

Журнал аналитической химии, 2019, T. 74, № 8, стр. 595-605
Сравнительное исследование способов подготовки проб донных отложений при определении компонентов нефтепродуктов хроматографическими методами
В. Е. Котова a, b, *, Ю. А. Андреев a, b, М. С. Черновьянц b
a Гидрохимический институт
344090 Россия, Ростов-на-Дону, просп. Стачки, 198
b Южный федеральный университет, химический факультет
344090 Россия, Ростов-на-Дону, ул. Р. Зорге, 7
* E-mail: Valentina.E.Kotova@gmail.com
Поступила в редакцию 16.02.2018
После доработки 04.05.2018
Принята к публикации 29.11.2018
Аннотация
Разработан способ определения алифатических углеводородов и полициклических ароматических углеводородов (ПАУ) в донных отложениях методами газовой хроматографии–масс-спектрометрии и ВЭЖХ со спектрофлуориметрическим детектированием соответственно. Изучены различные способы извлечения ПАУ из матрицы твердого образца: экстракция органическими растворителями при механическом перемешивании, ультразвуковой обработке и в аппарате Сокслета. Установлено, что степень извлечения зависит от способа обработки проб, она максимальна при механическом перемешивании и экстракции в аппарате Сокслета (до 60–80%). Наиболее простой способ механического перемешивания является оптимальным. Предложена схема количественного разделения алифатических углеводородов и ПАУ в широком интервале содержаний методом колоночной хроматографии с применением силикагеля. Пределы обнаружения ПАУ составили 0.1–3 нг/г в пересчете на сухой остаток, значения sr = 0.04 для бензо[а]пирена при содержании 1 нг/г в пересчете на сухой остаток. Разработанная методика применена для определения алифатических углеводородов и ПАУ в реальных образцах донных отложений. Предложенный вариант подготовки проб позволяет определить из одного образца две группы веществ компонентов нефтепродуктов и тем самым расширить возможности установления происхождения углеводородного загрязнения водного объекта.
Нефть и продукты ее переработки представляют собой чрезвычайно сложную смесь переменного состава веществ различных классов, обладающих разной устойчивостью, токсичностью, физико-химическими свойствами. Основными являются углеводороды: алифатические (индикаторные и часто максимальные по содержанию компоненты нефтепродуктов), нафтеновые и ароматические, в том числе и ПАУ, влияние которых особенно вредно и опасно [1]. Полициклические ароматические углеводороды как компоненты нефтепродуктов выделяют особо; они являются объектом пристального внимания уже многие годы. Агентство по охране окружающей среды США и ЕС включили некоторые ПАУ в перечень особо опасных загрязняющих веществ, что объясняется их мутагенными и канцерогенными свойствами [1–3].
Полициклические ароматические углеводороды распространены повсеместно, поскольку они образуются и поступают в окружающую среду за счет любых высокотемпературных процессов как в природе (лесные пожары, вулканическая активность), так и в сфере деятельности человека (выбросы отдельных видов промышленности, включая сжигание топлива, а также транспортные выхлопы). Основным фактором загрязнения ПАУ окружающей среды остается добыча все нарастающих объемов нефти [4].
Содержание ПАУ в различных объектах окружающей среды нормировано и подлежит контролю. Для некоторых ПАУ установлены ПДК для разных типов вод: 10 мкг/л для нафталина и 5 нг/л для бензо[а]пирена в питьевой воде [5], 10 мкг/л для нафталина и 10 нг/л для бензо[а]пирена в воде хозяйственно-питьевого и культурно-бытового водопользования [6 ] и 4 мкг/л для нафталина в воде рыбохозяйственного значения [7]. Следует отметить, что бензо[а]пирену во всех нормативах присвоен 1 класс опасности. Российскими нормативными документами содержание загрязняющих веществ в донных отложениях не регламентируется, существуют лишь нормы содержания бензо[a]пирена и суммарного содержания нефтепродуктов в почве. Обобщенные выборочные значения ПДК, принятые в различных государствах, приведены в табл. 1.
Таблица 1.
| Вещество | Германия | США | Финляндия | Голландия | Россия |
|---|---|---|---|---|---|
| Почва | |||||
| Бензо[a]пирен | 2–12 | 0.7–100 | 2–15 | – | 0.02 |
| Нефтепродукты | – | 200–10 000 | – | – | 180* (1000)** |
| Донные отложения | |||||
| ПАУ (сумма) | 10–100 | – | – | 1–40 | – |
Исследование донных отложений актуально, поскольку взвешенные частицы аккумулируют бόльшую часть неорганических и органических загрязняющих веществ, в том числе наиболее опасных и токсичных, происходит седиментация и интенсивное накопление этих частиц на дне, где процессы биохимического окисления протекают гораздо медленнее, чем в водной среде. При определенных условиях (ветровое взмучивание, изменение значения рН, минерализации и др.) эти сорбированные вещества могут переходить в водную среду, вызывая ее вторичное загрязнение [14]. Загрязненные донные отложения могут содержать токсичные вещества, которые накапливаются в бентосных организмах, влияют на видовое разнообразие бентофауны, нарушают пищевые цепи биоценоза. Таким образом, изучение процессов загрязнения донных отложений является важной и неотъемлемой частью мониторинга водных объектов, а информация о содержании загрязняющих веществ в донных отложениях – наиболее информативной для определения природы и возможного источника загрязнения.
Поскольку отдельные ПАУ проявляют различные канцерогенные и/или мутагенные свойства, очевидна необходимость определения каждого индивидуального вещества. В настоящее время для определения ПАУ применяют разные хроматографические методы анализа: газовую хроматографию, ВЭЖХ с несколькими вариантами детектирования (табл. 2) [15–19]. Наиболее часто для определения ПАУ применяют ВЭЖХ со спектрофлуориметрическим детектированием (ВЭЖХ-ФЛД) и газовую хромато-масс-спектрометрию (ГХ–МС). В силу своей чувствительности и селективности метод ВЭЖХ-ФЛД нашел более широкое распространение.
Таблица 2.
Методики определения ПАУ
| Методика | Способ регистрации |
Предел обнаружения | |
|---|---|---|---|
| мкг/кг (донные отложения) |
мкг/л (вода) |
||
| EPA 8100 | ГХ-ПИД | – | 0.1–425 |
| EPA 8270d | ГХ–МС | 660 | 10 |
| EPA 8275a | ТЭ/ГХ–МС | 0.01–0.5 | – |
| EPA 8310 | ВЭЖХ-УФ | – | 0.2–2 |
| ВЭЖХ-ФЛД | – | 0.01–0.1 | |
| EPA8410 | ГХ-ИК | 2–25 | – |
Для извлечения ПАУ из донных отложений используют различные способы: традиционные (экстракция в аппарате Сокслета [20–24], в том числе и автоматизированная [25]; жидкостная экстракция при механическом перемешивании [26, 27] или ультразвуковой обработке [20, 28–31]) и более современные (экстракция в микроволновом поле [32], сверхкритическая флюидная экстракция [28, 33, 34] и ускоренная экстракция растворителями [35–38]).
Цель данной работы – изучение различных способов извлечения наноколичеств ПАУ из донных отложений для последующего определения методом ВЭЖХ-ФЛД.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и аппаратура. Применяли индивидуальные ПАУ производства Supelco (США) с содержанием основного вещества от 97.3 до 99.9%: нафталин (Naph), аценафтилен (Acn), аценафтен (Ace), флуорен (Fl), фенантрен (Phe), антрацен (An), флуорантен (Flu), пирен (Py), бензо[a]антрацен (B[a]A), хризен (Chry), бензо[b]флуорантен (B[b]F), бензо[k]флуорантен (B[k]F), бензо[a]пирен (B[a]P), индено[1,2,3-cd]пирен (In[cd]P), дибензо[a,h]антрацен (DB[a,h]A), бензо[g,h,i]перилен (B[g,h,i]P); алифатические углеводороды (C10H22–C30H62). В качестве растворителей использовали гексан, ацетон, метиленхлорид отечественного производства квалификации не ниже х. ч. после очистки двукратной перегонкой. Ацетонитрил (Криохром, Россия и Lab-Scan, Польша) для ВЭЖХ и изооктан (Sigma-Aldrich, США) применяли без дополнительной очистки.
Разделение методом колоночной хроматографии проводили на силикагеле Davisil (Grade 635, 60−100 меш, размер пор 60 Å, США). Экстракты концентрировали в ротационном испарителе RV8 (IKA, Германия) при температуре водяной бани 45°C (с минимальным разрежением в системе).
Вещества взвешивали на весах специального класса точности МВ 210-А (Сартогосм, Россия) и B 210S (Sartorius, Германия) с дискретностью отсчета 0.00001 и 0.0001 г соответственно, растворяли в соответствующем растворителе в мерной колбе емк. 5.0 или 10.0 мл. Градуировочные и рабочие растворы готовили последовательным разбавлением.
Навески анализируемых проб донных отложений взвешивали на весах высокого класса точности ВЛТ-150 (Сартогосм, Россия) с дискретностью отсчета 0.001 г. Для сопоставления результатов анализа донных отложений разных типов и влажности расчет проводили на единицу массы сухого остатка (с.о.).
Влажность образцов определяли как классическим высушиванием до постоянной массы в сушильном шкафу SNOL 67/350 (Umega, Литва) при 105 ± 2°С, так и с применением анализатора влажности ML-50 (A&D, Япония).
Сульфат натрия прокаливали в муфельной печи SNOL 8.2/1100 (Umega, Литва) при 600 ± 10°С в течение 4 ч.
Для экстракции использовали механический встряхиватель LS 110 (LOIP, Россия) с частотой перемешивания 150–200 мин–1 и ультразвуковую ванну ПСБ-9535-05М (ПСБ-ГАЛС, Россия) мощностью 200 Вт.
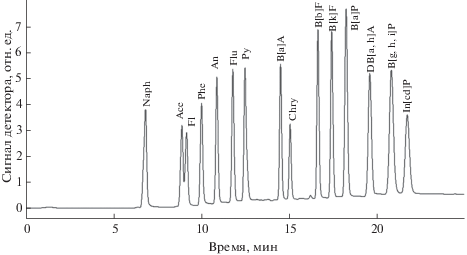
Определение методом ВЭЖХ выполняли на жидкостном хроматографе Agilent Technologies 1260 Infinity (Agilent Technologies, Германия) со спектрофлуориметрическим детектором. Использовали аналитическую колонку (250 × 3.0 мм) ZORBAX Eclipse PAH (Agilent Technologies, США) с неподвижной фазой – октадецилсиланом (С18), химически связанным с пористым силикагелем (размер зерна 5 мкм). В качестве подвижной фазы использовали смесь ацетонитрила (В) и особо чистой воды (А) I типа по ГОСТ Р 52501-2005 (ISO 3696:1987), полученной при помощи системы Simplicity-UV (MilliPore-Merck, Франция). Для измерения максимального аналитического сигнала каждого из исследуемых веществ группы ПАУ нами разработан наиболее селективный режим регистрации по длинам волн возбуждения (λвозб), испускания (λисп) с градиентом подвижной фазы (табл. 3), что позволяет использовать малые навески проб донных отложений [39, 40]. Общая продолжительность хроматографического анализа составила 25 мин. Аликвоту пробы объемом 50 мкл вводили микрошприцем, объем петли дозатора хроматографа составлял 20 мкл. Хроматограммы обрабатывали при помощи программного обеспечения Agilent Chemstation (rev. B.04.03-SP2). Для количественных расчетов применяли метод внешнего стандарта. Градуировочные зависимости линейны в диапазоне нескольких порядков величины концентрации с коэффициентом корреляции не ниже 0.999. Нижняя граница определяемых концентраций варьировалась для разных веществ и составила 0.12 нг/мл для An, верхняя – 2000 нг/мл для In[cd]P. Хроматограмма градуировочного раствора ПАУ с концентрацией В[a]P на уровне минимального значения ПДК [10] в качестве примера представлена на рис. 1.
Таблица 3.
Хроматографический режим детектирования и элюирования ПАУ
| Соеди-нение | λвозб, нм | λисп, нм | Время регистра-ции, мин | Состав элюента А : В, % |
|---|---|---|---|---|
| Naph | 270 | 340 | 6.87 | 55 : 45 |
| Ace | 280 | 325 | 8.93 | |
| Fl | 9.21 | |||
| Phe | 250 | 363 | 10.06 | |
| An | 405 | 10.89 | ||
| Flu | 460 | 11.85 | ||
| Py | 265 | 380 | 12.54 | |
| B[a]A | 270 | 400 | 14.58 | |
| Chry | 15.13 | 0 : 100 | ||
| B[b]F | 290 | 420 | 16.73 | |
| B[k]F | 17.53 | |||
| B[a]P | 270 | 415 | 18.35 | |
| DB[a,h]A | 290 | 400 | 19.73 | |
| B[g,h,i]P | 420 | 20.96 | 60 : 40 | |
| In[cd]P | 250 | 495 | 21.91 |
Хромато-масс-спектрометрическое определение алифатических углеводородов выполняли на газовом хроматографе с масс-селективным детектором Agilent Technologies 6850/5975C. Использовали кварцевую капиллярную колонку HP-5MS с неподвижной фазой 5%-дифенил-95%-диметилполисилоксан длиной 30 м, внутренним диаметром 0.25 мм и толщиной пленки фазы 0.25 мкм (Agilent Technologies, США). Для расшифровки и идентификации масс-спектров использовали библиотеку NIST 2011. В качестве газа-носителя применяли гелий (расход 1.5 мл/мин) без деления потока в режиме постоянной линейной скорости. Температура испарителя (инжектора) – 300°С; температурная программа термостата колонки – изотермический режим 55°С в течение 1 мин, линейное увеличение температуры до 320°С со скоростью 25 град/мин и выдержка в течение 3 мин. Масс-селективный детектор применяли в режиме сканирования диапазона масс 30–450 а. е. м. Температура источника ионов 300°С, температура квадруполя 180°С, задержка включения детектора 4 мин (время на выход растворителя). Общее время хроматографического анализа составило 17.5 мин.
Все оборудование периодически поверяли (средства измерений) и аттестовали (испытательное оборудование) в установленном порядке с обязательным подтверждением метрологических характеристик.
Общая схема анализа донных отложений представлена на рис. 2. Влажные донные отложения массой 2 г (точная навеска) экстрагировали три раза в условиях механического перемешивания в течение 10 мин [39] следующей последовательностью растворителей: 10 мл ацетона, 10 мл смеси ацетон–гексан–метиленхлорид (2 : 1 : 1, по объему), 10 мл смеси ацетон–гексан–метиленхлорид–изооктан (5 : 2 : 2 : 1, по объему). Суспендированные частицы удаляли центрифугированием или пропусканием экстракта через бумажный фильтр, предварительно очищенный в аппарате Сокслета метиленхлоридом в течение 6 ч и промытый ацетоном непосредственно перед использованием. Объединенный экстракт очищали встряхиванием с водой в делительных воронках, органическую фазу при необходимости отделяли центрифугированием и пропускали через слой сульфата натрия для обезвоживания, водную фазу повторно экстрагировали гексаном, общий объем концентрировали в ротационном испарителе и доводили до 1 мл в сосуде с V-образным дном емк. 2–5 мл. Полученный экстракт разделяли на фракции (по полярности веществ) методом колоночной хроматографии с использованием силикагеля [40], предварительно высушенного при 160 ± 5°С в течение 6 ч. Первую фракцию, содержащую только алифатические углеводороды, элюировали гексаном и анализировали методом ГХ–МС; вторую фракцию с ПАУ – смесью гексан–метиленхлорид (1 : 1, по объему), затем заменяли растворитель на 1 мл ацетонитрила и анализировали методом ВЭЖХ.

РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выбор способа и условий извлечения. Определение ПАУ в донных отложениях основано на их извлечении из матрицы твердого анализируемого образца трехкратной экстракцией смесью растворителей, очистке встряхиванием с водой, концентрировании экстрактов, выделении из них фракции ПАУ методом колоночной хроматографии на силикагеле и анализе экстракта методом ВЭЖХ. Ранее нами предложен состав экстракционной смеси для оптимального извлечения определяемых веществ из донных отложений различного типа [39–41].
Исследовали несколько способов экстракции ПАУ из донных отложений: механическое перемешивание, ультразвуковую обработку, экстракцию в аппарате Сокслета. Использовали донные отложения разного типа: мелкозернистые пески с примесью среднезернистых (далее – песчаные) и мелкоалевритовые илы (далее – илистые) из нескольких водных объектов (р. Дон, Азовское море и оз. Байкал). Поскольку реальные образцы всегда содержат некоторое количество ПАУ, изучали полноту их извлечения из проб одной и той же экстракционной системой, но разными способами обработки проб с целью получения максимальных содержаний.
Для сокращения продолжительности извлечения ПАУ использовали ультразвуковую трехкратную экстракцию по 3 мин согласно процедуре, описанной в методе EPA 3550C [31]. С целью уменьшения числа этапов подготовки проб предприняли попытку исключить стадию очистки экстрактов водой (рис. 2). Для этого экстракцию донных отложений проводили механическим перемешиванием, очищенный от суспендированных частиц экстракт помещали непосредственно в колбу ротационного испарителя. Полученные результаты приведены в табл. 4. Найдено, что все изученные способы экстракции позволяют извлекать и определять ПАУ в образцах донных отложений. Лучшая воспроизводимость результатов получена при анализе илистых донных отложений как более однородных образцов по сравнению с песчаными, поэтому для дальнейших исследований использовали только илистые донные отложения.
Таблица 4.
Результаты определения ПАУ (нг/г с.о.) в донных отложениях при различных способах извлечения (n = 4, P = 0.95)
| Соединение | Илистые | Песчаные | |||
|---|---|---|---|---|---|
| механическое перемешивание | ультразвуковая обработка | механическое перемешивание, без воды | механическое перемешивание | ультразвуковая обработка | |
| Naph | 5.5 (0.2) | 6.5 (0.2) | 5.1 (0.2) | 3.4 (0.01) | 2.6 (0.3) |
| Ace | 0.7 (0.3) | 0.5 (0.2) | 0.3 (0.1) | 0.1 (0.1) | 0.1 (0.3) |
| Fl | 1.7 (0.1) | 1.5 (0.2) | 1.1 (0.1) | 0.2 (0.1) | 0.2 (0.3) |
| Phe | 14.9 (0.2) | 11.6 (0.2) | 9.8 (0.1) | 3.6 (0.1) | 3.5 (0.3) |
| An | 1.7 (0.2) | 1.4 (0.2) | 1.1 (0.08) | 0.1 (0.4) | 0.1 (0.4) |
| Flu | 24.5 (0.1) | 17.2 (0.4) | 16.6 (0.07) | 1.5 (0.2) | 1.7 (0.5) |
| Py | 17.6 (0.3) | 16.5 (0.2) | 11.0 (0.08) | 0.8 (0.3) | 1.1 (0.7) |
| B[a]A | 12.3 (0.3) | 10.1 (0.2) | 7.9 (0.09) | 0.5 (0.4) | 0.8 (0.6) |
| Chry | 11.7 (0.4) | 9.4 (0.2) | 5.2 (0.04) | 0.4 (0.3) | 0.6 (0.5) |
| B[b]F | 23.3 (0.2) | 19.5 (0.2) | 13.8 (0.2) | 1.5 (0.2) | 1.6 (0.4) |
| B[k]F | 10.1 (0.3) | 8.0 (0.4) | 6.0 (0.08) | 0.4 (0.3) | 0.6 (0.5) |
| B[a]P | 13.1 (0.3) | 11.6 (0.4) | 8.6 (0.1) | 0.5 (0.4) | 1.0 (0.6) |
| DB[a,h]A | 1.5 (0.1) | 1.4 (0.4) | 1.1 (0.1) | 0.1 (0.4) | 0.2 (0.6) |
| B[g,h,i]P | 12.9 (0.3) | 11.0 (0.4) | 8.2 (0.08) | 0.6 (0.3) | 1.5 (0.6) |
| In[cd]P | 10.9 (0.2) | 7.8 (0.1) | 9.1 (0.2) | 0.7 (0.2) | 1.1 (0.3) |
Извлечение ПАУ путем механического перемешивания без очистки экстрактов водой (табл. 4) характеризуется более низкими содержаниями по сравнению с другими способами (для песчаных донных отложений воспроизводимые значения не получены). Такой результат можно связать с остаточным количеством более полярных растворителей в экстракте, за счет чего варьируется элюирующая сила подвижной фазы при разделении методом колоночной хроматографии, и часть аналитов попадает во фракцию 1, что приводит к их неколичественному выделению. Данный недостаток полностью устраняется заменой всех растворителей на неполярный (гексан) в разработанном нами способе подготовки пробы на стадии 2 (рис. 2), что не влияет на разделение колоночной хроматографией и позволяет количественно выделить обе группы аналитов.
Содержания ПАУ, полученные при использовании ультразвуковой обработки проб, оказались несколько ниже результатов после механического перемешивания. Возможно, продолжительность экстракции в течение 3 мин [31] недостаточна для количественного извлечения. На следующем этапе ПАУ извлекали из илистых донных отложений при механическом перемешивании и ультразвуковой обработке в течение одинакового времени (3 раза по 10 мин). Для сравнения провели экстракцию ПАУ в аппарате Сокслета в течение 6 ч со скоростью 10 циклов/ч. В качестве экстрагента использовали суммарную смесь растворителей, применяемых для извлечения при механическом перемешивании или ультразвуковой обработке. Результаты представлены в табл. 5. Наиболее высокие степени извлечения большинства аналитов получены для экстракции в аппарате Сокслета, однако этот способ – самый длительный из изученных вариантов подготовки проб. При ультразвуковой обработке степень извлечения оказалась наименьшей, а значения sr максимальными. Результаты, близкие к извлечению в аппарате Сокслета, получены при механическом перемешивании. Этот способ подготовки проб при экстракции из твердой матрицы образца является наиболее простым и не требует применения специального оборудования, потому его выбрали для дальнейших исследований.
Таблица 5.
Степени извлечения (%) ПАУ из донных отложений с использованием механического перемешивания (I), ультразвуковой обработки (II) и экстракции в аппарате Сокслета (III) (n = 5, P = 0.95)
| Соединение | I | II | III |
|---|---|---|---|
| Naph | 25 ± 7 (0.2) | 19 ± 9 (0.4) | 39 ± 12 (0.2) |
| Ace | 35 ± 5 (0.1) | 30 ± 5 (0.1) | 39 ± 5 (0.1) |
| Fl | 42 ± 6 (0.1) | 34 ± 5 (0.1) | 54 ± 6 (0.1) |
| Phe | 88 ± 18 (0.3) | 36 ± 15 (0.3) | 84 ± 16 (0.2) |
| An | 58 ± 18 (0.2) | 32 ± 10 (0.2) | 67 ± 23 (0.3) |
| Flu | 65 ± 14 (0.2) | 42 ± 13 (0.2) | 66 ± 17 (0.2) |
| Py | 80 ± 22 (0.4) | 46 ± 15 (0.3) | 59 ± 22 (0.3) |
| B[a]A | 55 ± 15 (0.2) | 46 ± 22 (0.4) | 64 ± 22 (0.3) |
| Chry | 53 ± 21 (0.3) | 47 ± 22 (0.4) | 65 ± 17 (0.2) |
| B[b]F | 74 ± 14 (0.2) | 57 ± 11 (0.2) | 79 ± 23 (0.2) |
| B[k]F | 75 ± 20 (0.2) | 43 ± 19 (0.4) | 68 ± 23 (0.3) |
| B[a]P | 69 ± 23 (0.3) | 30 ± 16 (0.4) | 70 ± 26 (0.3) |
| DB[a,h]A | 64 ± 6 (0.1) | 57 ± 9 (0.1) | 79 ± 13 (0.1) |
| B[g,h,i]P | 75 ± 15 (0.2) | 43 ± 13 (0.2) | 80 ± 16 (0.2) |
| In[cd]P | 65 ± 7 (0.1) | 51 ± 9 (0.1) | 66 ± 9 (0.1) |
Оптимизация процедуры выделения фракций алифатических углеводородов и ПАУ. Хроматографический анализ экстрактов донных отложений, загрязненных нефтепродуктами, осложнен наличием предельных углеводородов в концентрациях, часто превышающих суммарное содержание ПАУ на несколько порядков. Для повышения селективности анализа (во избежание перекрывания хроматографических пиков веществ этих классов) использовали колоночную хроматографию для количественного выделения фракций алифатических и полициклических ароматических углеводородов. Экстракты разделяли на фракции на микроколонке длиной 20 см и внутренним диаметром 0.3 см, обладающей рядом преимуществ по сравнению с колонками стандартных размеров: экономичность за счет малых количеств расходных материалов (силикагель, растворители) и малый диаметр, препятствующий дополнительному размыванию хроматографических зон разделяемых групп веществ. В обычно применяемых способах разделения необходимы десятки миллилитров особо чистых и дорогостоящих растворителей, большие количества сорбентов [23, 24, 26, 27].
Для выбора способа разделения стандартные растворы алифатических углеводородов (смесь алканов C10H22–C30H62 в гексане c концентрацией 1 мкг/мл каждого компонента), ПАУ (смесь в гексане c концентрацией компонентов от 2.4 до 40 нг/мл, концентрация бензо[a]пирена 4 нг/мл) и их смесь помещали в колонку с силикагелем, элюировали гексаном и смесью гексан–метиленхлорид (1 : 1, по объему) и отбирали фракции по 0.5 мл, которые затем анализировали. По результатам анализа строили кривые элюирования (рис. 3а) для определения объемов элюентов, необходимых для полного разделения групп исследуемых веществ [39]. Поскольку в некоторых смесях концентрации алифатических углеводородов на несколько порядков превышали концентрации ПАУ, на рис. 3 представлена зависимость относительной интенсивности сигналов детекторов, полученных методами ГХ–МС и ВЭЖХ соответственно. Установлено, что группа алифатических углеводородов количественно элюируется в первые 3.5 мл элюата (фракция 1), а ПАУ – в последующие 3.0 мл смеси гексан–метиленхлорид (фракция 2). В выбранных условиях группа ПАУ не элюируется с силикагеля гексаном объемом до 5 мл.
Рис. 3.
Элюирование алифатических углеводородов C10H22–C30H62 (фракция 1) и 15 приоритетных ПАУ (фракция 2) в колоночной хроматографии.
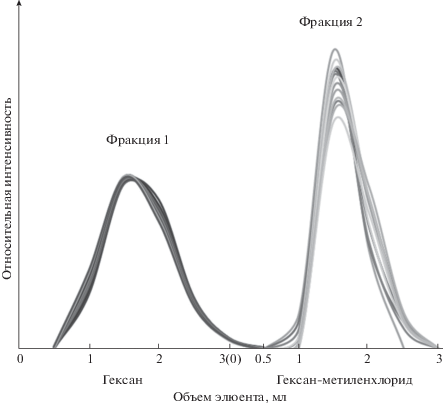
Построены кривые элюирования для различных концентраций фракции ПАУ в присутствии значительно бόльших количеств алифатических углеводородов (как в реальных загрязненных нефтепродуктами донных отложениях). Установлено, что при суммарных содержаниях ПАУ до 5 мкг/г с.о. (соответствует пятикратному превышению минимального значения ПДК для бензо[a]пирена) и алифатических углеводородов до 15 мг/г с.о. не наблюдается перекрестное загрязнение целевых фракций аналитов. Доля ПАУ, элюирующихся после фракции 2, составляет не более 4.5% от суммарного содержания для самой большой из изученных концентраций. Разработанная схема анализа позволяет количественно выделить из экстрактов фракции алифатических углеводородов и ПАУ в широком диапазоне концентраций (от единиц нанограммов до единиц микрограммов ПАУ при более чем тысячекратном избытке алифатических углеводородов) с небольшим расходом дорогостоящих силикагеля и высокочистых растворителей.
Оценка метрологических характеристик. В табл. 6 приведены результаты определения ПАУ в пробах донных отложений разного типа и соответствующие значения sr. Последние свидетельствуют о хорошей воспроизводимости в обоих случаях.
Таблица 6.
Результаты определения ПАУ (нг/г с.о.) в илистых (I) и песчаных (II) донных отложениях (n = = 10, P = 0.95)
| Соединение | I | II |
|---|---|---|
| Naph | 7.9 ± 0.9 (0.2) | 5.3 ± 0.5 (0.1) |
| Ace | 0.5 ± 0.1 (0.3) | 0.20 ± 0.01 (0.1) |
| Fl | 1.4 ± 0.2 (0.2) | 0.30 ± 0.04 (0.2) |
| Phe | 13 ± 1 (0.1) | 4.8 ± 0.4 (0.1) |
| An | 1.6 ± 0.2 (0.2) | 0.10 ± 0.01 (0.2) |
| Flu | 23 ± 2 (0.1) | 1.4 ± 0.2 (0.2) |
| Py | 15 ± 2 (0.2) | 0.7 ± 0.2 (0.3) |
| B[a]A | 11 ± 2 (0.2) | 0.7 ± 0.1 (0.3) |
| Chry | 8 ± 1 (0.2) | 0.4 ± 0.2 (0.6) |
| B[b]F | 18 ± 1 (0.1) | 0.9 ± 0.2 (0.3) |
| B[k]F | 8.0 ± 0.8 (0.1) | 0.40 ± 0.08 (0.3) |
| B[a]P | 12 ± 1 (0.2) | 0.5 ± 0.1 (0.4) |
| DB[a,h]A | 1.4 ± 0.2 (0.2) | 0.05 ± 0.01 (0.4) |
| B[g,h,i]P | 10 ± 1 (0.2) | 0.60 ± 0.08 (0.2) |
| In[cd]P | 10 ± 1 (0.2) | 0.7 ± 0.1 (0.3) |
Правильность разработанной методики анализа проб донных отложений оценивали способом введено–найдено для высоких и низких значений концентраций (табл. 7). Анализировали модельные образцы (прокаленные при 600°С в течение 6 ч донные отложения, которые не содержали определяемые вещества, что подтверждено их предварительным анализом), а также реальные пробы с добавками (с вычитанием содержания, найденного в пробе без добавки). Значения sr составили 0.03–0.4 для низких концентраций (на уровне единиц нанограммов) и 0.01–0.04 для высоких концентраций (на уровне сотен нанограммов).
Таблица 7.
Результаты определения ПАУ (нг/г с.о.) в донных отложениях методом введено–найдено (n = 10, P = = 0.95)
| Соединение | Введено | Найдено | sr | Введено | Найдено | sr |
|---|---|---|---|---|---|---|
| Naph | 5.1 | 4.6 ± 0.7 | 0.4 | 505 | 525 ± 18 | 0.03 |
| Ace | 1.05 | 1.08 ± 0.05 | 0.1 | 105 | 102 ± 2 | 0.04 |
| Fl | 1.0 | 1.1 ± 0.1 | 0.2 | 102 | 105 ± 3 | 0.02 |
| Phe | 1.02 | 1.09 ± 0.07 | 0.2 | 102 | 99 ± 4 | 0.03 |
| An | 0.61 | 0.62 ± 0.02 | 0.09 | 60.8 | 61.3 ± 0.4 | 0.02 |
| Flu | 7.0 | 6.9 ± 0.1 | 0.05 | 700 | 704 ± 5 | 0.02 |
| Py | 1.00 | 1.00 ± 0.04 | 0.1 | 100.2 | 100.6 ± 0.4 | 0.01 |
| B[a]A | 1.00 | 0.98 ± 0.02 | 0.1 | 100 | 103 ± 4 | 0.03 |
| Chry | 1.00 | 1.01 ± 0.03 | 0.1 | 100 | 101 ± 2 | 0.02 |
| B[b]F | 3.4 | 3.3 ± 0.1 | 0.04 | 340 | 338 ± 3 | 0.02 |
| B[k]F | 0.60 | 0.59 ± 0.01 | 0.03 | 60 | 61 ± 1 | 0.02 |
| B[a]P | 1.08 | 1.06 ± 0.02 | 0.04 | 108 | 112 ± 4 | 0.02 |
| DB[a,h]A | 1.01 | 0.99 ± 0.02 | 0.04 | 101 | 100 ± 1 | 0.02 |
| B[g,h,i]P | 1.60 | 1.54 ± 0.06 | 0.1 | 160 | 164 ± 4 | 0.02 |
| In[cd]P | 10.0 | 10.3 ± 0.4 | 0.05 | 1000 | 986 ± 21 | 0.03 |
Для оценки чувствительности методики рассчитаны пределы обнаружения как утроенное стандартное отклонение содержания каждого ПАУ, при котором хроматографические пики аналитов соизмеримы с мешающими пиками посторонних веществ. Значения пределов обнаружения (n = 10, P = 0.99), нг/г с.о., составили: 0.1 для An, 0.2 для B[a]A, B[a]P и DB[a,h]A, 0.3 для Ace, B[k]F и B[g,h,i]P, 0.4 для Chry, 0.5 для Fl, 0.6 для Py и B[b]F, 1 для In[cd]P, 2 для Flu, 3 для Naph и Phe.
Анализ реальных объектов. Предложенный вариант подготовки и анализа проб использован при изучении загрязнения различных типов донных отложений [42]. На рис. 4, 5 представлены хроматограммы обеих фракций экстрактов из двух образцов донных отложений мелкоалевритового ила (глубина отбора 50 м) и глинистого ила (глубина отбора 250 м).
Рис. 4.
Хроматограммы фракции алифатических углеводородов экстрактов мелкоалевритового (а) и глинистого (б) илов.
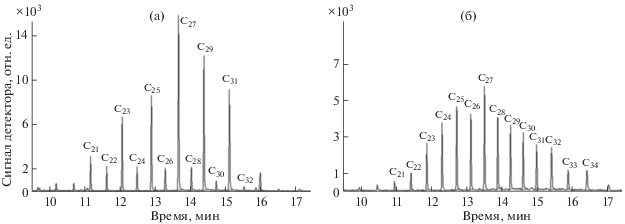
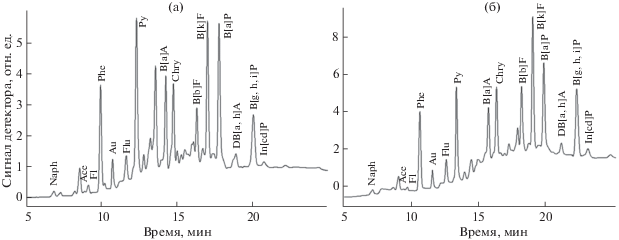
Для выявления источника загрязнения при поступлении компонентов нефтепродуктов в объекты окружающей среды обычно используют один из классических способов дифференциации углеводородов между биогенными или нефтяными, основанный на соотношении алифатических углеводородов, – индекс нечетности CPI (carbon preference index) [43, 44]. Поскольку разработанный способ анализа донных отложений позволяет определять из одной пробы и ПАУ, и алифатические углеводороды (потенциальные представители нефтепродуктов), становится возможным проводить идентификацию также по фракции ПАУ. Соотношение ПАУ позволяет дифференцировать источник их происхождения на петрогенный или пирогенный с бόльшими, чем по индексу CPI, вариантами градаций: петрогенный, смешанный (продукты переработки или сгорания нефти, выхлопные газы) или пирогенный (сжигание травы, древесины, угля). Возможность многостороннего подхода к выявлению источника происхождения углеводородов в донных отложениях важна при изучении хронического загрязнения водоемов [42]. Фракции алифатических углеводородов проанализированных проб имеют различные индексы нечетности. Значение CPI = = 5.493 указывает на преимущественный вклад биогенных углеводородов в мелкоалевритовом иле (CPI > 1, рис. 4а), а значение CPI = 1.048 – на бόльшее количество нефтяных углеводородов в глинистом иле (CPI ≈ 1, рис. 4б). Содержание углеводородов фракции ПАУ свидетельствуют о пирогенном источнике их происхождения в обеих пробах (рис. 5, табл. 6).
Список литературы
Yu Y., Wang P., Cui Y., Wang Y. Chemical analysis of DNA damage // Anal. Chem. 2018. V. 90. № 1. P. 556.
Майстренко В.Н., Клюев Н.А. Экологоаналитический мониторинг стойких органических загрязнителей. М.: БИНОМ. Лаборатория знаний, 2009. 323 с.
Ноллет Л.М.Л. Анализ воды. Справочник: пер. с англ. 2-го изд. / Под ред. Васильевой И.А., Пролетарской Е.Л. СПб: ЦОП “Профессия”, 2012. 920 с.
ИТС 28-2017. Добыча нефти. М.: Бюро НДТ, 2017. 273 с.
СанПин 2.1.4.1074-01. Питьевая вода. Гигиенические требования к качеству воды централизованных систем питьевого водоснабжения. Контроль качества. Зарегистрировано в Минюсте РФ 31.10.2001 г. № 3011. http://docs.cntd.ru/document/901798042 (24.11.2018 г.).
ГН 2.1.5.1315-03. Предельно-допустимые концентрации (ПДК) химических веществ в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования. В ред. Дополнений и изменений № 1, утв. Постановлением Главного государственного санитарного врача РФ от 28.09.2007 г. № 75, Постановления Главного государственного санитарного врача РФ от 28.09.2007 г. № 77, Изменений № 2, утв. Постановлением Главного государственного санитарного врача РФ от 16.09.2013 г. № 49, Постановлений Главного государственного санитарного врача РФ от 30.08.2016 г. № 147, от 13.07.2017 г. № 97. Зарегистрировано в Минюсте России 19.05.2003 г. № 4550. http://docs.cntd.ru/document/901862249 (24.11.2018 г.).
Приказ Минсельхоза России от 13.12.2016 г. № 552 “Об утверждении нормативов качества воды водных объектов рыбохозяйственного значения, в том числе нормативов предельно допустимых концентраций вредных веществ в водах водных объектов рыбохозяйственного значения”. http://docs.cntd.ru/document/420389120 (24.11.2018 г.).
Das neue BundesBodenschutzgesetz. (“Берлинские листы”, “Бранденбургские листы”). https://www.gesetze-im-internet.de/bbodschg/BBodSchG.pdf. (24.11. 2018 г.).
Leidraad Bodemsanering (1994): Mitteilung des Rijksinstituut voor Volksgezondheit en Milieuhygiene. (“Голландские листы”). https://lfu.brandenburg. de/ cms/media.php/lbm1.a.3310.de/lua_bd48.pdf. (24.11. 2018 г.).
ГН 2.1.7.2041-06. Предельно допустимые концентрации (ПДК) химических веществ в почве. В ред. Постановления Главного государственного санитарного врача РФ от 26.06.2017 г. № 89. Зарегистрировано в Минюсте России 7.02.2006 г. № 7470. http://docs.cntd.ru/document/901966754 (24.11.2018 г.).
Распоряжение мэра Санкт-Петербурга от 30.08.1994г. № 891-р. “Порядок определения размеров ущерба от загрязнения земель химическими веществами”. Утв. Роскомземом 10.11.1993 г. и Минприроды РФ 18.11.1993 г. http://docs.cntd.ru/document/9033369 (24.11.2018 г.).
Распоряжение мэра-председателя Правительства Санкт-Петербурга А.А. Собчака от 30.08.1994 г. № 891-р. “О введении регионального норматива по охране почв в Санкт-Петербурге”. http://docs.cntd. ru/document/9102762 (29.01.2018 г.).
Капелькина Л.П. Гармонизация экологических стандартов II (ГЭС II). Промежуточный технический отчет. Нормативы качества окружающей среды. Санкт-Петербург, 2008. 18 с. http://www.libed. ru/knigi-nauka/ 870198-6-es-rossiya-programma-sotrudnichestva-garmonizaciya-ekologicheskih-standartov-ges-ii-zaklyuchitelniy- tehnicheskiy-ot.php (25.01.2018 г.).
Никаноров А.М., Иваник В.М. Словарь-справочник по гидрохимии и качеству вод суши (понятия и определения). Ростов-на-Дону: ООО “Центр Печатных Технологий АртАртель”, 2014. 548 с.
US EPA Method 8100. Polynuclear aromatic hydrocarbons. 1986. 10 p. https://www.epa.gov/sites/production/files/2015-12/documents/8100.pdf (29.01.2018 г.).
US EPA Method 8270D. Semivolatile organic compounds by gas chromatography/mass spectrometry. 2014. 71 p. https://www.epa.gov/sites/production/files/2015-12/documents/8270d.pdf (29.01.2018 г.).
US EPA Method 8275A. Semivolatile organic compounds (PAHs and PCBs) in soils/sludges and solid wastes using thermal extraction/gas chromatography/mass spectrometry (TE/GC/MS). 1996. 23 p. https://www.epa.gov/sites/production/files/2015-12/ documents/8275a.pdf (29.01.2018 г.).
US EPA Method 8310. Polynuclear aromatic hydrocarbons. 1986. 13 p. https://www.epa.gov/sites/production/files/2015-12/documents/8310.pdf (29.01.2018 г.).
US EPA Method 8410. Gas chromatography/fourier transform infrared spectrometry for semivolatile organics: capillary column. 2014. 21 p. https://www.epa. gov/sites/production/files/2015-12/documents/8410.pdf (29.01.2018 г.).
Kicinski H.G., Adamek S., Kettrup A. Trace enrichment and HPLC analysis of polycyclic aromatic hydrocarbons in environmental samples, using solid phase extraction in connection with UV/VIS diode-array and fluorescence detection // Chromatographia. 1989. № 34. V. 28. P. 203.
Domine D., Devillers J., Garrigues P., Budzinski H., Chastrette M., Karcher W. Chemometrical evaluation of the PAH contamination in the sediments of the Gulf of Lion (France) // Sci. Total Environ. 1994. V. 155. P. 9.
US EPA Method 3540C. Soxhlet extraction. 1996. 8 p. https://www.epa.gov/sites/production/files/2015-12/ documents/3540c.pdf (30.01.2018 г.).
Tolosa I., de Mora S., Sheikholeslami M.R., Villeneuve J.-P., Bartocci J., Cattini C. Aliphatic and aromatic hydrocarbons in coastal Caspian Sea sediments // Mar. Pollut. Bull. 2004. № 48. P. 44.
Wang Y., Tian Z., Zhu H., Cheng Z., Kang M., Luo C., Li J., Zhang G. Polycyclic aromatic hydrocarbons (PAHs) in soils and vegetation near an e-waste recycling site in South China: Concentration, distribution, source, and risk assessment // Sci. Total Environ. 2012. V. 439. P. 187.
US EPA Method 3541. Automated Soxhlet extraction. 1994. 10 p. https://www.epa.gov/sites/production/files/ 2015-06/documents/epa-3541.pdf (30.01.2018 г.).
Leite N.F., Peralta-Zamora P., Grassi M.T. Multifactorial optimization approach for the determination of polycyclic aromatic hydrocarbons in river sediments by gas chromatography-quadrupole ion trap selected ion storage mass spectrometry // J. Chromatogr. A. 2008. V. 1192. P. 273.
Gaspare L., Machiwa J.F., Mdachi S.J.M., Streck G., Brack W. Polycyclic aromatic hydrocarbon (PAH) contamination of surface sediments and oysters from the inter-tidal areas of Dares Salaam, Tanzania // Environ. Pollut. 2009. V. 157. P. 24.
Berg B.E., Lund H.S., Kringstad A., Kvernheim A.L. Routine analysis of hydrocarbons, PCB and PAH in marine sediments using supercritical CO2 extraction // Chemosphere. 1999. V. 38. № 3. P. 587.
Banjoo D.R., Nelson P.K. Improved ultrasonic extraction procedure for the determination of polycyclic aromatic hydrocarbons in sediments // J. Chromatogr. A. 2005. V. 1066. P. 9.
Filipkowska A., Lubecki L., Kowalewska G. Polycyclic aromatic hydrocarbon analysis in different matrices of the marine environment // Anal. Chim. Acta. 2005. V. 547. №. 2. P. 243.
US EPA Method 3550C. Ultrasonic extraction. 2007. 17 p. https://www.epa.gov/sites/production/files/2015-12/documents/3550c.pdf (30.01.2018 г.).
US EPA Method 3546. Microwave extraction. 2007. 13 p. https://www.epa.gov/sites/production/files/ 2015-12/documents/3546.pdf (30.01.2018 г.).
US EPA Method 3560. Supercritical fluid ext raction of total recoverable petroleum hydrocarbons. 1996. 8 p. https:/0,512/ documents/3560.pdf (30.01.2018 г.).
US EPA Method 3561. Supercritical fluid extraction of polynuclear aromatic hydrocarbons. 1996. 14 p. https://www.epa.gov/sites/production/files/2015-12/ documents/3561.pdf (30.01.2018 г.).
Amador-Munoza O., Villalobos-Pietrini R., Aragyn-Pina A., Tran T.C., Morrison P., Marriott P.J. Quantification of polycyclic aromatic hydrocarbons based on comprehensive two-dimensional gas chromatography-isotope dilution mass spectrometry // J. Chromatogr. A. 2008. V. 1201. P. 161.
Witt G., Liehr G.A., Borck D., Mayer P. Matrix solid-phase microextraction for measuring freely dissolved concentrations and chemical activities of PAHs in sediment cores from the western Baltic Sea // Chemosphere. 2009. V. 74. P. 522.
Qiao M., Qia W., Liua H., Qua J. Simultaneous determination of typical substituted and parent polycyclic aromatic hydrocarbons in water and solid matrix by gas chromatography-mass spectrometry // J. Chromatogr. A. 2013. V. 1291. P. 129.
Andersson M., Klug M., Eggen O.A., Ottesen R.T. Polycyclic aromatic hydrocarbons (PAHs) in sediments from lake Lille Lungegårdsvannet in Bergen, western Norway; appraising pollution sources from the urban history // Sci. Total Environ. 2014. V. 470–471. P. 1160.
Котова В.Е., Андреев Ю.А., Черновьянц М.С. Хроматографическое изучение компонентного состава нефтепродуктов в донных отложениях // Сорбционные и хроматографические процессы. 2016. Т. 16. № 6. С. 885.
Котова В.Е., Андреев Ю.А. Способ подготовки проб для определения алифатических и полициклических ароматических углеводородов в донных отложениях. Патент № 2646402 РФ. Заявка № 2017106715 от 23.02.2017, опубл. 5.03.2018 г.
Андреев Ю.А., Котова В.Е., Черновьянц М.С. Сравнительное исследование способов пробоподготовки для ВЭЖХ определения приоритетных ПАУ в донных отложениях / Тез. докл. “Третьего съезда аналитиков России”. Москва, 8–13 октября 2017 г. http://www.wssanalytchem.org/car2017/Publications/ 2017-Abstracts.pdf. 2017. М.: ГЕОХИ РАН, 2017. С. 330.
Котова В.Е., Андреев Ю.А., Черновьянц М.С. Идентификация источников поступления полициклических ароматических углеводородов в донные отложения озера Байкал // Вода: химия и экология. 2017. № 4. С. 71.
Никаноров А.М., Страдомская А.Г. Проблемы нефтяного загрязнения пресноводных экосистем. Ростов-на-Дону: НОК, 2008. 222 с.
Peters K.P., Walters C.C., Moldowan J.M. The biomarker guide. V. 2. Biomarkers and isotopes in petroleum exploration and Earth history. 2nd ed. New York: Cambridge University Press, 2005. 1156 p.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии