

Журнал аналитической химии, 2019, T. 74, № 8, стр. 580-587
Определение приоритетных полициклических ароматических углеводородов в воде на следовом уровне концентраций
А. Г. Горшков a, *, О. Н. Изосимова a, О. В. Кустова a
a Лимнологический институт Сибирского отделения Российской академии наук
664033 Иркутск, ул. Улан-Баторская, 3, Россия
* E-mail: gorchkov_ag@mail.ru
Поступила в редакцию 19.07.2018
После доработки 04.02.2019
Принята к публикации 04.02.2019
Аннотация
Предложена методика определения приоритетных полициклических ароматических углеводородов (ПАУ) в воде методом газовой хроматографии–тандемной масс-спектрометрии. Методика отличается от известных низким уровнем предела определения 0.1–1.0 нг/л и внутрилабораторной прецизионностью 15–20%. Простую и быструю подготовку проб – однократную экстракцию ПАУ н-гексаном (объем пробы 0.1 л, коэффициент концентрирования 102–103) – легко реализовать в серийном анализе при мониторинговых исследованиях. Прямой анализ экстрактов устраняет риск потерь легколетучих ПАУ. Кроме того, преимуществами методики являются небольшие объемы экстрагента (1 мл) и соответственно органического растворителя для его последующей утилизации. Методика апробирована при мониторинге ПАУ в водах озера Байкал, его притоков и анализе атмосферных осадков на городских и фоновых территориях.
Полициклические ароматические углеводороды поступают в окружающую среду в результате неполного сгорания органических веществ, разливов нефти при ее добыче, транспортировке и переработке. К отличительным свойствам ПАУ относят устойчивость и, как следствие, сохранение в природных объектах в течение длительного времени, гидрофобность и сорбцию на частицах взвешенного вещества в воде или на частицах атмосферного аэрозоля; токсичность, мутагенность и канцерогенность, с которыми связывают биологическое действие ПАУ на живую природу и здоровье человека [1]. Полициклические ароматические углеводороды встречаются во всех типах вод, включены Стокгольмской конвенцией (2001 г.) в ряд стойких органических загрязняющих веществ (СОЗ). Агентством по охране окружающей среды США (EPA) для системы контроля ПАУ в природных объектах в качестве приоритетных выбраны 16 соединений [2], Европейской комиссией ЕС – 8 аренов [3]. В России в водных объектах хозяйственно-питьевого и культурно-бытового водопользования, в питьевой воде нормируется одно соединение этого класса – бенз[a]пирен, который рассматривается как маркер других ПАУ [4, 5].
В воде ПАУ определяют методами ВЭЖХ или хромато-масс-спектрометрии (ГХ–МС). В зависимости от сложности органической матрицы анализируемых образцов и уровня содержания в них ПАУ разработаны различные способы подготовки проб. Широко применяемый метод US EPA 3510с основан на отборе проб объемом 1 л, жидкостно–жидкостной экстракции ПАУ в хлористый метилен или н-гексан. Последующие стадии подготовки проб – концентрирование экстрактов под вакуумом или в токе азота, выделение узкой фракции ПАУ методом хроматографии – характеризуются потенциальным риском потерь легких аренов и неэкономичны в связи с необходимостью утилизации больших объемом органических растворителей [6–9]. Применение методов твердофазной экстракции и различных вариантов микроэкстракции в рамках поиска эффективных, быстрых и надежных способов подготовки проб дает возможность проводить анализ с минимальным объемом проб (от 2 до 50 мл) и пределами обнаружения от 0.1 до 60 нг/л [10–15], однако они не нашли широкого применения в серийном анализе, судя по данным мониторинговых исследований органических микрозагрязнений в водных экосистемах [7–9, 16].
Физико-химические свойства ПАУ из приоритетного ряда (в частности, их летучесть и растворимость в воде) варьируются в широких диапазонах, что необходимо учитывать как в методологии их контроля в водных объектах, так и при выборе оптимальных условий подготовки проб для анализа. Высокая гидрофобность высокомолекулярных гомологов ПАУ обусловливает минимальный уровень их содержания в водной фазе из-за ассоциации с частицами взвешенного вещества и перехода в донные отложения водоемов, в то время как во фракции аренов, присутствующих в воде, доминируют низкомолекулярные гомологи (нафталины), имеющие максимальную растворимость в воде. В водоемах, расположенных в чистых районах, уровень загрязнения – фоновый. Например, в озере Байкал содержание нафталинов достигает 7.0–20 нг/л, что ниже, чем пределы их определения, установленные в нормативных документах государственного экологического контроля.
По методике ПНД Ф 14.1:2:4.70-96 [17] с применением метода ВЭЖХ с флуоресцентным детектированием контроль нафталина, флуорантена и пирена в природных и питьевых водах возможен при их содержании в пробе выше, чем 20 нг/л, а в случае аценафтена, флуорена и фенантрена – 6 нг/л. В методике РД 52.44.590-2016 [18] предел определения бенз[a]пирена, бенз[g,h,i]перилена и дибенз[a,h]антрацена в поверхностных водах и атмосферных осадках составляет 0.5 нг/л, для аренов, имеющих четыре–пять циклов в структуре, – 1.0 нг/л. В рамках международного стандарта ISO 17993:2002 [19] определение в поверхностной и питьевой водах 15 приоритетных ПАУ, в том числе нафталинов, возможно с нижней границей диапазона определяемых содержаний (сн) 10 нг/л.
Следует отметить, что методики с применением метода ВЭЖХ отличаются низким уровнем точности результатов определения ПАУ (δ = 45–65%, объем пробы 1 л, содержание ПАУ от 20 до 5000 нг/л [17, 18]). Такие характеристики методик связаны, очевидно, с применением колонок с обращенно-фазовыми сорбентами для разделения ПАУ, хроматография на которых предполагает смену растворителя (экстрагента, как правило, н-гексана) в образце, вводимом в хроматограф, на полярный органический модификатор подвижной фазы (метанол, ацетонитрил). Стадия смены растворителя вносит, по-видимому, значимый вклад в суммарную погрешность определения, учитывая отсутствие внутренних стандартов на стадии измерения. Включение в методики [20, 21] метода ГХ–МС снизило сн приоритетных ПАУ до 10 нг/л (поверхностные воды) и повысило точность измерения (δ = 25%).
Снижение сн ПАУ путем увеличения объема анализируемых проб практически нереализуемо при мониторинге водных объектов с большими объемами водных масс (в оз. Байкал размер водного тела достигает 23 000 км3), так как требуется отбор и анализ статистически значимого числа проб больших объемов. Для определения ПАУ в водных объектах с фоновым уровнем загрязнения необходимы новые аналитические решения, которые могут быть включены в современные системы мониторинга СОЗ.
Цель настоящей работы – разработка методики определения приоритетных ПАУ в воде на уровне следовых концентраций методом газовой хроматографии–тандемной масс-спектрометрии (ГХ–МС/МС) и ее апробация при мониторинге СОЗ в уникальной водной экосистеме оз. Байкал, отличающейся чистотой своих вод.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реагенты. Пробы воды в пелагиали оз. Байкал, прибрежной зоне и в его притоках, в поверхностном микрослое отбирали в ходе экспедиций весной и осенью 2017 г. Пробы воды верхнего водного слоя (горизонт 5 м) отбирали с помощью кассетного пробоотборника SBE-32 (CarouselWaterSampler, Sea-BirdElectronics). На каждой станции отбирали две пробы в стеклянные бутыли емк. 1 л, к которым добавляли 0.5 мл 1 М водного раствора азида натрия (MERCK, ос. ч.) в качестве консерванта. Бутыли с водой закрывали крышкой с прокладкой из алюминиевой фольги и хранили при +5°С до анализа в лаборатории. Пробы воды притоков Южного Байкала (устья рек, май и сентябрь 2017 г.), речной воды (р. Ангара в пределах городской территории, сентябрь 2017 г., март 2018 г.) отбирали в стеклянные бутыли емк. 1 л и доставляли в лабораторию для анализа. Пробы атмосферных осадков отбирали в стеклянные стаканы в парковой зоне г. Иркутска: дождь в мае 2018 г., свежевыпавший снег в декабре 2017 г. В лаборатории снег таял, затем анализировали снеговую воду. Пробы поверхностного микрослоя отбирали по методике [22].
Пробы байкальской и речной воды, атмосферных осадков анализировали без фильтрования. ПАУ экстрагировали н-гексаном (КРИОХРОМ®, сорт 1), который перегоняли перед анализом над КОН. Сульфат натрия (НЕВАРЕАКТИВ®, х. ч.) прокаливали при 600°С в течение 6 ч.
Хроматографический анализ. Для определения приоритетных ПАУ в воде в мерную колбу емк. 100 мл отбирали аликвоту пробы и добавляли суррогатный внутренний стандарт в 0.5 мл ацетона. Внутренний стандарт содержал смесь дейтерированных ПАУ (по 10 нг каждого): нафталин-d8, аценафтен-d10, фенантрен-d10, хризен-d12, перилен-d12, (Supelco, США). Раствор встряхивали и выдерживали при комнатной температуре в течение не менее 30 мин, затем добавляли 1 мл н-гексана, смесь энергично встряхивали в течение 3–5 мин и оставляли до полного расслоения фаз. Верхний слой н-гексана переносили в пробирку Эппендорфа объемом 2.0 мл, добавляли безводный Na2SO4, встряхивали и центрифугировали. Надосадочную жидкость переносили во флакон автодозатора хромато-масс-спектрометра. Для определения высокомолекулярных ПАУ, присутствующих на уровне следов (≥0.1 нг/л), экстракты концентрировали в токе аргона во флаконе автодозатора хромато-масс-спектрометра до объема ~0.1 мл.
Подготовленные образцы анализировали на хромато-масс-спектрометре Agilent Technologies 7890B GC System 7000C GC-MS Triple Quad с капиллярной колонкой OPTIMA® 17 MS Macherey-Nagel (30 м × 0.25 мм × 0.25 мкм) в режиме программирования температуры колонки: 50°С в течение 0.5 мин, затем нагрев от 50 до 310°С со скоростью 20 град/мин с последующим выдерживанием при 310°С в течение 15–20 мин (в зависимости от конфигурации хроматографа: с/без обратной продувки). Температура дозатора 280°С, температура источника 230°С, энергия ионизации 70 эВ. Образец вводили в колонку хроматографа в режиме без деления потока, объем образца 0.002–0.004 мл. Пики ПАУ и внутренних стандартов регистрировали в режиме мониторинга выбранных ионов (МВИ) и мониторинга заданных реакций (МЗР), идентифицировали по относительным временам удерживания, гомогенность аналитических пиков оценивали по соотношению площадей пиков двух переходов R = Sмзр-1/Sмзр-2 (табл. 1).
Таблица 1.
Режимы детектирования аналитических ионов компонентов
| Компонент | ГХ–МС-МВИ | ГХ–МС/МС-МЗР | ||||
|---|---|---|---|---|---|---|
| аналити-ческий ион, m/z | подтвержда-ющий ион, m/z | переходы | время выдержки, мс | энергия соударений, В | ||
| МЗР-1 | МЗР-2 | |||||
| Приоритетные ПАУ | ||||||
| Нафталин | 128 | 129, 127 | 128→128 | 128→102 | 10 | 20 |
| 2-Метилнафталин | 142 | 141, 115 | 142→141 | 141→115 | 10 | 20 |
| 1-Метилнафталин | 142 | 141, 115 | 142→141 | 141→115 | 10 | 20 |
| Аценафтилен | 152 | 151, 76 | 152→152 | 152→126 | 10 | 30 |
| Аценафтен | 154 | 153, 152 | 154→153 | 152→151 | 10 | 30 |
| Флуорен | 166 | 165, 82 | 166→165 | 165→163 | 10 | 20 |
| Фенантрен | 178 | 176, 179 | 178→178 | 178→152 | 10 | 25 |
| Антрацен | 178 | 176, 179 | 178→178 | 178→152 | 10 | 25 |
| Флуорантен | 202 | 200, 203 | 202→202 | 202→201 | 10 | 15 |
| Пирен | 202 | 200, 203 | 202→202 | 202→201 | 10 | 15 |
| Бенз[a]антрацен | 228 | 226, 229 | 228→228 | 228→226 | 10 | 20 |
| Хризен | 228 | 226, 229 | 228→228 | 228→226 | 10 | 20 |
| Бенз[b]флуорантен | 252 | 253,126 | 252→252 | 252→250 | 10 | 35 |
| Бенз[k]флуорантен | 252 | 253,126 | 252→252 | 252→250 | 10 | 35 |
| Бенз[e]пирен | 252 | 250, 125 | 252→252 | 252→250 | 10 | 35 |
| Бенз[a]пирен | 252 | 253, 126 | 252→252 | 252→250 | 10 | 35 |
| Перилен | 252 | 250, 125 | 252→252 | 252→250 | 10 | 35 |
| Индено[1,2,3-c,d]пирен | 276 | 138, 278 | 276→276 | 276→274 | 10 | 30 |
| Бенз[g,h,i]перилен | 276 | 138, 274 | 276→276 | 276→274 | 10 | 30 |
| Дибенз[a,h]антрацен | 278 | 139, 279 | 278→278 | 278→276 | 10 | 30 |
| Суррогатные внутренние стандарты | ||||||
| Нафталин-d8 | 136 | 137, 108 | 136→136 | 128→108 | 10 | 20 |
| Аценафтен-d10 | 164 | 162, 160 | 164→162 | 162→160 | 10 | 20 |
| Фенантрен-d10 | 188 | 189, 184 | 188→188 | 188→160 | 10 | 25 |
| Хризен-d12 | 240 | 236, 241 | 240→236 | 240→240 | 10 | 30 |
| Перилен-d12 | 264 | 260, 132 | 264→260 | 264→264 | 10 | 35 |
Количественное определение ПАУ. Количество ПАУ в пробах воды рассчитывали по методу внутреннего стандарта. Хроматограф градуировали в интервале ожидаемых концентраций ПАУ в экстрактах в диапазоне 0.05–50 нг/мл. Градуировочные растворы готовили разбавлением аттестованной смеси ПАУ Supelco смесью н-гексан–ацетон (1 : 1, по объему). Содержание ПАУ в пробах рассчитывали как среднее значение для двух результатов определения в одной пробе воды. Внутрилабораторную прецизионность определения ПАУ оценивали путем расчета относительных дисперсий отклонений параллельных результатов измерений от средних значений (при числе степеней свободы не менее 30) [23]. Однородность дисперсий для выделенных подгрупп концентраций ПАУ проверяли по критерию Кохрена. Правильность определения ПАУ оценивали методом введено–найдено, при этом добавку к пробам воды вводили в виде растворов ПАУ в ацетоне. Чистоту растворителей и оборудования оценивали проведением холостых опытов. Присутствие в н-гексане следов ПАУ рассматривали как систематическую погрешность определения, которую вычитали из результата анализа. Предел обнаружения ПАУ оценивали по соотношению сигнал/шум (S/N) ≥ 3 на регистрируемых хроматограммах.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для определения приоритетных ПАУ в воде при содержании индивидуальных аренов на уровне следов нами разработана методика, основанная на экстракции аналитов в н-гексан (Vпробы = = 0.1 л, коэффициент концентрирования 102–103). Найдено, что при прямом анализе экстрактов, полученных однократной экстракцией с извлечением аналитов до ~80%, регистрации пиков ПАУ методом ГХ–МС-МВИ с соотношением S/N ≥ 10 возможна при содержании целевых компонентов не менее чем 1.0 нг/л, например, при анализе экстрактов байкальской воды (рис. 1а). При содержании ПАУ в воде на более низком уровне высота пиков аналитических и (или) подтверждающих ионов сопоставима с уровнем фона (например, для пиков аценафтилена, аценафтена, рис. 1а) и, как следствие, надежность идентификации пиков ПАУ снижается или они не регистрируются с S/N > 3 (пики ионов ПАУ с m/z 252, 276 и 278). Из-за наложения пиков ПАУ и мешающих компонентов экстракта результаты определения могут быть завышены, например, для антрацена и аценафтилена до трех раз. Исключением является пик пирена, регистрируемый при содержании в пробе ~0.4 нг/л, очевидно, вследствие отсутствия фона на масс-роматограмме при m/z 202.
Рис. 1.
Масс-фрагментограммы экстрактов байкальской воды, полученные в режимах МВИ (а) и МЗР (б, в). Пики компонентов (нг/л): 1 – нафталин (14), 2 – 2-метилнафталин (19), 3 – 1-метилнафталин (11), 4 – аценафтилен (1.1), 5 – аценафтен (1.2), 6 – флуорен (3.9), 7 – фенантрен (5.3), 8 – антрацен (0.23), 9 – флуорантен (2.1), 10 – пирен (0.76), 11 – бенз[a]антрацен (0.41), 12 – хризен (0.58), 13 – бенз[b]флуорантен (0.25), 14 – бенз[k]флуорантен (0.15), 15 – бенз[e]пирен (0.12), 16 – бенз[а]пирен, 17 – перилен, 18 – дибенз[a,h]антрацен, 19 – индено[1,2,3-c,d]пирен, 20 – бенз[g,h,i]перилен (концентрации компонентов 16–20 ниже предела определения). Масс-спектрометрические параметры регистрации аналитических пиков в режимах МВИ и МЗР приведены в табл. 1.
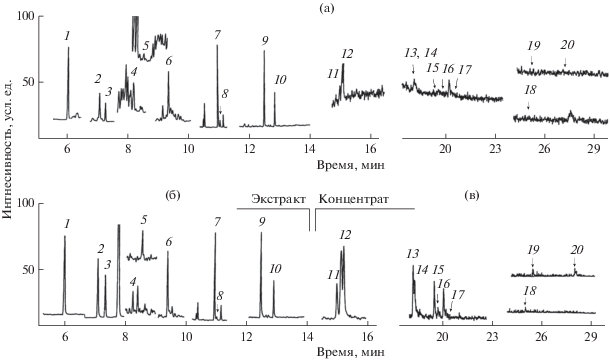
Так как концентрирование экстракта сопровождается ростом не только интенсивности аналитических пиков, но и фона, анализ сконцентрированных образцов методом ГХ-МС-МВИ неэффективен в рамках надежности идентификации и точности определения. Применение метода ГХ–МС/МС с детектированием аналитических пиков в режиме МЗР решает проблему надежности идентификации и снижает сн. Выбор соответствующих переходов обеспечивает практическое отсутствие фона и гомогенность пиков, для которых по данным ГХ–МС-МВИ значение S/N < 10 (рис. 1б).
Для высокомолекулярных ПАУ (ряд бенз[a]антрацен–бенз[a]пирен) снижение уровня определения достигнуто путем концентрирования экстрактов до объема ~ 0.1 мл (рис. 1в). Показано, что стадия концентрирования не приводит к значимым потерям аналитов и снижению точности их определения. При таком варианте подготовки пробы определение бенз[a]пирена на уровне 0.1 нг/л соответствует 0.02 ПДК, установленной для питьевой воды.
В случае нафталинов, фенантрена, флуорантена и пирена концентрирование экстракта снижает точность результата определения вследствие потерь летучих ПАУ и накопления в образце аналитов, присутствующих в экстрагенте на уровне следов. По этой причине в методике предложен способ прямого анализа экстрактов и Сн определяется чистотой используемого экстрагента, а также объем аликвоты пробы не превышает 100 мл и объем экстрагента 1 мл. Увеличение объема пробы воды (до 1 л и более) и ее многократная экстракция большим объемом экстрагента (до 60 мл и более) с последующим концентрированием экстракта не приводит к снижению предела определения данной группы ПАУ с требуемой точностью определения.
Ограниченное число мешающих и сопутствующих компонентов в экстрактах проб воды с загрязнением на фоновом уровне дает возможность регистрировать пики ПАУ, доминирующих в выделенных фракциях, за короткий хроматографический цикл – до 17 мин (рис. 1а, 1б). Для определения высокомолекулярных ПАУ, в том числе бенз[a]пирена, необходимо увеличение времени хроматографии до 30 мин (рис. 1в).
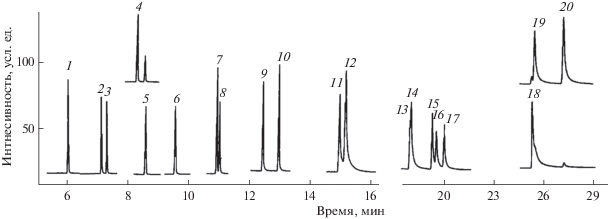
Для хроматографирования экстрактов предложена колонка Optima® 17 ms с фазой средней полярности (50% фенил–50% метилполисилоксан), на которой достигается разделение всех ПАУ из приоритетного ряда (рис. 2). Однако ресурс данных колонок ограничен (~1000 образцов), и после его окончания разрешение пиков бенз[b]флуорантена и бенз[k]флуорантена снижается. Разделение выделенных фракций ПАУ на колонках с фазами низкой полярности (HP-5 ms, DB-5 ms, Rxi-5 Sil ms) [7–9, 11, 12, 14, 15, 24] отличается близкими временами удерживания у пар аренов: фенантрен – антрацен, бенз[a]антрацен – хризен, бенз[b]флуорантен – бенз[k]флуорантен и индено[1,2,3-c,d]пирен – дибенз[a,h]антрацен. Разделение приоритетных ПАУ на специализированных колонках, например колонке НТ-8 SGE Inc. с карборановой фазой, используемой при определении хлорорганических загрязнителей, характеризуется разделением всех ПАУ, за исключением бенз[b]флуорантен и бенз[k]флуорантен, имеющих равные времена удерживания.
Рис. 2.
Масс-фрагментограммы градуировочного раствора аттестованной смеси ПАУ с концентрацией 10 нг/мл (20 пг/пик) каждого компонента, полученные в режиме МВИ: 1 – нафталин, 2 – 2-метилнафталин, 3 – 1-метилнафталин, 4 – аценафтилен, 5 – аценафтен, 6 – флуорен, 7 – фенантрен, 8 – антрацен, 9 – флуорантен, 10 – пирен, 11 – бенз[a]антрацен, 12 – хризен, 13 – бенз[b]флуорантен, 14 – бенз[k]флуорантен, 15 – бенз[e]пирен, 16 – бенз[а]пирен, 17 – перилен, 18 – дибенз[a,h]антрацен, 19 – индено[1,2,3-c,d]пирен, 20 – бенз[g,h,i]перилен.
Разработанная методика апробирована при анализе проб воды оз. Байкал, характеризующихся следовым уровнем концентраций ПАУ, а также проб, отличающихся более высоким содержанием аренов (табл. 2): образцы воды прибрежной зоны озера; пробы воды, собранные в период весеннего таяния льда и снега; пробы поверхностного водного микрослоя, в котором аккумулируются ПАУ, поступающие из атмосферы; образцы атмосферных осадков в городской зоне. Образцы осадков отличались высоким содержанием суммы ПАУ (ΣПАУ) – до 600 нг/л, качественным составом аренов (присутствовали индено[1,2,3-c,d]пирен, бенз[g,h,i]перилен и бенз[a]пирен).
Таблица 2.
Результаты (нг/л) определения ПАУ в водных образцах
| Район отбора проб | Число проб | ΣПАУ | Σнафталинов (% от ΣПАУ) |
Бенз[a]пирен |
|---|---|---|---|---|
| Пелагиаль оз. Байкал, 5 м, центральные точки в трех котловинах | 9 | 9.3–40 | 5.5–17 (47–79) | <0.1 |
| Прибрежная зона, п. Листвянка, 100 м от берега | 8 | 18–220* | 12–130 (58–68) | <0.1 |
| Притоки Южного Байкала | 6 | 7.3–20 | 1.1–4.6 (15–40) | <0.1 |
| Поверхностный водный микрослой оз. Байкал, южная котловина | 12 | 90–290 | 70–260 (78–89) | 0.2–0.5 |
| р. Ангара в черте г. Иркутска | 2 | 36–95 | 28–75 (75) | <0.1 |
| Свежевыпавший снег в парковой зоне г. Иркутска | 2 | 410–610 | 210–220 (34–55) | 0.2–4.1 |
| Дождь в парковой зоне г. Иркутска | 2 | 340–420 | 63–150 (18–37) | 1.7–5.8 |
По результатам апробации методики сн нафталинов составила 1.0 нг/л, приоритетных ПАУ с m/z 152, 154, 166, 178 и 202 – 0.2 нг/л, высокомолекулярных ПАУ – 0.1 нг/л. Оценка правильности определения (табл. 3) показала отсутствие значимой систематической погрешности, внутрилабораторная прецизионность (σR) составила 15–20% для индивидуальных ПАУ.
Таблица 3.
Оценка правильности определения ПАУ в воде прибрежной зоны Байкала (п. Лиственничное) методом введено–найдено (Р = 0.95)
| ПАУ | Содержание в пробе воды, нг/л (n = 4) | Введено, нг | Найдено, нг/л (n = 2) | Добавка, нг | |||
|---|---|---|---|---|---|---|---|
| I | II | I | I | I | II | ||
| Нафталин | 1.9 ± 0.3 | 2.8 | 1.4 | 4.7 ± 0.7 | 3.6 ± 0.5 | 2.8 ± 0.4 | 1.6 ± 0.2 |
| 2-Метилнафталин | 0.34 ± 0.05 | 2.2 | 1.1 | 2.5 ± 0.4 | 1.6 ± 0.2 | 2.2 ± 0.3 | 1.3 ± 0.2 |
| 1-Метилнафталин | 0.25 ± 0.04 | 2.2 | 1.1 | 2.4 ± 0.4 | 1.5 ± 0.2 | 2.2 ± 0.3 | 1.2 ± 0.2 |
| Аценафтилен | 0.23 ± 0.03 | 2.5 | 0.57 | 2.7 ± 0.4 | 0.8 ± 0.1 | 2.5 ± 0.4 | 0.54 ± 0.08 |
| Аценафтен | <0.2 | 2.6 | 0.59 | 2.6 ± 0.4 | 0.63 ± 0.09 | 2.6 ± 0.4 | 0.63 ± 0.09 |
| Флуорен | 0.26 ± 0.04 | 2.8 | 0.65 | 2.9 ± 0.4 | 0.8 ± 0.1 | 2.7 ± 0.4 | 0.58 ± 0.09 |
| Фенантрен | 0.7 ± 0.1 | 2.3 | 0.53 | 3.0 ± 0.5 | 1.3 ± 0.2 | 2.3 ± 0.3 | 0.6 ± 0.1 |
| Антрацен | 0.20 ± 0.03 | 1.9 | 0.44 | 2.2 ± 0.3 | 0.60 ± 0.09 | 2.0 ± 0.3 | 0.40 ± 0.06 |
| Флуорантен | 0.40 ± 0.06 | 2.3 | 0.54 | 3.2 ± 0.5 | 1.0 ± 0.2 | 2.8 ± 0.4 | 0.6 ± 0.1 |
| Пирен | <0.2 | 2.5 | 0.62 | 3.0 ± 0.5 | 0.7 ± 0.1 | 3.0 ± 0.5 | 0.7 ± 0.1 |
| Бенз[a]антрацен | <0.1 | 1.6 | 0.36 | 2.0 ± 0.4 | 0.41 ± 0.08 | 2.0 ± 0.4 | 0.41 ± 0.08 |
| Хризен | <0.1 | 2.0 | 0.47 | 2.3 ± 0.5 | 0.5 ± 0.1 | 2.3 ± 0.5 | 0.5 ± 0.1 |
| Бенз[b]флуорантен Бенз[k]флуорантен* | 0.10 ± 0.02 | 2.3 | 0.54 | 2.7 ± 0.5 | 0.6 ± 0.1 | 2.6 ± 0.5 | 0.5 ± 0.1 |
| Бенз[e]пирен | 0.20 ± 0.04 | 0.92 | 0.22 | 1.2 ± 0.2 | 0.39 ± 0.08 | 1.0 ± 0.2 | 0.19 ± 0.04 |
| Бенз[а]пирен | <0.1 | 0.87 | 0.21 | 0.9 ± 0.2 | 0.28 ± 0.06 | 0.9 ± 0.2 | 0.28 ± 0.06 |
| Перилен | <0.1 | 0.77 | 0.20 | 0.8 ± 0.2 | 0.22 ± 0.04 | 0.8 ± 0.2 | 0.22 ± 0.04 |
| Индено[1,2,3-c,d]пирен | <0.1 | 0.67 | 0.22 | 0.7 ± 0.1 | 0.23 ± 0.05 | 0.7 ± 0.1 | 0.23 ± 0.05 |
| Бенз[g,h,i]перилен | <0.1 | 0.84 | 0.20 | 0.9 ± 0.2 | 0.24 ± 0.05 | 0.9 ± 0.2 | 0.24 ± 0.05 |
| Дибенз[a,h]антрацен | <0.1 | 0.80 | 0.20 | 0.8 ± 0.2 | 0.24 ± 0.05 | 0.8 ± 0.2 | 0.24 ± 0.05 |
Установлено, что предложенная методика подготовки проб легко реализуется в серийном анализе при мониторинговых исследованиях. Прямой анализ экстрактов устраняет риск потерь легколетучих ПАУ, минимальный объем экстрагента (1 мл) сокращает расход органического растворителя и его объемы для последующей утилизации. Объем воды, требуемый для проведения анализа, не превышает 0.1 л и дает возможность контролировать содержание ПАУ в крупных водных объектах с отбором статистически значимого числа проб, а также проводить исследования, при которых объем отбираемых проб воды ограничен (поверхностный микрослой, атмосферные осадки, пробы из отдаленных и труднодоступных районов). Применение метода ГХ–МС/МС соответствует нормам Европейского законодательства по выполнению химических анализов и представления результатов [25].
Исследование выполнено в рамках государственного задания Министерства науки и высшего образования РФ (проект № 0345–2016–0008, № гос. рег. АААА-А16-116122110065-4) на оборудовании приборного центра коллективного пользования физико-химического ультрамикроанализа ФГБУН Лимнологический институт СО РАН. Авторы выражают искреннюю благодарность сотрудникам ФГБУН Лимнологический институт СО РАН с.н.с., к.б.н. С.М. Шишлянникову и м.н.с., к.б.н. И.С. Михайлову за помощь в отборе проб воды озера Байкал и его притоков; с.н.с., к.б.н, М.Ю. Сусловой за отбор проб воды поверхностного микрослоя.
Список литературы
IARC. Monographs on the evaluation of carcinogenic risks to humans: some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. International Agency for Research on Cancer, France. V. 92. Some Non-heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. Lyon. France, 2010. 868 p.
EPA USA. Environmental Protection Agency. Priority Pollutants, 2002.
Directive 2013/39/EU of the European parliament and of the council. August 12, 2013.
ГН 2.1.5.2280-07. Предельно допустимые концентрации (ПДК) химических веществ в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования, 2007.
СанПиН 2.1.4.1074-01. Питьевая вода и водоснабжение населенных мест, 2010.
US EPA Method 3510C Separatory funnel liquid-liquid extraction, 1996. 8 p.
Net S., Dumoulin D., El-Osmani R., Rabodonirina S., Ouddane B. Case study of PAHs, Me-PAHs, PCBs, Phthalates and pesticides contamination in the somme river water, France // Int. J. Environ. Res. 2014. V. 8. № 4. P. 1159.
Aziz F., Husain J., Riffat S., Malik N., Katsoyiannis A., Mahmood A., Li J., Zhang G., Jone K.C. Occurrence of polycyclic aromatic hydrocarbons in the Soan River, Pakistan: Insights into distribution, composition, sources and ecological risk assessment // Ecotoxicol. Environ. Saf. 2014. V. 109. P. 77.
Munyengabe A., Mambanda1 A., Moodley B. Polycyclic aromatic hydrocarbons in water, soils and surface sediments of the Msunduzi river // J. Environ. Anal. Chem. 2017. V. 4. № 4. https://doi.org/10.4172/2380-2391.1000227
King A.J., Readmanb J.W., Zhou J.L. Determination of polycyclic aromatic hydrocarbons in water by solid-phase microextraction–gas chromatography–mass spectrometry // Analytica Chimica Acta. 2004. V. 523. P. 259.
Sánchez-Avila J., Fernandez-Sanjuan M., Vicente J., Lacorte S. Development of a multi-residue method for the determination of organicmicropollutants in water, sediment and mussels using gas chromatography–tandem mass spectrometry // J. Chromatogr. A. 2011. V. 1218. P. 6799.
Fu S., Fan J., Hashi Y., Chen Z. Determination of polycyclic aromatic hydrocarbons in water samples onine microextraction by packed sorbent coupled with gas chromatography–mass spectrometry // Talanta. 2012. V. 94. P. 152.
Helvécio C., Menezes A.C., Paiva M.J.N., Santos R.R., Sousa L.P., Resende S.F., Saturnino J.A., Paulo B.P., Cardeal Z.L. A sensitive GC/MS method using cold fiber SPME to determine polycyclic aromatichydrocarbons in spring water // Microchem. J. 2013. V. 110. P. 209.
Sánchez-Avila J.I., Kretzschmar T. Simultaneous determination of polycyclic aromatic hydrocarbons, alkylphenols, phthalate esters and polychlorinated biphenyls in environmental waters based on headspace–solid phase microextraction followed by gas chromatography–tandem mass spectrometry // J. Environ. Anal. Chem. 2017. V. 4. № 4. https://doi.org/10.4172/2380-2391.1000226
Yan J., Kim M., Haberl M., Kwok H., Brunswick P., MacInnis C., van Aggelena G., Shang D. Determination of polycyclic aromatic hydrocarbons in surface water using simplified liquid–liquid micro-extraction and pseudo-MRM GC/MS/MS // Anal. Methods. 2018. V. 10. P. 405.
Sánchez-Avila J., Tauler R., Lacorte S. Organic micropollutants in coastal waters from NW Mediterranean Sea: Sources distribution and potential risk // Environt. Int. 2012. V. 46. P. 50.
ПНД Ф 14.1:2:4.70-96. Методика выполнения измерений массовой концентрации полициклических ароматических углеводородов в пробах питьевых, природных и сточных вод хроматографическим методом ВЭЖХ. М.: Росгидромет, 1996 (издание 2004). 25 с.
РД 52.44.590. Массовая концентрация приоритетных компонентов полициклических ароматических углеводородов в пробах атмосферных осадков и поверхностных вод. Методика измерений методом высокоэффективной жидкостной хроматографии. М.: Росгидромет, 2016. 49 с.
International standard. Water quality. Determination of 15 polycyclic aromatic hydrocarbons (PAH) in water by HPLC with fluorescence detection after liquid-liquid extraction, ISO 17993:2002. 22 c.
PBM. Polycyclic Aromatic Hydrocarbons in Water by GS/MS. Revision (2017). https://www2.gov.bc.ca/assets/ gov/environment/research-monitoring-and-reporting/ monitoring/emre/methods/pah_in_water_pbm_7mar 2017_draft.pdf (22.05.2018).
International standard. Water quality. Determination of 16 polycyclic aromatic hydrocarbons (PAH) in water. Method using gas chromatograph with mass spectrometric detection, ISO 28540:2011. 29 c.
Galachyants A.D., Belkova N.L., Sukhanova E.V., Blinov V.V., Parfenova V.V. Methods of Neuston sampling for the quantitative characteristic of microbial communities of lake Baikal // Inland Water Biology. 2016. V. 9. № 3. P. 329.
Смагунова А.Н., Карпунова О.М. Методы математической статистики в аналитической химии. Иркутск: Иркутский государственный университет, 2008. 340 с.
Sadeghi R., Kobarfard F., Yazdanpanah H., Eslamizad S., Bayat M. Validation of an analytical method for determination of 13 priority polycyclic aromatic hydrocarbons in mineral water using dispersive liquid-liquid microextraction and GC-MS // Iran. J. Pharm. Res. 2016. V. 15. № 1. P. 157.
Commission Decision 2002/657/EC, August 12, 2002, implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results // Off. J. Eur. Union. 2002. 36 p.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии