

Журнал аналитической химии, 2020, T. 75, № 11, стр. 1014-1020
Жидкостная микроэкстракция тетрациклинов из биологических жидкостей для их последующего определения методом высокоэффективной жидкостной хроматографии с УФ-детектированием
К. Д. Черкашина a, *, А. И. Сумина a, К. С. Вах a, А. В. Булатов a
a Санкт-Петербургский государственный университет, Институт химии
198504 Петергоф, Санкт-Петербург, Университетский просп., 26, Россия
* E-mail: ksenya_cherkashina@list.ru
Поступила в редакцию 03.12.2019
После доработки 18.02.2020
Принята к публикации 17.03.2020
Аннотация
Предложена схема жидкостной микроэкстракции для выделения и концентрирования антибиотиков тетрациклинового ряда из биологических жидкостей. Схема предполагает образование in situ мицеллярной фазы первичного амина из водного раствора пробы при введении полярного органического растворителя. Возможности разработанной схемы продемонстрированы на примере определения тетрациклина, окситетрациклина и доксициклина в плазме, сыворотке крови и моче человека методом ВЭЖХ-УФ. Достигнуты пределы обнаружения (3σ) для окситетрациклина и доксициклина 0.03 мг/л, для тетрациклина 0.08 мг/л. Время микроэкстракции не превышает 5 мин.
Одним из эффективных методов разделения и концентрирования является жидкостная микроэкстракция из гомогенного раствора (ЖМЭГР), которая предполагает образование in situ фазы экстрагента непосредственно в растворе пробы [1, 2]. В качестве эктрагентов в ЖМЭГР используют полярные органические растворители (одноатомные спирты [3, 4], ацетонитрил [5, 6]), способные неограниченно смешиваться с водным раствором пробы. Для образования фазы экстрагента и последующего фазового разделения требуется снижение энергии гидратации органического растворителя, которое может достигаться за счет эффектов “высаливания” [7] или “высахаривания” [8] при растворении в водной фазе “высаливающих” или “высахаривающих” агентов [9–12]. Метод обеспечивает большую площадь контакта водной и органической фаз и, как следствие, высокую скорость массопереноса аналитов [1, 2]. Основным ограничением метода является низкая эффективность с точки зрения достигаемых коэффициентов концентрирования. Как правило, разделение фаз наблюдается при их объемном соотношении 1 : 1.
Для повышения эффективности ЖМЭГР предложены растворители с “переключаемой полярностью”. Установлено, что жидкие высшие вторичные и третичные амины (гидрофобная форма) в присутствии диоксида углерода образуют водорастворимые гидрокарбонаты (гидрофильная форма). При введении в гомогенный раствор гидрокарбоната амина сильной минеральной кислоты или щелочи протекают реакции ионного обмена и выделяется гидрофобная форма амина во всем объеме раствора с последующим разделением фаз [13–15]. При растворении высшей карбоновой кислоты в водном щелочном растворе также наблюдается образование гомогенного раствора за счет ионизации экстрагента (образуется гидрофильная форма). Последующее подкисление пробы инициирует протонирование гидрофильной формы и образование фазы высшей карбоновой кислоты. Низкая растворимость растворителей с “переключаемой полярностью” в водной фазе обеспечивает возможность достижения высоких коэффициентов концентрирования в схемах ЖМЭГР (объем экстрагента меньше 100 мкл) [13, 16].
Антибиотики тетрациклинового ряда находят широкое применение для лечения различных заболеваний дыхательных путей, мочевыделительной системы, кожи и слизистых оболочек [17]. Важной задачей является разработка экспрессных и простых схем определения тетрациклинов в биологических жидкостях для регулирования режимов приема лекарственных препаратов в процессе лечения. В схемах хроматографического определения тетрациклинов в сложных матрицах в первую очередь внимание уделяют пробоподготовке для устранения матричных эффектов [18].
Поиск и изучение новых экстракционных систем – важные задачи современной аналитической химии [19–21]. В данной работе для реализации ЖМЭГР антибиотиков тетрациклинового ряда изучены высшие первичные амины. При их выборе опирались на явление образования гидратов аминов в водном растворе [22]. В работе [23] установлено, что при введении неорганической соли в водный раствор гидрата первичного амина происходит быстрое выделение фазы амина, однако этот вариант ЖМЭГР реализован только в схеме выделения тетрациклина из водных растворов мочи, другие антибиотики тетрациклинового ряда (окситетрациклин и доксициклин) эффективно не извлекались в образующуюся фазу амина.
В данной работе установлено явление образования in situ мицеллярной фазы при введении полярного органического растворителя в водный раствор гидрата первичного амина. С целью подтверждения эффективности предложенного варианта ЖМЭГР изучена возможность выделения и концентрирования различных тетрациклинов (тетрациклина, окситетрациклина и доксициклина) из биологических жидкостей (плазма, сыворотка крови и моча человека) для последующего их определения методом ВЭЖХ-УФ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растворы окситетрациклина (10 г/л), тетрациклина (10 г/л) и доксициклина (1 г/л) готовили путем растворения точных навесок антибиотиков в метаноле. Приготовленные растворы тетрациклинов хранили при +4°C в темном месте. Рабочие растворы тетрациклинов готовили непосредственно перед экспериментом разбавлением исходного раствора деионизованной водой. В работе использовали тетрациклин, окситетрациклин, доксициклин, н-октиламин, н-гептиламин, н-дециламин, н-нониламин, трилон Б, ацетат натрия, хлорид кальция, муравьиную, уксусную и трихлоруксусную кислоты, этанол и изопропанол (Sigma-Aldrich, Германия). Для приготовления подвижных фаз применяли ацетонитрил и (J.T. Baker Chemical Company, США).
Для определения тетрациклинов использовали систему ВЭЖХ LC-20 Prominence (Shimadzu, Япония) с УФ-детектором. Хроматографическое разделение тетрациклинов выполняли на колонке SUPELCO C18 (250 мм × 4.6 мм × 5 мкм) в соответствии с рекомендациями [23]. Подвижную фазу, состоящую из раствора А (0.5%-ная HCOOH) и раствора Б (смесь ацетонитрила с метанолом в соотношении 2 : 1), подавали со скоростью 0.75 мл/мин. Элюирование осуществляли в градиентном режиме: с 0 по 9 мин 80% А, с 9 по 16 мин линейное уменьшение до 20% А, с 16 по 19 мин 20% А, с 19 по 21 мин линейное увеличение до 80% А, с 21 по 23 мин 80% А. Детектирование выполняли при длине волны 355 нм.
Многоугловой спектрометр динамического и статического рассеяния света Photocor Complex (Photocor Instruments Inc., Россия), оснащенный оптическим коррелятором реального времени (угол рассеяния θ диапазон 40°–100°, температура 20 ± 0.1°C, длина волны лазерных источников света 445 и 654 нм), использовали для установления коэффициентов трансляционной диффузии.
Для определения содержания изопропанола и н-октиламина применяли газовый хроматограф Хроматэк-Кристал 5000 (Хроматек, Россия) с пламенно-ионизационным детектором и капиллярные колонки BPX 1 (10 м × 0.53 мм × 2.65 мкм) и OPTIMA WAXplus (30 м × 0.32 мм × 0.25 мкм) соответственно. Режимы программирования температуры для изопропанола: 40°С в течение 5 мин, нагрев до 200°С со скоростью 40 град./мин, 250°С в течение 5 мин; для н-октиламина: 120°С в течение 8 мин. Расход газа-носителя (азота) составлял 2.3 и 1.0 мл/мин для колонок BPX 1 и OPTIMA WAXplus соответственно.
Концентрацию воды в экстрактах определяли методом Фишера с использованием кулонометрического титратора 831 КФ (Metrohm, Швейцария).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
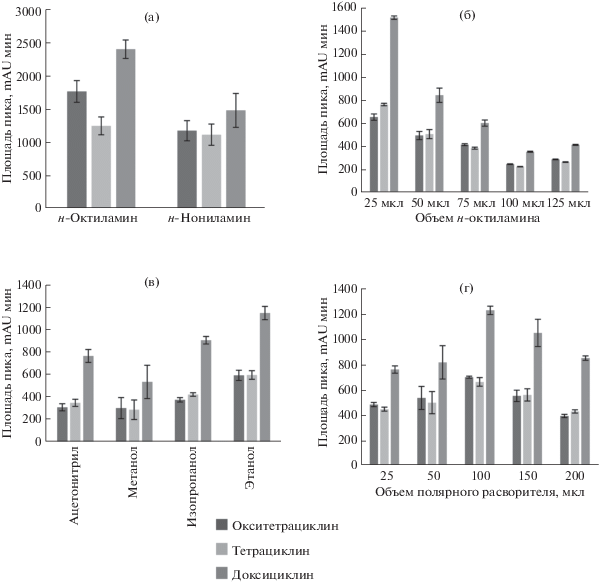
С целью оптимизации условий выделения тетрациклинов из водной фазы изучали возможность их экстракции различными первичными аминами (н-гептиламин, н-октиламин, н-нониламин и н-дециламин). Для этого 100 мкл первичного амина смешивали с 1 мл водного раствора тетрациклинов, смесь встряхивали до образования гомогенного раствора гидрата амина (2 мин). При введении полярного органического растворителя (200 мкл ацетонитрила) в растворы гидратов н-гептиламина, н-октиламина и н-нониламина происходило разделение фаз при центрифугировании (1 мин, 9788 × g) и экстракция тетрациклинов. Результаты анализа образующихся фаз методом ВЭЖХ-УФ показали, что в присутствии н-гептиламина хроматографическое разделение тетрациклина и окситетрациклина не достигается, а н-октиламин обеспечивает более эффективное извлечение аналитов по сравнению с н-нониламином (рис. 1а). Для дальнейших исследований выбрали н-октиламин.
Рис. 1.
Влияние типа амина (а), объема н-октиламина (б), типа полярного растворителя (в) и объема полярного растворителя (г) на эффективность микроэкстракции тетрациклинов (cTЦ = 25 мг/л, n = 3).
Для уточнения природы процессов, протекающих в экстракционной системе, исследовали фазу экстракта методом динамического рассеяния света. Цилиндрическую стеклянную ячейку (внутренний диаметр 10 мм) с исследуемым образцом погружали в иммерсионную жидкость (н-декан). Автокорреляционные функции интенсивности получали с использованием обратного преобразования Лапласа с помощью программного обеспечения DynaLS. Установленные значения коэффициентов трансляционной диффузии раствора гидрата н-октиламина в воде (20 × 10–8 см2/с) и образующейся фазы (85 × 10–8 см2/с) оказались значительно более низкими, чем значения тех же коэффициентов для н-октиламина (3.7 × 10–5 см2/с [24]) и воды (2.3 × 10–5 см2/с [25]). Низкие значения коэффициентов трансляционной диффузии можно объяснить образованием супрамолекулярных структур. При растворении н-октиламина в водной фазе образуется мицеллярный раствор (гидрат амина играет роль поверхностно-активного вещества), а при введении полярного органического растворителя происходит коацервация с выделением насыщенной мицеллярной фазы (рис. 2).
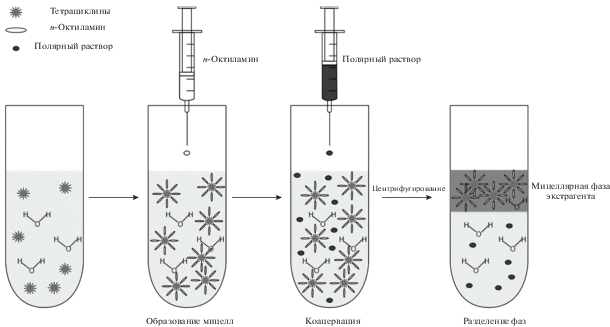
Методами Фишера и газовой хроматографии с пламенно-ионизационным детектированием установили, что мицеллярная фаза экстракта содержит воду (39%), н-октиламин (34%) и полярный органический растворитель (26%). В водной фазе пробы присутствуют также полярный органический растворитель (10%) и н-октиламин (0.7%), но в более низких концентрациях, чем в фазе экстракта. Полученные результаты подтвердили факт образования мицеллярной фазы, насыщенной мицеллами н-октиламина.
Для достижения максимальной эффективности микроэкстракции аналитов изучилиено влияние объема н-октиламина в диапазоне от 25 до 200 мкл при постоянном объеме водного раствора тетрациклинов (1 мл). С увеличением объема н-октиламина площади хроматографических пиков уменьшаются (рис. 1б). Максимальные аналитические сигналы наблюдаются при минимальном объеме н-октиламина (25 мкл), который выбрали в качестве оптимального.
Мицеллярная фаза образуется при ведении полярного растворителя. Природа полярного растворителя влияет на процессы разделения фаз и массопереноса аналитов. Исследовали влияние различных полярных растворителей (ацетонитрил, метанол, этанол и изопропанол) на экстракцию тетрациклинов. При введении в экстракционную систему этанола объем фазы экстракта (30 мкл) был минимальным, при этом получены максимальные площади пиков (рис. 1в). Однако в этом случае объем отбираемой фазы оказался невоспроизводимым. Удовлетворительную эффективность экстракции и высокую прецизионность анализа обеспечил изопропанол.
Объем полярного растворителя влияет на эффективность концентрирования. Варьировали объем изопропанола в диапазоне от 25 до 200 мкл. С одной стороны, при увеличении содержания изопропанола в водной фазе уменьшается количество свободных молекул воды, доступных для взаимодействия с амином и, как следствие, происходит коацервация. С другой стороны, изопропанол увеличивает растворимость полярных аналитов в водной фазе и снижает степень их извлечения. Установлено, что максимальные площади пиков достигаются при введении в водную фазу 100 мкл изопропанола (рис. 1г).
На основании полученных результатов разработана следующая схема ЖМЭГР. 1 мл водного раствора пробы и 25 мкл н-октиламина помещают в пластиковую пробирку и интенсивно встряхивают в течение 2 мин. После этого добавляют 100 мкл изопропанола, перемешивают смесь в течение 1 мин и центрифугируют (1 мин, 9788 × g). После центрифугирования мицеллярную фазу отбирают с помощью шприца и разбавляют метанолом в соотношении 1 : 1 для последующего определения тетрациклинов методом ВЭЖХ-УФ.
Для построения градуировочных графиков использовали водные растворы тетрациклинов. Градуировочные графики для определения окситетрациклина и доксициклина линейны в диапазоне от 0.1 до 10.0 мг/л (y = 273 878x – 14 331, y = = 471 441x – 19 328 соответственно), для тетрациклина – в диапазоне от 0.25 до 10.0 мг/л (y = = 273 860x + 8892.7). Достигнуты пределы обнаружения (3σ-критерий) для окситетрациклина и доксициклина составили 0.03 мг/л, для тетрациклина – 0.08 мг/л. Относительное стандартное отклонение составило не более 10% (n = 5).
Разработанную схему ЖМЭГР использовали для определения окситетрациклина, тетрациклина и доксициклина в пробах мочи, плазмы и сыворотки крови. Правильность результатов подтверждали методом введено–найдено и сравнением с данными референтных методов [26, 27].
Для устранения мешающего влияния мочевины и фосфатов при анализе мочи в пробу (5 мл) последовательно вводили 250 мкл 0.1 М раствора ZnSO4 и 100 мкл 1.5 М раствора NaOH. После центрифугирования (5000 × g в течение 3 мин) супернатант дополнительно отфильтровывали через мембранный фильтр (0.45 мкм), разбавляли в 50 раз деионизованной водой и проводили микроэкстракцию.
Тетрациклины могу связываться с белками крови, а также образовывать хелатные комплексы с ионами металлов [28, 29]. Для устранения матричных эффектов к пробам плазмы и сыворотки крови (0.25 мл) добавляли 100 мкл ацетатного буферного раствора (0.1 М, рН 6.5), содержащего 35 ммоль/л CaCl2, 25 ммоль/л трилона Б и 300 мкл изопропанола. После центрифугирования (10 000 × g в течение 5 мин) супернатант разбавляли в 8 раз и выполняли микроэкстракцию. Концентрации аналитов в пробах мочи, плазмы и сыворотки крови устанавливали с помощью градуировочных графиков с учетом разбавления проб деионизованной водой (в 50 раз).
Результаты определения тетрациклинов в биологических жидкостях представлены в табл. 1. При определении тетрациклинов в пробах мочи, плазмы и в сыворотке получены заниженные результаты, что связано с образованием водорастворимых устойчивых комплексов аналитов с ионами металлов, которые присутствуют в биологических жидкостях [30]. Результаты оценивали с помощью F- и t-тестов. Полученные значения F ≤ 19.00 указывают на незначительное различие в величинах стандартных отклонений, а значения t ≤ 2.78 указывают на то, что нет статистически значимого различия между результатами разработанной и референтных методик.
Таблица 1.
Результаты (мг/л) определения тетрациклинов в образцах мочи, сыворотки и плазмы крови (n = 3, P = 0.95, Fкр = 19, tкр = 2.78)
| Образец | Введено | Разработанная схема | Реферетная методика | F-тест | t-тест | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OТЦ | ТЦ | ДЦ | ОТЦ | ТЦ | ДЦ | ОТЦ | ТЦ | ДЦ | ОТЦ | ТЦ | ДЦ | ОТЦ | ТЦ | ДЦ | |
| Моча 1 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 35 | 35 | 35 | 37 ± 2 | 34 ± 1 | 30 ± 2 | 37.5 ± 0.8 | 35.8 ± 0.9 | 34 ± 2 | 3.81 | 2.09 | 1.38 | 0.47 | 1.44 | 2.61 | |
| 75 | 75 | 75 | 76 ± 1 | 60 ± 4 | 83 ± 3 | 74 ± 1 | 59 ± 4 | 85 ± 4 | 2.30 | 1.14 | 1.74 | 2.15 | 0.27 | 0.72 | |
| Моча 2 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 35 | 35 | 35 | 36 ± 1 | 30 ± 2 | 37 ± 1 | 34 ± 1 | 30.0 ± 0.4 | 36 ± 0.7 | 1.12 | 15.58 | 2.50 | 2.63 | 0.06 | 1.08 | |
| 75 | 75 | 75 | 74 ± 1 | 65 ± 4 | 78 ± 4 | 76 ± 1 | 63 ± 4 | 80 ± 3 | 1.38 | 1.14 | 3.50 | 2.77 | 0.52 | 1.18 | |
| Моча 3 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 35 | 35 | 35 | 38 ± 2 | 28 ± 2 | 37 ± 2 | 35 ± 2 | 30 ± 1 | 32 ± 3 | 1.68 | 1.35 | 4.69 | 2.03 | 1.79 | 2.38 | |
| 75 | 75 | 75 | 73 ± 1 | 55 ± 2 | 74 ± 1 | 71 ± 2 | 55 ± 4 | 76 ± 1 | 3.00 | 4.89 | 1.03 | 1.94 | 0.11 | 2.00 | |
| Сыворотка крови 1 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.1 ± 0.5 | 2.7 ± 0.6 | 3.59 ± 0.08 | 3.2 ± 0.6 | 2.8 ± 0.4 | 3.6 ± 0.8 | 1.21 | 2.76 | 2.83 | 0.80 | 1.00 | 2.65 | |
| 7.5 | 7.5 | 7.5 | 6.4 ± 0.7 | 6.6 ± 0.3 | 5.7 ± 0.7 | 6.1 ± 0.4 | 6.4 ± 0.6 | 6.5 ± 0.9 | 3.65 | 3.19 | 6.37 | 1.58 | 1.67 | 2.05 | |
| Сыворотка крови 2 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.1 ± 0.6 | 2.7 ± 0.2 | 3.8 ± 0.1 | 3.7 ± 0.7 | 2.7 ± 0.3 | 3.6 ± 0.9 | 1.29 | 3.30 | 2.20 | 2.49 | 0.46 | 2.53 | |
| 7.5 | 7.5 | 7.5 | 5.4 ± 0.9 | 5.8 ± 0.4 | 5.1 ± 0.5 | 6.1 ± 0.8 | 6.2 ± 0.9 | 5.8 ± 0.9 | 1.33 | 5.40 | 3.53 | 2.38 | 2.02 | 2.50 | |
| Сыворотка крови 3 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.2 ± 0.4 | 2.7 ± 0.5 | 4.1 ± 0.5 | 3.7 ± 0.6 | 2.8 ± 0.5 | 3.8 ± 0.6 | 1.62 | 1.04 | 1.44 | 2.65 | 0.21 | 2.48 | |
| 7.5 | 7.5 | 7.5 | 5.9 ± 0.6 | 5.7 ± 0.9 | 5.6 ± 0.5 | 5.7 ± 0.6 | 5.7 ± 0.2 | 5.8 ± 0.8 | 1.09 | 16.16 | 2.82 | 1.38 | 0.26 | 0.91 | |
| Плазма крови 1 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.2 ± 0.6 | 3.2 ± 0.4 | 2.8 ± 0.5 | 3.2 ± 0.2 | 2.9 ± 0.7 | 3.3 ± 0.8 | 10.72 | 2.93 | 3.45 | 0.32 | 1.38 | 1.85 | |
| 7.5 | 7.5 | 7.5 | 6.7 ± 1.5 | 6.6 ± 0.7 | 7.2 ± 0.8 | 6.1 ± 0.4 | 6.4 ± 0.6 | 6.5 ± 0.6 | 17.9 | 1.59 | 1.43 | 2.74 | 2.25 | 2.06 | |
| Плазма крови 2 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.1 | 3.0 ± 0.4 | 3.0 ± 0.6 | 3.2 ± 0.4 | 3.2 ± 0.7 | 3.6 ± 0.7 | 9.92 | 6.40 | 1.84 | 1.25 | 1.04 | 2.61 | |
| 7.5 | 7.5 | 7.5 | 5.4 ± 0.5 | 6.1 ± 0.7 | 6 ± 1 | 6.1 ± 0.8 | 6.2 ± 0.9 | 5.8 ± 0.8 | 3.77 | 1.41 | 2.54 | 2.47 | 0.26 | 2.50 | |
| Плазма крови 3 | 0 | 0 | 0 | < ПО | < ПО | < ПО | < ПО | < ПО | < ПО | – | – | – | – | – | – |
| 3.5 | 3.5 | 3.5 | 3.7 ± 0.5 | 2.9 ± 0.3 | 2.9 ± 0.5 | 3.2 ± 0.6 | 3.5 ± 0.8 | 3.4 ± 0.7 | 1.47 | 12.63 | 1.68 | 2.33 | 2.26 | 2.37 | |
| 7.5 | 7.5 | 7.5 | 5.6 ± 0.9 | 5.9 ± 0.6 | 5.5 ± 0.8 | 5.7 ± 0.6 | 5.7 ± 0.2 | 5.8 ± 0.7 | 4.95 | 7.95 | 2.82 | 0.87 | 1.07 | 0.91 | |
* * *
Разработана схема жидкостной микроэкстракции, предполагающая образование in situ мицеллярной фазы экстрагента непосредственно в растворе пробы. Схема микроэкстракции успешно применена для выделения тетрациклинов из проб биологических жидкостей для их последующего ВЭЖХ-УФ-определения. Изучена эффективность применения различных первичных аминов для выделения тетрациклинов. Предлагаемый подход исключает необходимость применения “высаливающих” агентов для разделения фаз и обеспечивает возможность эффективного концентрирования целевых аналитов. Разработанную. схему можно использовать в практике персонализированной медицины для определения тетрациклинов в биологических жидкостях. Достигнутые пределы обнаружения тетрациклинов в моче (0.03–0.08 мг/л) лежат в диапазоне терапевтических концентраций (для мочи до 300 мг/л, для сыворотки и плазмы крови 1–10 мг/л [31–33]).
Авторы выражают благодарность РФФИ (19-33-90007\19) за финансовую поддержку. Научные исследования проводились с использованием оборудования Научного парка Санкт-Петербургского государственного университета, а именно ресурсных центров “Методы анализа состава вещества” и “Центр диагностики функциональных материалов для медицины, фармакологии и наноэлектроники”.
Список литературы
Çabuk H., Köktürk M., Ata Ş. pH-assisted homogeneous liquid–liquid microextraction using dialkylphosphoric acid as an extraction solvent for the determination of chlorophenols in water samples // J. Sep. Sci. 2014. V. 37. P. 1343.
Yazdanfar N., Yamini Y., Ghambarian M. Homogeneous liquid–liquid microextraction for determination of organochlorine pesticides in water and fruit samples // Chromatographia. 2014. V. 77. P. 329.
Gupta M., Pillai A. K., Singh, A., Jain A., Verma K. K. Salt-assisted liquid–liquid microextraction for the determination of iodine in table salt by high-performance liquid chromatography-diode array detection // Food Chem. 2011. V. 124. № 4. P. 1741
Chen X., Li J., Zhang Y., Hu S., Du Y., Bai X. Double salting-out effect assisted heat-shrinkable tubing liquid phase microextraction followed by high performance liquid chromatography for determination of flavonoids in human plasma // J. Chromatogr. A. 2019. V. 1603. P. 44.
Rashidipour M., Heydari R., Maleki A.A., Mohammadi E.A., Davari B. Salt-assisted liquid–liquid extraction coupled with reversed-phase dispersive liquid–liquid microextraction for sensitive HPLC determination of paraquat in environmental and food samples // J. Food Meas. Charact. 2019. V. 13. № 1. P. 269.
Xu X.-Y.A, Ye J.-Q.A, Nie J.A., Li Z.-G., Lee M.-R. A new liquid-liquid microextraction method by ultrasound assisted salting-out for determination of triazole pesticides in water samples coupled by gas chromatography-mass spectrometry // Anal. Methods. 2015. V. 7. № 3. P. 1194.
Hey M.J., Jackson D.P., Yan H. The salting-out effect and phase separation in aqueous solutions of electrolytes and poly (ethylene glycol) // Polymer. 2005. V. 46. № 8. P. 2567.
Wang B., Ezejias T., Feng H., Blaschek H. Sugaring-out: A novel phase separation and extraction system // Chem. Eng. Sci. 2008. V. 63. № 9. P. 2595.
Ahmed S., Mahmoud A.M. A novel salting-out assisted extraction coupled with HPLC-fluorescence detection for trace determination of vitamin K homologues in human plasma // Talanta. 2015. V. 144. P. 480.
Song S., Ediage E.N., Wu A., De Saeger S. Development and application of salting-out assisted liquid/liquid extraction for multi-mycotoxin biomarkers analysis in pig urine with high performance liquid chromatography/tandem mass spectrometry // J. Chromatogr. A. 2013. V. 1292. P. 111.
Nugbienyo L., Malinina Y., Garmonov S., Kamencev M., Salahov I., Andruch V., Bulatov A. Automated sugaring-out liquid-liquid extraction based on flow system coupled with HPLC-UV for the determination of procainamide in urine // Talanta. 2017. V. 167. P. 709.
Zhang J., Myasein F., Wu H., El-Shourbagy T.A. Sugaring-out assisted liquid/liquid extraction with acetonitrile for bioanalysis using liquid chromatography–mass spectrometry // Microchem. J. 2013. V. 108. P. 198.
Vakh C., Pochivalov A., Andruch V., Moskvin L., Bulatov A. A fully automated effervescence-assisted switchable solvent-based liquid phase microextraction procedure: liquid chromatographic determination of ofloxacin in human urine samples // Anal. Chim. Acta. 2016. V. 907. P. 54.
Shih H.K., Shu T.Y., Ponnusamy V.K., Jen J.F. A novel fatty-acid-based in-tube dispersive liquid-liquid microextraction technique for the rapid determination of nonylphenol and 4-tert-octylphenol in aqueous samples using high-performance liquid chromatography–ultraviolet detection // Anal. Chim. Acta. 2015. V. 854. P. 70.
Jessop P.G., Kozycz L., Rahami Z.G., Schoenmakers D., Boyd A.R., Wechsler D., Holland A.M. Tertiary amine solvents having switchable hydrophilicity // Green Chem. 2011. V. 13. № 3. P. 619.
Kokosa, J.M. Advances in solvent-microextraction techniques // Trends Anal. Chem. 2013. V. 43. P. 2.
Klein N. C., Cunha B. A., Tetracyclines // Med. Clin. North Am. 1995. V. 79. № 4. P. 789.
Anderson C.R., Rupp H.S., Wu W.H. Complexities in tetracycline analysis—chemistry, matrix extraction, cleanup, and liquid chromatography // J. Chromatogr. A. 2005. V. 1075. № 1–2. P. 23.
Kocúrová L., Balogh I.S., Šandrejová J., Andruch V. Recent advances in dispersive liquid–liquid microextraction using organic solvents lighter than water. A review // Microchem. J. 2012. V. 102. P. 11.
Anthemidis A.N., Ioannou K.I.G. Recent developments in homogeneous and dispersive liquid–liquid extraction for inorganic elements determination. A review // Talanta. V. 80. № 2. P. 413.
Moradi M., Yamini Y., Ebrahimpour B. Emulsion-based liquid-phase microextraction: A review // J. Iran. Chem. Soc. 2014. V. 11. № 4. P. 1087.
Pettersson A.B.A., Rosenholm J.B. A calorimetric investigation of the adsorption of octylamine on titanium dioxide from aqueous solutions // Prog. Coll. Polym. Sci. 1990. V. 82. P. 38.
Cherkashina K., Vakh C., Lebedinets S., Pochivalov A., Moskvin L., Lezov A., Bulatov A. An automated salting-out assisted liquid-liquid microextraction approach using 1-octylamine: On-line separation of tetracycline in urine samples followed by HPLC-UV determination // Talanta. 2018. V. 184. P. 122.
Winkelmann M., Schneider L., Gerlinger W., Sachweh B., Miller R., Schuchmann H.P. Mass transport characteristics of alkyl amines in a water/n-decane system // J. Coll. Interface Sci. 2012. V. 372 № 1. P. 164.
Krynicki K., Green C.D., Sawyer D.W. Pressure and temperature dependence of selfdiffusion in water // Faraday Disc. Chem. Soc. 1978. V. 66. P. 199.
Fernandez-Torres R., Consentino M.O., Lopez M.B., Mochon M.C. Simultaneous determination of 11 antibiotics and their main metabolites from four different groups by reversed-phase high-performance liquid chromatography–diode array–fluorescence (HPLC–DAD–FLD) in human urine samples // Talanta. 2010. V. 81. № 3. P. 871.
Kazuo I., Norio O., Mitsuru Y. Rapid determination of tetracycline antibiotics in serum by reversed-phase high-performance liquid chromatography with fluorescence detection // J. Chromatogr. 1993. V. 619. № 2. P. 319.
Chi Z., Liu R. Phenotypic characterization of the binding of tetracycline to human serum albumin // Biomacromolecules. 2010. V. 12. № 1. P. 203.
Jezowska-Bojczuk M., Lambs L., Kozlowski H., Berthon G. Metal ion-tetracycline interactions in biological fluids. 10. Structural investigations on copper(II) complexes of tetracycline, oxytetracycline, chlortetracycline, 4-(dedimethylamino) tetracycline, and 6-desoxy-6-demethyltetracycline and discussion of their binding modes // Inorg. Chem. 1993. V. 32. № 4. P. 428.
Brion M., Berthon G., Fourtillan J.B. Metal ion–tetracyclines interactions in biological fluids. Potentiometric study of calcium complexes with tetracycline, oxytetracycline, doxycycline and minocycline and simulation of their distributions under physiological conditions // Inorg. Chim. Acta. 1981. V. 55. P. 47.
Calixto C.M.F., Cavalheiro E.T.G., Determination of tetracyclines in bovine and human urine using a graphite-polyurethane composite electrode // Anal. Lett. 2015. V. 48. № 9. P. 1454.
Sonntag O., Scholer A., Drug interference in clinical chemistry: recommendation of drugs and their concentrations to be used in drug interference studies // Ann. Clin. Biochem. 2001. V. 38. № 4. P. 376.
Regenthal R., Krueger M., Koeppel C., Preiss R., Drug levels: therapeutic and toxic serum/plasma concentrations of common drugs // J. Clin. Monit. Comput. 1999. V. 15. № 7. P. 529.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии