ЭС 0 эВ).

Описанные выше исследования повторили для дейтерированной формы хлорамфеникола, которую в дальнейшем использовали в качестве внутреннего стандарта при определении хлорамфеникола в пищевых продуктах животного происхождения. Максимальный сигнал установленного исходного иона хлорамфеникола-D5 с m/z 326.0 получен при НФ 120 В, наибольшие сигналы выбранных ионов-продуктов с m/z 157.0 и 262.0 зафиксированы при ЭС 5 и 0 эВ соответственно.
Далее в режиме хроматографирования оптимизировали параметры источника ионизации: температура газа для десольватации 350°С, расход газа для десольватации 600 дм3/ч, давление на распылителе 45 psi, напряжение на капилляре 4000 В. Параметры масс-спектрометрического детектирования хлорамфеникола в режиме мониторинга заданных реакций приведены в табл. 1.
Таблица 1.
Параметры масс-спектрометрического детектирования хлорамфеникола в режиме мониторинга заданных реакций с регистрацией отрицательно заряженных ионов
| Соединение | Исходный ион, m/z |
Ионы-продукты, m/z | Напряжение на фрагментаторе, В | Энергия соударений, эВ |
|---|---|---|---|---|
| Хлорамфеникол | 321.0 | 152.0/257.0 | 120 | 5/0 |
| Хлорамфеникол-D5 | 326.0 | 157.0/262.0 | 120 | 5/5 |
Выбор условий хроматографического разделения. Для разработки методики выбрана хроматографическая колонка с обращенной фазой Zorbax SB C18, поскольку она является универсальной, устойчивой в широком диапазоне рН и пригодной при проведении серийных анализов. Для выбора органического компонента подвижной фазы изучили величину отклика и время удерживания хлорамфеникола при элюировании подвижными фазами метанол–вода и ацетонитрил–вода (3 : 7, по объему). Найдено, что при использовании ацетонитрила время удерживания хлорамфеникола составило 3.4 мин, а при использовании метанола – 8.4 мин, при этом увеличилась ширина хроматографического пика у основания и практически в 3 раза уменьшилась высота пика. Поэтому в качестве органического компонента подвижной фазы использовали ацетонитрил.
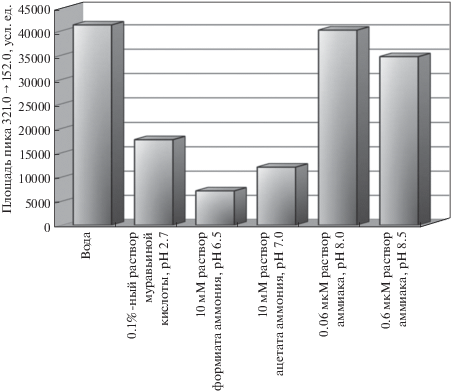
Описано [10–13, 15–22] использование водной фазы с различными модификаторами, поэтому мы изучили влияние состава и рН водного раствора на величину отклика определяемого соединения (рис. 4). Наибольший отклик для m/z 321.0 → → 152.0 получен при использовании воды и растворов аммиака с рН 8.0 и 8.5, поскольку в нейтральной и слабощелочной средах часть молекул хлорамфеникола депротонирована по атому азота амидной группы (схема). Применение 0.1%-ного раствора муравьиной кислоты привело к значительному снижению площади пика аналита, так как при низких значениях рН хлорамфеникол находится в нейтральной молекулярной форме и не регистрируется масс-спектрометром. При использовании растворов ацетата и формиата аммония площадь пика хлорамфеникола снизилась в 3.4 и 5.8 раз соответственно по сравнению с сигналом, полученным с чистой водой. Вероятно, несмотря на нейтральное значение рН данных водных элюентов, природа катионов и анионов солевых добавок повлияла на характер ионизации аналита, что и привело к снижению сигнала m/z 321.0 → 152.0. В качестве водного компонента элюента выбрали воду.
Рис. 4.
Зависимость площади пика водного раствора хлорамфеникола (10 нг/мл) от состава водного компонента.
Экстракция хлорамфеникола и очистка экстрактов. При анализе методом ВЭЖХ–МС/МС на отклик хлорамфеникола сильно влияют соединения, соэкстрагируемые из пищевой матрицы, поскольку они могут как подавлять его ионизацию, так и способствовать ей, искажая результаты количественного определения. Чтобы минимизировать указанный эффект и учесть потери целевого соединения в результате пробоподготовки, в методе ВЭЖХ–МС/МС принято использовать матричную градуировку: в “чистые” пищевые матрицы, соответствующие по составу исследуемым образцам и не содержащие целевого соединения, перед началом процедуры пробоподготовки вносят необходимые аликвоты растворов стандартов. Однако при большом разнообразии исследуемых групп пищевых продуктов, с одной стороны, возникает проблема поиска “чистых” пищевых матриц, а с другой стороны, возрастает стоимость и продолжительность исследований. Решением этой проблемы при наличии внутреннего стандарта, отличающегося от аналита лишь изотопным составом, является использование внешней градуировки и применение эффективной процедуры подготовки проб, обеспечивающей максимальное извлечение аналита и наиболее полное отделение его от компонентов матрицы.
Описаны [10–13, 15–22] методики экстракции хлорамфеникола с использованием различных по природе и полярности экстрагентов, однако данные по коэффициентам распределения антибиотика практически отсутствуют. В табл. 2 приведены значения коэффициентов распределения и степени извлечения хлорамфеникола из водных растворов различными экстрагентами. Установлено, что наибольшая степень извлечения хлорамфеникола достигается экстракцией этилацетатом и н-бутанолом. Высокая экстрагирующая способность данных полярных растворителей обусловлена образованием ассоциатов молекул растворителя и антибиотика по гидратно-сольватному и донорно-акцепторному механизмам благодаря присутствию в молекуле хлорамфеникола полярных функциональных групп: гидроксильных, амидной группы и нитрогруппы (схема). Экстрагирующая сила малополярного хлороформа невелика, в то время как смесь хлороформа с ацетоном (2 : 1) характеризуется хорошей экстрагирующей способностью (табл. 2). Неполярный н-гексан показал самую слабую экстрагирующую способность.
Таблица 2.
Коэффициенты распределения хлорамфеникола между водной и органической фазой и степени его извлечения (n = 3)
| Органический растворитель | D | R, % | Voрг/Vвод |
|---|---|---|---|
| Этилацетат | 42 ± 3 | 97.6 ± 0.1 | 1.0 |
| Хлороформ | 0.17 ± 0.01 | 15 ± 1 | 1.0 |
| Хлороформ–ацетон (2 : 1, по объему) |
6.3 ± 0.6 | 83 ± 1 | 0.75 |
| н-Бутанол | 6.8 ± 0.6 | 89.3 ± 0.9 | 1.2 |
| н-Гексан | 0.015 ± 0.002 | 1.5 ± 0.2 | 1.0 |
Таким образом, наиболее подходящими растворителями для извлечения хлорамфеникола являются этилацетат и н-бутанол. Использование данных плохо смешивающихся с водой органических растворителей в пробоподготовке пищевых продуктов позволяет также очистить пробу от электролитов, сахаров и белковых компонентов, поскольку они не экстрагируются в органическую фазу. Учитывая меньшую температуру кипения этилацетата (77.1°С) по сравнению с н-бутанолом (117.4°С), в качестве экстрагента для дальнейших исследований выбрали этилацетат, что позволило концентрировать аналит упариванием при более низкой температуре и сократить общую продолжительность пробоподготовки. Поскольку при экстракции хлорамфеникола из водных растворов проб пищевых продуктов этилацетатом может образовываться устойчивая эмульсия, к пробе вместо воды добавляли 4 М водный раствор сульфата аммония, который уменьшает взаимное растворение воды и этилацетата, подавляет образование эмульсии и способствует скорейшему установлению межфазового равновесия.
Этилацетат из пищевых продуктов извлекает также и липиды. Поскольку из водных растворов н-гексан практически не экстрагирует хлорамфеникол (табл. 2), для последующей очистки от липидов этилацетатный экстракт упаривали досуха, полученный остаток заново растворяли в воде и экстрагировали липиды в н-гексан.
Для дополнительной очистки полученных обезжиренных водных экстрактов и концентрирования аналита перед инструментальным определением использовали метод ТФЭ на полимерных полиамидных патронах SampliQ OРT. Показано, что хлорамфеникол при пропускании водного раствора хорошо удерживается на сорбенте и затем полностью элюируется метанолом: выход хлорамфеникола составил 99.3 ± 0.9% (n = 3).
Методика определения хлорамфеникола. На основании проведенных исследований разработана методика определения содержания остаточных количеств хлорамфеникола в сырье животного происхождения и пищевых продуктах. Методика включает экстракцию аналита этилацетатом из раствора пробы в 4 М растворе сульфата аммония, упаривание органического растворителя, растворение полученного сухого остатка в воде, обезжиривание н-гексаном, концентрирование методом ТФЭ и количественное определение методом ВЭЖХ–МС/МС.
Для построения градуировочного графика готовили водно-ацетонитрильные (7 : 3, по объему) растворы с концентрацией хлорамфеникола 0.1, 0.2, 0.4, 0.8 и 1.0 нг/мл, содержащие 1 нг/мл хлорамфеникола-D5. Использовали свежеприготовленные растворы.
Зависимость относительной площади пика (площадь пика аналита, деленная на площадь пика внутреннего стандарта) от относительной концентрации (концентрация аналита, деленная на концентрацию внутреннего стандарта), построенная с помощью программного обеспечения Agilent MassHunter, линейна и описывается уравнением y = 0.7758x – 0.0088 с коэффициентом детерминации R2 = 0.999.
Валидация. Для валидации методики использовали молоко сырое, консервы мясные для детского питания, биологически активную добавку “Гематоген”, сухую адаптированную молочную смесь для питания детей раннего возраста, сыр, свинину с шестью уровнями содержания внесенного хлорамфеникола в диапазоне концентраций от 0.3 до 0.9 мкг/кг. Для каждого продукта проведено по 15 определений, выполненных в условиях промежуточной прецизионности с двумя изменяющимися факторами: время, оператор. Каждое определение включало 2 единичных результата измерения, полученных в условиях повторяемости. Значения относительного стандартного отклонения повторяемости sr составили 0.028–0.052, значения промежуточной прецизионности sl(TO) составили 0.031–0.060, смещение метода незначимо. Максимальная расширенная неопределенность U составила 18.4% при Р = 0.95.
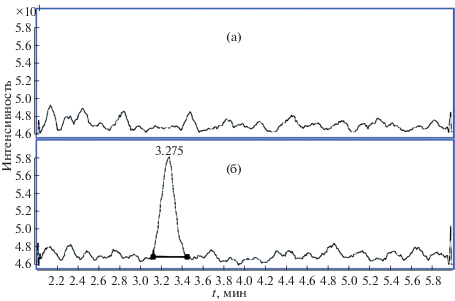
На рис. 5 представлены типичные хроматограммы образца молока сырого без внесения хлорамфеникола и того же образца молока с внесением хлорамфеникола в количестве 0.3 мкг/кг продукта. Пик аналита интенсивен (соотношение сигнал/шум 24.4), что делает возможным определение хлорамфеникола на регламентируемом уровне. Предел определения методики составляет 0.2 мкг/кг, что соответствует требованиям [3–7 ].
Список литературы
Егоров Н.С. Основы учения об антибиотиках. М.: Изд-во МГУ. Высш. школа, 2004. 528 с.
http://www.msdvetmanual.com/pharmacology/antibacterial-agents/phenicols (14.11.2017).
Commission decision 181/2003/EC of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin // Off. J. Eur. Communities. 2003. № L 71. P. 17.
Показатели безопасности и безвредности для человека продовольственного сырья и пищевых продуктов : гигиенический норматив : утв. постановлением Министерства здравоохранения Республики Беларусь 21.06.2013 № 52 / Сб. нормативных документов по продовольственному сырью и пищевым продуктам. Минск: Респ. центр гигиены, эпидемиологии и общественного здоровья, 2014.
Технический регламент Таможенного союза “О безопасности пищевой продукции” (ТР ТС 021/2011): прият Решением Комиссии Таможенного союза 9 октября 2011 г. № 880. Минск: БелГИСС, 2015. 150 с.
Технический регламент Таможенного союза “О безопасности молока и молочной продукции” (ТР ТС 033/2013): принят Решением Совета Евразийской экономической комиссии 9 октября 2013 г. № 67. Минск: БелГИСС, 2015. 91 с.
Технический регламент Таможенного союза “О безопасности мяса и мясной продукции” (ТР ТС 034/2013): принят Решением Совета Евразийской экономической комиссии от 9 октября 2013 г. № 68. Минск: БелГИСС, 2013. 43 с.
Молоко и молочные продукты. Микробиологические методы определения наличия антибиотиков. ГОСТ 31502-2012. М.: Стандартинформ, 2013. 10 с.
Мясо и мясные продукты. Качественный метод определения остаточных количеств антибиотиков и других антимикробных химиотерапевтических веществ. ГОСТ Р 55481-2013. М.: Стандартинформ, 2014. 13 с.
Определение остаточных количеств левомицетина (хлорамфеникола, хлормицетина) в продуктах животного происхождения методом высокоэффективной жидкостной хроматографии и иммуноферментного анализа: Методические указания МУК 4.1.1912-04. М.: Федеральный центр Госсанэпиднадзора Минздрава России, 2004. 24 с.
Мясо и мясные продукты. Метод определения содержания хлорамфеникола (левомицетина) с помощью жидкостной хроматографии. ГОСТ ISO 13493. М.: Федеральный центр Госсанэпиднадзора Минздрава России, 2015. 7 с.
Молоко и продукты переработки молока. Методика определения содержания антибиотиков методом высокоэффективной жидкостной хроматографии. ГОСТ 33526-2015. М.: Стандартинформ, 2016. 9 с.
Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания сульфаниламидов, нитроимидазолов, пенициллинов, амфениколов с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором. ГОСТ Р 54904-2012. М.: Стандартинформ, 2013. 15 с.
Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results // Off. J. Eur. Communities. 2002. № L 221. P. 8.
Douny C., Widart J., de Pauw E., Maghuin-Rogister G., Scippo M.-L. Determination of chloramphenicol in honey, shrimp, and poultry meat with liquid chromatography–mass spectrometry: validation of the method according to commission decision 2002/657/EC // Food Anal. Methods. 2013. V. 6. № 5. P. 1458.
Borràs S.,Companyó R., Guiteras J., Bosch J., Medina M., Termes S. Multiclass method for antimicrobial analysis in animal feeds by liquid chromatography–tandem mass spectrometry // Anal. Bioanal. Chem. 2013. V. 405. № 26. P. 8475.
Schneider M.J., Lehotay S.J., Lightfield A.R. Validation of a streamlined multiclass, multiresidue method for determination of veterinary drug residues in bovine muscle by liquid chromatography–tandem mass spectrometry // Anal. Bioanal. Chem. 2015. V. 407. № 15. P. 4423.
Moragues F., Igualada C., León N. Validation of the determination of chloramphenicol residues in animal feed by liquid chromatography with an ion trap detector based on european decision 2002/657/EC // Food Anal. Methods. 2012. V. 5. № 3. P. 416.
Moragues F., Igualada C., León N. Confirmatory method for the determination of amphenicols in muscle and kidney of several animal species // Food Anal. Methods. 2017. V. 10. № 3. P. 610.
Ozcan N., Aycan O. Determination of chloramphenicol in honey, milk, and egg by liquid chromatography/mass spectrometry: Single-laboratory validation // J. AOAC Int. 2013. V. 96. № 5. P. 1158.
Zhong G., Liu X., Wang C. Study on pretreatment during simultaneous determination of sulfonamides and chloramphenicols in honey // Agric. Biotechnol. 2017. V. 6. № 2. P. 51.
Уланова Т.С., Карнажицкая Т.Д., Пшеничникова Е.О., Нахиева Э.А. Разработка методики определения хлорамфеникола в мясных продуктах // Анализ риска здоровью. 2013. № 4. С. 82.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии