

Журнал аналитической химии, 2019, T. 74, № 9-app, стр. 38-47
Экспрессное определение аминогликозидов в молоке по точным массам ионов методом ультравысокоэффективной жидкостной хроматографии–квадруполь времяпролетной масс-спектрометрии высокого разрешения
В. Г. Амелин a, b, *, Д. С. Большаков a
a Федеральный центр охраны здоровья животных
600901 Владимир, мкрн. Юрьевец, Россия
b Владимирский государственный университет им. Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 12.07.2018
После доработки 19.11.2018
Принята к публикации 08.03.2019
Аннотация
Предложен способ экспрессного определения 10 аминогликозидов (амикацина, апрамицина, гентамицина, гигромицина В, дигидрострептомицина, канамицина А, неомицина, паромомицина, спектиномицина, стрептомицина) в молоке по точным массам ионов методом ультравысокоэффективной жидкостной хроматографии в сочетании с квадруполь-времяпролетной масс-спектрометрией высокого разрешения. Предложен простой способ пробоподготовки. Определение аминогликозидов возможно совместно с другими ветеринарными препаратами в молоке в условиях обращенно-фазовой хроматографии с добавлением ион-парного реагента (гептафтормасляной кислоты) в анализируемый экстракт из пробы. Установлен значительный матричный эффект при определении аминогликозидов в молоке (79–96%), который нивелирован разбавлением экстракта в 10 раз деионированной водой. Количественное определение проводили методом абсолютной градуировки. Диапазоны определяемых содержаний аминогликозидов составили 10–1000 нг/г. Относительное стандартное отклонение результатов анализа не превышало 0.17. Продолжительность идентификации аминогликозидов в молоке 15 мин, продолжительность определения обнаруженных аналитов 30–40 мин.
Аминогликозиды (аминогликозидные аминоциклитолы) – природные и полусинтетические антибиотики широкого спектра действия, имеющие в своем составе молекулы аминосахаридов, соединенные гликозидной связью с аминоциклитоловым кольцом (схема 1 ). В настоящее время выделено более 10 природных антибиотиков, продуцируемых некоторыми штаммами грибов Actinomyces, Micromonospora, Streptomyces и Bacillus. К группе полусинтетических аминогликозидов (наряду с амикацином) относят и природный аминоциклитольный антибиотик спектиномицин, не содержащий аминосахаридов, но имеющий сходную структуру [1].
Аминогликозидные антибиотики нашли широкое применение как в медицине, так и в ветеринарии. В ветеринарии их используют для лечения мастита у молочных животных, лептоспироза у крупного рогатого скота, овец, коз и свиней, а также при лечении различных грамотрицательных бактериальных инфекций [1]. Неправильное и/или избыточное применение антибактериальных препаратов может приводить к их накоплению в различных тканях животных и соответственно в продукции животного происхождения, употребление которой в пищу может приводить к развитию антибиотикорезистентности.
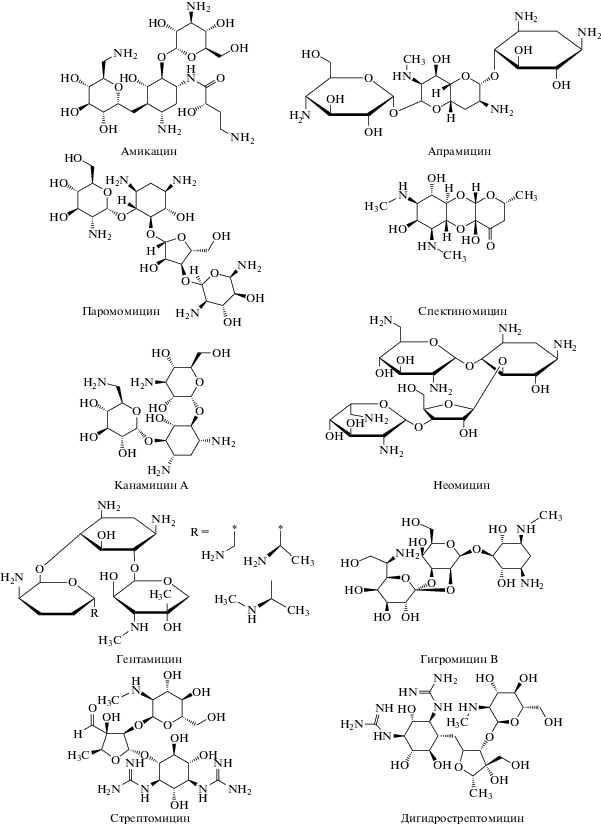
Схема 1 . Структурные формулы аминогликозидов.
Высокополярные аминогликозиды практически не удерживаются на обращенно-фазовом сорбенте в жидкостной хроматографии. Тем не менее, ранее нами показана возможность определения аминогликозидов наряду с другими ветеринарными препаратами в пищевых продуктах методом обращенно-фазовой ВЭЖХ–квадруполь времяпролетной масс-спектрометрии высокого разрешения [2]. Определение проводили по точным массам ионов, образующихся в электрораспылительном устройстве. Методика разработана для одновременного определения 492 токсикантов в пищевых продуктах (в том числе и аминогликозидов), однако оказалось, что определение аминогликозидов в данных условиях малочувствительно, время их выхода составляло 1–2 мин при общей продолжительности хроматографирования 25 мин.
В настоящее время аминогликозиды в пищевых продуктах определяют отдельно от других ветеринарных препаратов методом гидрофильной хроматографии (ГФХ) с тандемным масс-спектрометрическим детектированием (МС/МС) [3, 4], ион-парной ВЭЖХ–МС/МС [5–11], либо обращенно-фазовой ВЭЖХ–МС/МС после предварительной дериватизации их фенилизоцианатом [12]. Гидрофильная хроматография является альтернативой нормально-фазовой хроматографии (НФХ) для разделения сильнополярных соединений. Подобно НФХ, в ГФХ элюированию способствует использование полярных неподвижных фаз, но ГФХ уникальна тем, что присутствие воды в подвижной фазе способствует образованию на полярном адсорбенте полимолекулярного адсорбционного слоя, в котором происходит высокоселективное разделение аналитов [3]. Подобные работы еще не нашли широкого распространения, очевидно, в силу большей специфичности используемых полярных фаз. При использовании ион-парной ВЭЖХ в качестве ион-парного реагента применяли гептафтормасляную кислоту, вводимую в подвижную фазу [5–11].
Следует отметить, что пробоподготовка для определения аминогликозидов сложна, длительна и вкючает очистку экстракта методом твердофазной экстракции (ТФЭ). При очистке методом ТФЭ применяли сорбенты различной природы: Oasis HLB на основе сополимера дивинилбензола и N-винилпирролидона [5], Baker Bond CBX на основе катионообменной смолы [4, 6], Oasis MCX на основе смеси катионообменной смолы и обращенно-фазового сорбента [3, 9]. Использование ТФЭ позволяет эффективно устранить матричный эффект, но существенно влияет на степень извлечения целевых компонентов [6].
Ранее нами предложено определение 150 ветеринарных препаратов (без аминогликозидов) в молоке методом обращенно-фазовой ультра-ВЭЖХ с масс-спектрометрическим детектором высокого разрешения [13]. Цель данной работы – использование этих условий для определения аминогликозидов, а также значительное упрощение пробоподготовки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Использовали ультравысокоэффективный жидкостный хроматограф UltiMate 3000 (Thermo Scientific, США) в сочетании с квадруполь-времяпролетным масс-спектрометрическим детектором maXis 4G и ионизацию электрораспылением в устройстве ionBooster (Bruker Daltonics, Германия). Разделение проводили на колонке 30 × 2.1 мм (1.7 мкм) ACQUITY UPLC® BEN C18 (Thermo Scientific, США) в режиме градиентного элюирования.
Применяли весы аналитические Sartorius TE214S I специального класса точности (Sartorius, Германия), центрифугу MPW-260R (MPW Med. Instruments, Польша), фильтры мембранные политетрафторэтиленовые 0.20 мкм диаметром 25 мм (Corning Incorporated, Германия).
Реактивы. Использовали стандартные образцы аминогликозидов (дисульфат амикацина; сульфат апрамицина; сульфат 1-водный гентамицина, гигромицина Б; сульфат дигидрострептомицина; дисульфат 1-водный канамицина А; трисульфат 1-водный неомицина; сульфат паромомицина; дигидрохлорид 5-водный спектиномицина; 1.5-сульфат 1-водный стрептомицина) с содержанием основного вещества 69.9–100% (Sigma, Sigma-Aldrich, Fluka, USP reference standard, США). Все растворы готовили с использованием деионированной воды (15–18 МОм ∙ см, ТУ 2638-263-00209792-00). Исходные стандартные растворы (1 мг/мл) готовили растворением навески препаратов в метаноле или ацетонитриле. Рабочие растворы готовили разбавлением исходных растворов водой. Использовали метанол (Fisher Scientific UK, Великобритания), н-гексан, ацетонитрил (Scharlab, S.L., Испания), муравьиную кислоту, гептафтормасляную кислоту (Aldrich, США), изопропанол, трихлоруксусную кислоту (ООО “Компонент-реактив”, Россия).
Идентификация и определение. Аминогликозиды идентифицировали по полученным хроматограммам с использованием программного продукта DataAnalysis-4.1, TargetAnalysis (Bruker Daltonics, Германия), картину изотопного распределения аналитов составляли с использованием IsotopePattern (Bruker Daltonics, Германия). Неизвестную концентрацию аналита в пробе определяли по внешней градуировке после разбавления экстракта водой в 10 раз.
Оценка матричного эффекта. Для оценки матричного эффекта (МЭ) использовали площади хроматографических пиков аналитов с концентрацией 10 и 100 нг/г, полученные в условиях анализа экстракта молока, не содержащего исследуемых соединений, и воды. Кроме того, матричный эффект рассчитывали с использованием градуировочных графиков (по тангенсу углов наклона), построенных с использованием воды и экстракта из молока. Матричный эффект рассчитывали по формуле:
Условия хроматографического разделения и де-тектирования. Подвижная фаза состояла из 0.1%-ной муравьиной кислоты в воде (А) и 0.1%-ной муравьиной кислоты в ацетонитриле (В). Осуществляли градиентное элюирование: 0 мин – 5% В, 0.5 мин – 5% В, 2 мин – 50% В, 5 мин – 100% В, 6 мин – 5% В, 8 мин – 5% В. Скорость потока подвижной фазы 0.4 мл/мин. Температура хроматографической колонки 50°С, объем вводимой пробы 50 мкл. Температура термостата автоматического дозатора 10°С. Выбор оптимальных условий определения описан в работе [13].
Использовали ионизацию электрораспылением в устройстве ionBooster (Bruker Daltonics, Германия). Установлены следующие оптимальные значения параметров: напряжение на щите капилляра 400 В, на капилляре 1000 В, давление газа-распылителя азота 4.76 атм, поток газа-осушителя азота 6 л/мин, температура газа-осушителя азота 200°С, поток газа-испарителя азота 250 л/ч, температура газа-испарителя азота 250°C.
Диапазон регистрируемых масс ионов 100–1000 Да. Для градуировки использовали раствор формиата натрия 10 мМ в смеси вода–изопропанол (1 : 1, по объему) в интервале хроматографирования 7.5–8 мин.
Пробоподготовка. В центрифужную пробирку емк. 15 мл вносили 1.00 мл перемешанного молока, добавляли 0.25 мл 20%-ной трихлоруксусной кислоты, 0.1 г ЭДТА и перемешивали, центрифугировали в течение 5 мин при 2700 об./мин. Надосадочную жидкость переливали в другую пробирку емк. 15 мл, добавляли 2 мл н-гексана, встряхивали вручную в течение 5 мин.
Для идентификации отбирали нижний слой экстракта с помощью шприца, добавляли 50 мкл 10%-ной гептафтормасляной кислоты и фильтровали через мембранный фильтр 0.20 мкм в микрофлакон для хроматографирования, по полученным хроматограммам проводили идентификацию с использованием соответствующего программного обеспечения.
Для количественного анализа отбирали 0.5 мл нижнего слоя экстракта в пробирку емк. 15 мл, добавляли 4.5 мл воды, перемешивали, отбирали 1 мл полученного раствора, добавляли 50 мкл 10%-ной гептафтормасляной кислоты и фильтровали через мембранный фильтр 0.20 мкм в микрофлакон для хроматографирования. Концентрацию определяли по градуировочному графику, построенному с использованием воды.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Использование ион-парной добавки. Аминогликозиды в своей структуре не содержат хромоформных групп, поэтому их прямое спектральное определение в УФ или видимой областях спектра невозможно (схема 1 ). Кроме того, антибиотики данного класса являются высокополярными соединениями, практически не удерживающимися на обращенно-фазовом сорбенте С18 (рис. 1). Это может приводить к погрешностям при идентификации антибиотиков по временам удерживания. Даже при использовании режима сканирования ионов–продуктов (МС/МС-режим) в этом случае идентификация аминогликозидов затруднена в силу близости их структур.
Рис. 1.
Масс-хроматограммы однозарядных [M + H]+ (а) и двухзарядных [M + 2H]2+ (б) протонированных молекул аминогликозидов без добавки (I) и в присутствии добавки ион-парного реагента (II): 1 – спектиномицин, 2 – дигидрострептомицин, 3 – гигромицин В, 4 – стрептомицин, 5 – канамицин А, 6 – амикацин, 7 – парамомицин, 8 – неомицин, 9 – апрамицин, 10 – гентамицин.
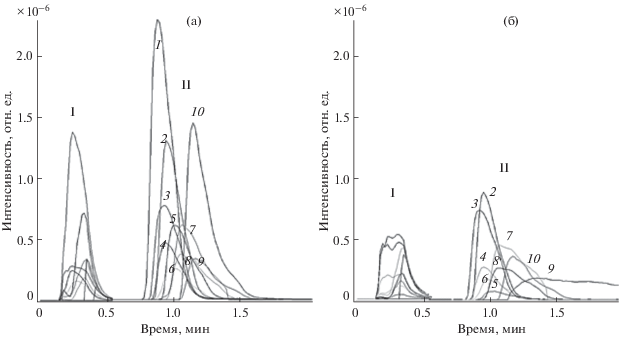
Использование ион-парных реагентов может в значительной степени повысить удерживание заряженных компонентов пробы на обращенно-фазовом сорбенте. Однако известно, что введение ион-парной добавки в состав подвижной фазы для динамического модифицирования колонки неприемлемо при использовании градиентного режима элюирования и масс-спектрометрического детектирования. По этой причине ион-парную добавку вносили непосредственно в раствор для хроматографирования. В качестве ион-парной добавки использовали гептафтормасляную кислоту. Применение высокополярного соединения способствует образованию ионной пары с молекулами аминогликозидов, что приводит к увеличению гидрофобности молекул разделяемых компонентов. При этом происходит дифференцирование времен удерживания аналитов (рис. 1).
Условия хроматографического разделения. Установлено, что использование части экстракта из молока, разбавление его деионированной водой и анализ в выбранных ранее условиях определения 150 препаратов [13] позволяет значительно сократить продолжительность анализа и определять уже 160 ветеринарных препаратов в молоке.
Идентификация. В условиях ионизации электрораспылением определяемые аминогликозиды образуют как однозарядные ионы [M + H]+, так и двухзарядные протонированные молекулы [M + 2H]2+ (рис. 1). Исключением является спектиномицин, который образует только однозарядные ионы. Спектиномицин и стрептомицин образуют протонированные аддукты с молекулой воды [M + H2O + H]+ (табл. 1, рис. 1).
Таблица 1.
Основные характеристики аминогликозидов в присутствии гептафтормасляной кислоты
| Аналит | Брутто-формула | Ион | tR, мин | m/z | МДУ ТС*, нг/г | cmin, нг/г | сн, нг/г |
|---|---|---|---|---|---|---|---|
| Амикацин | C22H43N5O13 | [M + H]+ [M + 2H]2+ |
1.00 | 586.2930 293.6501 | – | 0.5 | 1.0 |
| Апрамицин | C21H41N5O11 | [M + H]+ [M + 2H]2+ |
1.15 | 540.2875 270.6474 | – | 0.2 | 0.5 |
| Гентамицин | C21H43N5O7 | [M + H]+ | 1.25 | 478.3235 | 100 | 0.5 | 1.0 |
| Гигромицин В | C20H37N3O13 | [M + H]+ [M + 2H]2+ |
0.91 | 528.2399 264.6236 | – | 0.2 | 0.5 |
| Дигидрострептомицин | C21H41N7O12 | [M + H]+ [M + 2H]2+ | 1.05 | 584.2885 292.6479 | – | 0.1 | 1.0 |
| Канамицин А | С18H36N4О11 | [M + H]+ [M + 2H]2+ |
1.02 | 485.2453 243.1263 | 150 | 0.3 | 0.5 |
| Неомицин | C23H46N6O13 | [M + H]+ [M + 2H]2+ |
1.28 | 615.3195 308.1634 | 1500 | 0.5 | 1.0 |
| Паромомицин | C23H45N5O14 | [M + H]+ [M + 2H]2+ |
1.30 | 616.3036 308.6554 | – | 0.5 | 1.0 |
| Спектиномицин | C14H24N2O7 | [M + H2O + H]+ | 0.90 | 351.1762 | 200 | 0.5 | 1.0 |
| Стрептомицин | C21H39N7O12 | [M + H]+ [M + 2H]2+ [M + H2O + H]+ |
1.00 | 582.2729 291.6401 600.2835 | 200 | 0.5 | 1.0 |
Аминогликозиды идентифицировали с использованием программы TargetAnalysis. Идентификационными параметрами служили времена удерживания, точность моноизотопной массы иона (m/z) и параметр mSigma (соответствие теоретического изотопного распределения практическому). Погрешность определения масс ионов не превышала ±5 млн–1 (n = 3). На рис. 2 представлены сгенерированный программой IsotopePattern масс-спектр гентамицина и спектр, полученный экспериментально. Как видно, характер и картина форм изотопных отношений полностью совпадают.
Рис. 2.
Масс-спектры гентамицина, сгенерированные программой IsotopePattern по брутто-формуле для [M + H]+ (а), [M + 2H]2+ (в) и полученные экспериментально для экстракта из молока [M + H]+ (б), [M + 2H]2+ (г).

Рис. 2.
Окончание.

Оценка матричного эффекта. Матричный эффект обусловлен влиянием присутствующих в экстракте компонентов матрицы на ионизацию аминогликозидов. В условиях ионизации электрораспылением они могут как усиливать (+), так и понижать (–) интенсивность сигнала аналита. В табл. 2 представлены значения МЭ для проб молока при определении десяти аминогликозидов в условиях до 15-кратного разбавления экстракта пробы водой. Согласно [14] МЭ можно пренебречь, когда его значения находятся в диапазоне ±20%. В соответствии с данными табл. 2 и 3 при определении аминогликозидов в молоке наблюдается значительный МЭ. Следует отметить, что МЭ в большей степени влияет на регистрацию двухзарядных протонированных форм аминогликозидов, что может быть связано с интенсификацией конкурентных реакций комплексо- и солеобразования при ионизации аналитов. Существует несколько способов нивелирования МЭ при анализе объектов биологического происхождения: уменьшение объема вводимой пробы, разбавление конечного экстракта водой, использование изотопно-меченного внутреннего стандарта, использование метода стандартной добавки, использование матричной градуировки [15]. Нами использован наиболее простой и эффективный вариант нивелирования МЭ – разбавление экстракта водой (использование внешней градуировки).
Таблица 2.
Матричный эффект (%) при разбавлении экстракта аминогликозидов из молока водой
| Крат-ность разбав-ления | Амика-цин | Апрами-цин | Гентами-цин | Гигроми-цин | Дигидро-стрепто-мицин | Канами-цин А | Нео-ми-цин | Паромо-мицин | Спек-тино-мицин | Стрепто-мицин | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [M + H]+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + 2H]2+ | [M + H]+ | [M + 2H]2+ | [M + H2O + H]+ | [M + H]+ | [M + 2H]2+ | [M + H2O + H]+ | |
| 0 | 76 | 98 | 34 | 98 | 36 | 71 | 80 | 99 | 89 | 99 | 67 | 91 | 0.8 | 32 | 95 | 95 | 72 | 96 | 80 |
| 2 | 70 | 94 | 15 | 94 | 18 | 67 | 72 | 97 | 85 | 98 | 67 | 69 | 10 | 10 | 92 | 94 | 72 | 95 | 83 |
| 4 | 50 | 93 | 13 | 87 | 99 | 54 | 71 | 96 | 80 | 98 | 56 | 71 | 16 | 25 | 88 | 91 | 70 | 96 | 83 |
| 8 | 30 | 57 | 75 | 58 | 48 | 34 | 37 | 62 | 44 | 61 | 44 | 37 | 34 | 56 | 25 | 39 | 30 | 93 | 36 |
| 10 | 15 | 20 | 105 | 20 | 17 | 20 | 16 | 19 | 20 | 12 | 16 | 18 | 18 | 58 | 18 | 20 | 19 | 89 | 12 |
| 15 | 15 | 20 | 169 | 15 | 15 | 13 | 18 | 14 | 29 | 17 | 18 | 19 | 20 | 78 | 19 | 20 | 20 | 86 | 13 |
Таблица 3.
Уравнения градуировочных графиков, коэффициенты корреляции (R2) для водных растворов аминогликозидов, экстрактов из молока в диапазоне 1−200 нг/г и наблюдаемый матричный эффект
| Аналит | Водные растворы | R2 | Экстракт из молока | R2 | МЭ, % |
|---|---|---|---|---|---|
| Амикацин | y = 15728x + 29427 | 0.9998 | y = 840x + 15331 | 0.9938 | −95 |
| Апрамицин | y = 23222x + 119209 | 0.9949 | y = 1723x + 3021 | 0.9941 | −93 |
| Гентамицин | y = 5568x + 31874 | 0.9899 | y = 3659x + 27040 | 0.9847 | −34 |
| Гигромицин В | y = 32266x + 96766 | 0.9981 | y = 1447x + 29679 | 0.9992 | −96 |
| Дигидрострептомицин | y = 33711x + 128008 | 0.9894 | y = 1351x – 6113 | 0.9879 | −96 |
| Канамицин А | y = 11208x + 76935 | 0.9961 | y = 2269x + 16437 | 0.9923 | −79 |
| Неомицин | y = 38404x – 33613 | 0.9962 | y = 1581x + 32832 | 0.9928 | −96 |
| Паромомицин | y = 36878x + 7190 | 0.9988 | y = 2559x – 76048 | 0.9856 | −93 |
| Спектиномицин | y = 38308x + 16106 | 0.9990 | y = 1900x + 21103 | 0.9999 | −95 |
| Стрептомицин | y = 8472x + 14320 | 0.9983 | y = 966x – 8296 | 0.9932 | −88 |
Количественное определение. Пределы обнаружения (сmin) и пределы определения (сн) рассчитывали при отношениях сигнал/шум, равном 3 и 10 соответственно для стандартных растворов. Пределы определения составили 0.5–1.0 нг/г для водных стандартных растворов. Учитывая такие низкие значения пределов обнаружения и достаточно высокие значения максимально допустимого уровня аминогликозидов в молоке (табл. 1), имеется возможность разбавления экстракта из молока водой для нивелирования матричного эффекта. Установлено, что нивелирование матричного эффекта (≤20%) наблюдается при 10-кратном разбавлении экстракта водой (табл. 2). В этом случае для определения концентрации аминогликозидов использовали метод абсолютной градуировки и градуировочные графики, построенные с использованием воды (табл. 3). Как видно из табл. 2, разбавление пробы по-разному влияет на одно- и двухзарядные ионы. Для количественного анализа не следует использовать ионы [M + H]+ апрамицина и паромомицина, ионы [M + 2H]2+ стрептомицина, поскольку при разбавлении пробы водой МЭ для этих ионов усиливается. Для остальных аналитов возможно использование как суммы интенсивностей сигналов одно- и двухзарядных ионов, так и интенсивностей отдельных ионов (табл. 4). Диапазоны определяемых содержаний в этом случае составили 10–1000 нг/г.
Таблица 4.
Правильность и воспроизводимость определения аминогликозидов в молоке методом введено–найдено (n = 3, P = 0.95)
| Аналит | Ион для количественного анализа | Введено, нг/г |
Найдено, нг/г | Отн. погреш-ность, % | sr |
|---|---|---|---|---|---|
| Амикацин | [M + H]+ [M + 2H]2+ |
0 | 0 | – | – |
| 10 | 9 ± 2 | −10 | 0.15 | ||
| 500 | 490 ± 15 | −2 | 0.02 | ||
| Апрамицин | [M + 2H]2+ | 0 | 0 | – | – |
| 10 | 11 ± 4 | 10 | 0.09 | ||
| 500 | 480 ± 50 | −4 | 0.03 | ||
| Гентамицин | [M + H]+ [M + 2H]2+ |
0 | 60 ± 10 | – | 0.09 |
| 10 | 68 ± 11 | −20 | 0.11 | ||
| 500 | 600 ± 60 | 8 | 0.02 | ||
| Гигромицин В | [M + H]+ [M + 2H]2+ |
0 | 0 | – | – |
| 10 | 8 ± 3 | −20 | 0.17 | ||
| 500 | 490 ± 12 | −2 | 0.02 | ||
| Дигидрострептомицин | [M + H]+ [M + 2H]2+ |
0 | 18 ± 6 | – | 0.11 |
| 10 | 27 ± 9 | −10 | 0.10 | ||
| 500 | 488 ± 60 | −6 | 0.04 | ||
| Канамицин А | [M + H]+ [M + 2H]2+ |
0 | 98 ± 15 | – | 0.08 |
| 10 | 116 ± 13 | −20 | 0.09 | ||
| 500 | 588 ± 39 | −2 | 0.03 | ||
| Неомицин | [M + 2H]2+ | 0 | 0 | – | – |
| 10 | 8 ± 4 | −20 | 0.17 | ||
| 500 | 480 ± 35 | −4 | 0.04 | ||
| Паромомицин | [M + 2H]2+ | 0 | 0 | – | – |
| 10 | 7 ± 3 | −30 | 0.17 | ||
| 500 | 590 ± 20 | 18 | 0.03 | ||
| Спектиномицин | [M + H2O + H]+ | 0 | 0 | – | – |
| 10 | 9 ± 2 | −10 | 0.09 | ||
| 500 | 490 ± 15 | −2 | 0.02 | ||
| Стрептомицин | [M + H]+ [M + H2O + H]+ |
0 | 110 ± 20 | – | 0.09 |
| 10 | 120 ± 10 | 0 | 0.08 | ||
| 500 | 600 ± 55 | −2 | 0.03 |
В табл. 4 представлены результаты оценки правильности и воспроизводимости предлагаемой методики. Установлено, что относительная погрешность анализа составляет не более 20%, относительное стандартное отклонение не превышает 0.17. Продолжительность идентификации аминогликозидов в молоке 15 мин, продолжительность определения обнаруженных аналитов 30–40 мин.
Список литературы
Mingeot-Leclercq M.P., Glupczynski Y., Tulkens P.M. Aminoglycosides: Activity and resistance // Antimicrob. Agents Chemother. 1999. V. 43(4). P. 727. https://doi.org/10.1159/000013861
Amelin V., Korotkov A., Andoralov A. Identification and determination of 492 contaminants of different classes in food and feed by high resolution mass spectrometry using the standard addition method // J. AOAC Int. 2016. V. 99. № 6. P. 1600.
Oertel R., Neumeister V., Kirch W. Hydrophilic interaction chromatography combined with tandem-mass spectrometry to determine six aminoglycosides in serum // J. Chromatogr. A. 2004. V. 1058. P. 197.
Ishii R., Horie M., Chan W., MacNeil J. Multi-residue quantitation of aminoglycoside antibiotics in kidney and meat by liquid chromatography with tandem mass spectrometry // Food Addit. Contam. 2008. V. 25. № 12. P. 1509. https://doi.org/10.1080/02652030802189740
Zhu W.-X., Yang J.-Z., Wei W., Liu Y.-F., Zhang S.-S. Simultaneous determination of 13 aminoglycoside residues in foods of animal origin by liquid chromatography-electrospray ionization tandem mass spectrometry with two consecutive solid-phase extraction steps // J. Chromatogr. A. 2008. V. 1207. P. 29.
Holthoon F.L., Essers M.L., Mulder P.J., Stead S.L., Caldow M., Ashwin H.M., Sharman M. A generic method for the quantitative analysis of aminoglycosides (and spectinomycin) in animal tissue using methylated internal standards and liquid chromatography tandem mass spectrometry // Anal. Chim. Acta. 2009. V. 637. P. 135.
Arsand J.B., Jank L., Martins M.T., Hoff R.B., Barreto F., Pizzolatto T.M., Sirtori C. Determination of aminoglycoside residues in milk and muscle based on a simple and fast extraction procedure followed by liquid chromatography coupled to tandem mass spectrometry and time of flight mass spectrometry // Talanta. 2016. V. 154. P. 38.
Plozza T., Trenerry V.C., Zeglinski P., Nguyen H., Johnstone P. The confirmation and quantification of selected aminoglycoside residues in animal tissue and bovine milk by liquid chromatography tandem mass spectrometry // Int. Food Res. J. 2011. V. 18. № 3. P. 1077.
Kaufmann A., Butcher P., Maden K. Determination of aminoglycoside residues by liquid chromatography and tandem mass spectrometry in a variety of matrices // Anal. Chim. Acta. 2012. V. 711. P. 46. https://doi.org/10.1016/j.aca.2011.10.042
Kaufmann A., Maden K. Determination of 11 aminoglycosides in meat and liver by liquid chromatography with tandem mass spectrometry // J. AOAC Int. 2005. V. 88. P. 1118.
Babin Y., Fortier S. A highthroughput analytical method for determination of aminoglycosides in veal tissues by liquid chromatography/tandem mass spectrometry with automated cleanup // J. AOAC Int. 2007. V. 90. P. 1418.
Turnipseed S.B., Clark S.B., Karbiwnyk C.M., Andersen W.C., Miller K.E., Madson M.R. Analysis of aminoglycoside residues in bovine milk by liquid chromatography electrospray ion trap mass spectrometry after derivatization with phenyl isocyanate // J. Chromatogr. B. 2009. V. 877. P. 1487.
Амелин В.Г., Федина Н.М., Подколзин И.В., Коротков А.И. Быстрый скрининг и определение остаточных количеств ветеринарных препаратов в молоке методом ультравысокоэффективной жидкостной хроматографии-квадруполь-времяпролетной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2018. Т. 73. № 6. С. 461. (Amelin V.G., Fedina N.M., Podkolzin I.V., Korotkov A.I. Rapid screening and determination of residual veterinary drugs in milk by ultrahigh performance liquid chromatography–high-resolution quadrupole time-of-flight mass spectrometry // J. Analyt. Chem. 2018. V. 73. № 6. P. 576.)
Ferrer C., Lozano A., Aguera A., Giron A.J., Fernandez A.R. Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables // J. Chromatogr. A. 2011. V. 1218. P. 7634. https://doi.org/10.1016/j.chroma.2011.07.033
Zhou W., Yang S., Wang P.G. Matrix effects and application of matrix effect factor // Bioanalysis. 2017. V. 9. № 23. P. 1839. https://doi.org/10.4155/bio-2017-0214
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии