

Журнал аналитической химии, 2020, T. 75, № 6, стр. 543-552
Определение метаболитов циклогексилметилфторфосфоната в плазме крови человека с использованием жидкостной хромато-масс-спектрометрии высокого разрешения
А. В. Браун a, *, Я. В. Ставитская a, Т. М. Байгильдиев b, Д. В. Орешкин a, И. В. Рыбальченко a, И. А. Родин b
a 27 Научный Центр Министерства Обороны РФ
105005 Москва, Бригадирский переулок, 13, Россия
b Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: avbraun@yandex.ru
Поступила в редакцию 30.04.2019
После доработки 22.05.2019
Принята к публикации 29.12.2019
Аннотация
Оптимизированы условия извлечения фосфонилированного нонапептида, фосфонилированного тирозина и циклогексилметилфосфоновой кислоты из плазмы крови человека с последующим определением методом ВЭЖХ с детектированием тандемной масс-спектрометрией высокого разрешения. Выбраны условия пробоподготовки образцов плазмы крови человека, масс-спектрометрического детектирования в режиме мониторинга выбранных реакций и градиентного элюирования при определении указанных маркеров экспозиции циклогексилметилфторфосфонатом. Предел обнаружения по внесенному в плазму крови исходному веществу составляет 0.05–0.1 нг/мл в зависимости от определяемого маркера.
Фосфорорганические вещества нервно-паралитического действия (ФВНПД) являются высокотоксичными соединениями, которые первоначально были разработаны в качестве высокоактивных пестицидов в 1930-е гг. С тех пор большое количество ФВНПД синтезировано и накоплено в военных целях. Несмотря на то, что применение этих веществ является прямым нарушением Конвенции по запрещению химического оружия [1], и запасы их планомерно уничтожаются, остаются опасения, что ФВНПД могут быть использованы в террористических целях. В этой связи в различных странах проводятся многочисленные исследования по разработке эффективных способов лечения людей, подвергшихся их воздействию [2, 3]. В рамках этой деятельности актуальной задачей является поиск комплексных подходов, позволяющих проводить диагностику в случаях воздействия ФВНПД на человека.
Известно, что в результате гидролиза ФВНПД в организме образуются специфические метаболиты – алкиловые эфиры метилфосфоновой кислоты (АМФК) [4, 5]. Разработаны методы количественной оценки продуктов гидролиза ФВНПД в моче, слюне и плазме методами газовой (ГХ) [6–10] и жидкостной хроматографии (ЖХ). Появление ионизации электрораспылением в варианте ЖХ позволило создать чувствительные методики обнаружения АМФК без использования трудоемких процедур дериватизации, которые применяются при подготовке биоматериалов для анализа методами ГХ [11–15]. В работе [16] предложен подход к определению изопропилметилфосфоновой кислоты и циклогексилметилфосфоновой кислоты в плазме крови свиней, подвергшихся ингаляционному воздействию ФВНПД. Авторы использовали метод изотопного разбавления, пределы обнаружения составили 2 нг/мл. Однако АМФК выводятся из организма в течение нескольких дней [17, 18], что снижает возможность их идентификации за пределами указанного срока.
Представляет интерес использование для диагностики аддуктов ФВНПД с одной из основных мишеней их воздействия – ферментом бутирилхолинэстеразой (БХЭ). Наиболее эффективный метод определения аддуктов ФВНПД с БХЭ предполагает использование техники иммунозахвата специфического белка из плазмы. При этом аддукт с БХЭ подвергается дополнительному ферментативному гидролизу пепсином, в результате образуется пептид, состоящий из девяти аминокислот (FGES*AGAAS, нонапептид), который содержит связанный ковалентной связью остаток ФВНПД [19–22]. Однако большинство производных ФВНПД с БХЭ подвергаются деалкилированию (данный процесс называется “старением”), в результате чего точная диагностика становится затруднительной [23].
Другим важным путем метаболизма ФВНПД в организме является образование аддуктов с альбумином плазмы, при этом взаимодействие происходит по тирозиновому остатку (Tyr-411) [24–27]. В работе [28] представлен способ одновременного обнаружения тирозиновых аддуктов шести различных ФВНПД с пределами обнаружения 0.027–0.097 нг/мл. Показано, что образование тирозиновых аддуктов затруднено для некоторых типов ФВНПД. Таким образом, выявление любого из рассмотренных биомаркеров ФВНПД имеет свои трудности: аддукты с БХЭ подвержены “старению”, АМФК достаточно быстро выводятся из организма, аддукты с альбумином для некоторых ФВНПД образуются в относительно небольшом количестве. В связи с этим актуальной остаeтся задача создания универсального подхода для установления типа ФВНПД, попавшего в организм.
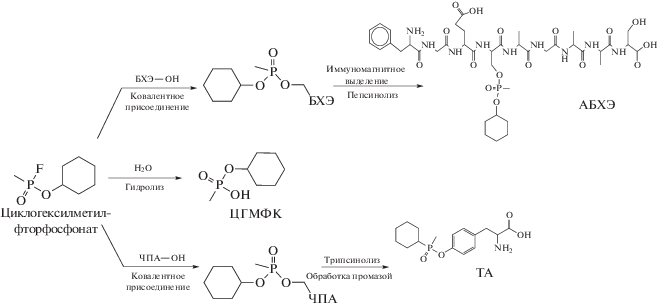
Цель данной работы состояла в разработке усовершенствованных процедур подготовки экспонированной ФВНПД плазмы крови человека к инструментальному анализу, оптимизации условий разделения методом ВЭЖХ с детектированием образующихся аддуктов ФВНПД тандемной масс-спектрометрией высокого разрешения (ВЭЖХ–МС/МС) для повышения чувствительности, селективности и надежности диагностики при ретроспективном мониторинге образцов плазмы крови человека на примере маркеров циклогексилметилфторфосфоната. В качестве объектов исследования выбрали три основных биомаркера циклогексиметилфторфосфоната: фосфорилированный нонапептид – аддукт циклогексиметилфосфоната с БХЭ (АБХЭ), альбуминовый аддукт циклогексиметилфосфоната с тирозином (ТА) и циклогексилметилфосфоновую кислоту (ЦГМФК), пути образования которых в организме иллюстрирует рис. 1.
Рис. 1.
Основные пути метаболизма циклогексилметилфторфосфоната при попадании в кровь живого организма и структуры биомаркеров при детектировании методом ВЭЖХ–МС/МС (ЧПА – альбумин человеческой плазмы).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растворы и реагенты. Ацетонитрил (Panreac, Испания), ацетат натрия (ГОСТ 199-78), уксусная кислота (99%, Sigma-Aldrich, кат. № 320 099); 0.25%-ный раствор трипсина (ООО “Биолот”, Санкт-Петербург, кат. № 1.2.2), муравьиная кислота (Fluka, кат. № 56 302), раствор проназы (Sigma, Protease Type XIV, P5147) с концентрацией 15 мг/мл в 25 мМ растворе NH4HCO3 (Reagentplus, A6141), раствор моноклональных антител (HAH 0020102, OG 19089, Thermo); 0.01 M фосфатно-солевой буферный раствор (pH 7.4) (ООО “Биолот”, Санкт-Петербург), пепсин (Sigma, № P6887); раствор циклогексилметилфосфоновой кислоты с концентрацией 1000 мкг/мл (Sigma, кат № ERC-034-1.2ML), тирозиновый аддукт циклогексилметилфторфосфоната с массовой долей основного вещества не менее 95% (определено методом ЯМР-спектроскопии) синтезировали по методикам [29]. Деионизированную воду очищали системой Milli-Q (Millipore, США). Образец донорской плазмы крови человека с внесением in vitro циклогексилметилфторфосфоната с конечной концентрацией в плазме 8 нг/мл предоставлен для использования в официальном биомедицинском тесте Организацией по запрещению химического оружия (ОЗХО). Образцы донорской плазмы человеческой крови, не содержащие ФВНПД, предоставлены Главным военным клиническим госпиталем имени Н.Н. Бурденко (партия № 111 045). Образцы хранили при −20°С.
Рабочие, буферные и другие необходимые растворы готовили растворением точных навесок в соответствующих растворителях в день проведения эксперимента.
Оборудование. Использовали аналитическую станцию, состоящую из жидкостного хроматографа Agilent Infinity 1260, оборудованного системой автоматического ввода пробы, с масс-селективным тандемным анализатором высокого разрешения Thermo Scientific Orbitrap Fusion Lumos (США) c источником ионов с ионизацией электрораспылением. Специфические биомаркеры циклогексилметилфторфосфоната выделяли на колонке 250 × 2.1 мм Thermo Acclaim 120 C18 с диаметром зерна сорбента 3 мкм (Thermo Scientific, США). Экспериментальные данные регистрировали и обрабатывали с помощью программных пакетов Xcalibur (ThermoScientific, США). Использовали набор для получения магнитных микросфер с привитыми антителами (143.11D, Invitrogen), магнитные микросферы (143.02D, Invitrogen), магнитный штатив DynaMagTM (Invitrogen, № 12321D), устройство для твердофазной экстракции (ТФЭ) (Supelco, кат. № 57030-U), систему для упаривания растворителей из пробирок TurboVap (Biotage TurboVap LV), фильтры Amicon Ultra-4 Centrifugal Filter Units (10k, Millipore), патроны для ТФЭ ISOLUTE, 100 мг/3 мл C8 (Biotage, кат. № 291-0010-B) и Chromabond SiOH, 100 мг/1 мл (Macherey-nagel, кат. № 730 071).
В ходе исследования выбрали и оптимизировали условия пробоподготовки образцов плазмы крови человека, масс-спектрометрического детектирования в режиме мониторинга выбранных реакций и градиентного элюирования при ВЭЖХ–МС/МС-анализе.
Подготовка проб для определения аддукта циклогексилметилфторфос-фоната с БХЭ. Приготовление иммуномагнитных микросфер с привитыми антителами проводили согласно протоколу к набору для получения магнитных микросфер с привитыми антителами (Dynabeads Antibody Coupling Kit), состоящему из буферных растворов C1, C2, HB, LB, SB. Магнитные микросферы отделяли от растворов путем помещения пробирок в магнитный штатив DynaMagTM на 30–60 с, надосадочную жидкость удаляли с помощью дозатора. Навеску микросфер Dynabeads M-270 Epoxy (143.02D) массой 11 мг промывали 1 мл буферного раствора C1, надосадочную жидкость отбрасывали. Микросферы суспендировали в 0.5 мл C1, смешанного с 0.05 мл раствора моноклональных мышиных антител Pierce BChE (HAH 0 020 102, OG 190 891, Thermo), затем добавляли 0.55 мл раствора C2. Полученный образец инкубировали при 37°C в течение ночи при перемешивании. Раствор, содержащий непрореагировавшие антитела, удаляли, а микросферы с привитыми антителами промывали сначала 0.8 мл раствора HB, затем 0.8 мл раствора LB. Иммуномагнитные микросферы промывали дважды по 0.8 мл раствора SB и суспендировали в 0.55 мл раствора SB.
Выделение бутилхолинэстеразы с иммуномагнитных микросфер с привитыми антителами. Суспензию иммуномагнитных микросфер тщательно перемешивали, аликвоты объемом 0.05 мл помещали в микропробирки емк. 1.5 мл, буферный раствор удаляли. Микросферы промывали 0.2 мл 0.01 M фосфатно-солевого буферного раствора (pH 7.4). Иммуномагнитные микросферы инкубировали с 0.25 мл образцов плазмы крови при комнатной температуре в течение 2 ч при перемешивании. Микросферы отделяли от плазмы с использованием магнитного штатива и промывали несколько раз 0.2 мл 0.01 M фосфатно-солевого буферного раствора. БХЭ элюировали с микросфер 0.1 мл 0.6%-ного водного раствора муравьиной кислоты. Для проведения ферментативного гидролиза отбирали 0.095 мл надосадочной жидкости.
Ферментативный гидролиз с использованием пепсина. 0.095 мл свежеприготовленного раствора пепсина в 0.6%-ной муравьиной кислоте с концентрацией 0.5 мг/мл добавляли в каждый образец, пробы инкубировали 2 ч при 37°C. Аликвоту объемом 0.020 мл анализировали методом ВЭЖХ–МС/МС.
Подготовка проб для определения аддукта циклогексилметилфосфоната с тирозином. К образцу плазмы крови объемом 1.0 мл добавляли 1.0 мл раствора проназы (15 мг/мл) в 25 мM растворе NH4HCO3 (Protease from Streptomycesgriseus, Sigma, P6911) и выдерживали пробу в течение 12 ч при 37°С. Для устранения избытка ферментов раствор фильтровали через фильтр Amicon Ultra-4 и центрифугировали в течение 15 мин при 10 000 g. Надосадочную жидкость пропускали через патрон ISOLUTE C8, предварительно кондиционированный ацетонитрилом (2 × 1.0 мл) и водой (2 × × 1.0 мл). Сорбент промывали водой (3 × 0.5 мл). Целевые вещества элюировали 50%-ным раствором ацетонитрила в воде (2 × 0.5 мл) и 60%-ным раствором ацетонитрила в воде (2 × 0.5 мл). Элюат упаривали досуха при 45°C в испарителе (TurboVap), остаток растворяли в 0.1 мл 5%-ного раствора ацетонитрила в воде. Аликвоту объемом 0.020 мл анализировали методом ВЭЖХ–МС/МС.
Подготовка проб для определения циклогексилметилфосфоновой кислоты. К 200 мкл плазмы крови добавляли 1 мл ацетонитрила и центрифугировали 5 мин при 10 000 g, после чего 1.2 мл надосадочной жидкости упаривали досуха при 70°С в токе азота на установке для концентрирования TurboVap. Сухой остаток экстрагировали дважды по 1 мл 5%-ной воды в ацетонитриле. Объединенный экстракт пропускали через патрон Chromabond SiOH, предварительно кондиционированный 1.0 мл 25%-ной воды в ацетонитриле и 1.0 мл ацетонитрила. Образец промывали 1.0 мл 10%-ной воды в ацетонитриле и элюировали 1.0 мл 25%-ной воды в ацетонитриле. Элюат упаривали досуха в токе азота при 70°С. Осадок растворяли в 150 мкл 5%-ного раствора ацетонитрила в воде. Аликвоту объемом 0.020 мл анализировали методом ВЭЖХ–МС/МС.
Хромато-масс-спектрометрический анализ. Использовали источник ионов с ионизацией электрораспылением в режиме регистрации выбранных реакций положительных ионов при обнаружении ТА и АБХЭ и отрицательных ионов в случае обнаружения ЦГМФК. Величина разрешения масс-анализатора составляла не менее 30 000, погрешность при определении величины m/z не превышала 3 млн–1. Температура переходного капилляра составляла 325°С, напряжение на распыляющем капилляре 3500 В (в случае обнаружения ТА и АБХЭ) и 2500 В (в случае обнаружения ЦГМФК), давление газа для распыления подвижной фазы в источнике ионов составляло 420 кПa.
Компоненты пробы разделяли в режиме градиентного элюирования, расход подвижной фазы 0.35 мкл/мин, температура термостата колонки 50°C. Подвижная фаза А – 0.1 об. % НСООН в воде, подвижная фаза Б – 0.1 об. % НСООН в ацетонитриле. Программы градиентного элюирования, используемые для разделения каждого из биомаркеров циклогексилметилфторфосфоната, представлены в табл. 1. Объем вводимой пробы составлял 20 мкл.
Таблица 1.
Программы градиентного элюирования, используемые для разделения ЦГМФК, АБХЭ и ТА (подвижная фаза А – 0.1 об. % НСООН в воде, подвижная фаза Б – 0.1 об. % НСООН)
| Метаболит | Программа элюирования |
|---|---|
| ЦГМФК | 0–2 мин: 95% А; 2–10 мин: 5–95% Б; 10–11 мин: 95% Б; 11–15 мин: 95% A |
| АБХЭ | 0–5 мин: 95% А; 5–9 мин: 5–80% Б; 9–11 мин: 80% Б; 11–15 мин: 95% A |
| ТА | 0–1.5 мин: 95% А; 1.5–11 мин: 5–70% Б; 11–11.5 мин: 70% Б; 11.5–15 мин: 95% A |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Оптимизация методик подготовки проб. В рамках исследований БХЭ, модифицированной по серину остатком циклогексилметилфторфосфоната [30], характерный аддукт извлекали из плазмы крови методом иммуномагнитной сепарации c помощью антител, привитых на магнитные сферы. После обработки извлеченной БХЭ пепсином получали нонапептид, модифицированный остатками циклогексилметилфторфосфоната, – [FGES(CyclohexylMPA)AGAAS]–АБХЭ. Данный способ ранее успешно использован для выделения аддуктов зомана и VX с БХЭ [30]. В ранее выполненной нами работе впервые показано выделение АБХЭ из плазмы крови человека, экспонированной циклогексилметилфторфосфонатом, с последующим определением методом ВЭЖХ–МС/МС высокого разрешения [31, 32].
В случае извлечения альбуминового аддукта циклогексилметилфторфосфоната в виде ТА из плазмы также использовали способ, описанный в работе [30], где представлен подход с использованием двух последовательных ферментативных гидролизов с применением 0.25%-ного раствора трипсина и проназы (15 мг/мл) в 25 мМ растворе NH4HCO3 с последующим выделением тирозинового аддукта зомана. На первом этапе проводят трипсинолиз в течение 6 ч, далее добавляют раствор проназы и затем выделяют соответствующий тирозиновый аддукт с использованием ТФЭ. В настоящей работе исследовано влияние стадии проведения трипсинолиза на интенсивность аналитического сигнала ТА. Для этого образец плазмы с внесением in vitro 8 нг/мл циклогексилметилфторфосфоната (образец ОЗХО) подвергали ферментативному гидролизу проназой с использованием стадии трипсинолиза, а также без использования этой стадии. Площадь пика ТА выбранной ионой реакции для количественной оценки в случае проведения стадии трипсинолиза составила 6 300 000 усл. ед., а при отсутствии стадии трипсинолиза – 5 900 000 усл. ед. Видно, что различие аналитических сигналов в обоих случаях несущественно, поэтому стадия проведения трипсинолиза при выделении ТА из плазмы нами не применялась, что позволило сократить продолжительность пробоподготовки более чем на 20%.
При обнаружении ЦГМФК в плазме применяли подход, описанный в работе [33], однако для увеличения аналитического сигнала варьировали объем аликвоты в диапазоне 50–300 мкл. Оказалось, что при использовании аликвоты плазмы более 200 мкл аналитический сигнал ЦГМФК не возрастает, однако при этом увеличивается влияние матрицы на ионизацию аналита. Так, при объеме аликвоты 200 мкл величина матричного эффекта составляет 93 ± 7%, при 250 мкл – 70 ± 10%, при 300 мкл – 60 ± 10%. По-видимому, емкость сорбента патрона Chromabond SiOH (100 мг, 1 мл) ограничена, и при объеме плазмы более 200 мкл в конечную аликвоту попадает большее количество мешающих ионизации компонентов. В то же время переход от объема аликвоты 50 мкл к 200 мкл приводит к четырехкратному увеличению площади пика ЦГМФК на хроматограмме, что и обусловило выбор объема плазмы 200 мкл в качестве оптимального.
Оптимизация условий масс-спектрометрического детектирования. В ходе исследования использовали ионизацию электрораспылением в режиме регистрации положительных ионов при обнаружении ТА и АБХЭ, поскольку в состав данных соединений входят аминогруппы, отвечающие за образование протонированных положительно заряженных молекул (рис. 1). В случае ЦГМФК использовали регистрацию отрицательных ионов, поскольку данное соединение обладает ярко выраженными кислотными свойствами, и в растворе происходит диссоциация с образованием отрицательно заряженных ионов. Условия масс-спектрометрического детектирования с варьированием напряжения на распыляющем капилляре оптимизировали в режиме прямого ввода раствора стандарта исследуемого соединения непосредственно в источник ионов, минуя хроматографическую колонку. Для этих целей в случае ТА и ЦГМФК использовали растворы с концентрациями 1 мкг/мл в воде, а при исследовании АБХЭ – 250 мкл плазмы с внесенным in vitro циклогексилметилфторфосфонатом (образец ОЗХО) с концентрацией 8 нг/мл и проводили описанную выше процедуру пробоподготовки для выделения АБХЭ.
На первом этапе работы исследовали влияние напряжения источника ионизации на величину сигнала протонированных (для ТА и АБХЭ) и депротонированного (для ЦГМФК) молекулярных ионов исследуемых соединений. Для этого использовали параметры работы масс-спектрометрического детектора, рекомендуемые производителем и прописанные в файле автоматической настройки системы – автотюнинга. Напряжение источника ионизации варьировали в диапазоне 1000–6000 В. Установлено, что при увеличении напряжения источника ионизации с 3500 до 4500 В (в режиме регистрации положительных ионов) и с 2500 до 3500 В (в режиме регистрации отрицательных ионов) интенсивности сигналов молекулярных ионов увеличиваются на 10–30%, однако при этом величина шума возрастает более чем на 50%, поэтому в дальнейшем устанавливали значения напряжения источника ионизации 3500 В при регистрации положительных ионов и 2500 В при регистрации отрицательных ионов.
На следующем этапе выбирали оптимальные пары ионных реакций при определении исследуемых соединений. Поскольку в масс-спектрах высокого разрешения наблюдали интенсивные сигналы соответствующих протонированных и депротонированных молекул, изучали их фрагментации. На рис. 2 представлены масс-спектры фрагментации ионов АБХЭ, ТА и ЦГМФК с m/z 956.41, 342.15 и 177.07 соответственно, полученные в режиме высокого разрешения. На основании полученных данных выбрали наиболее интенсивные ионные переходы, которые в дальнейшем использовали для определения метаболитов циклогексилметилфторфосфоната (табл. 2). После выбора характерных пар фрагментных ионов исследовали влияние энергии фрагментаций на величину аналитического сигнала этих ионов. Установлено, что при детектировании ЦГМФК и АБХЭ максимальная интенсивность сигналов выбранных ионных реакций достигается при 25 В, в случае ТА – при 10 В. Характеристики выбранных ионных реакций, используемых при обнаружении метаболитов циклогексилметилфторфосфоната, представлены в табл. 2. Время сканирования при детектировании выбранных метаболитов циклогексилметилфторфосфоната составило 100 мс.
Рис. 2.
Масс-спектры фрагментации ионов биомаркеров, выбранных для обнаружения циклогексилметилфторфосфоната в режиме регистрации положительно заряженных ионов: (а) – АБХЭ, (б) – ТА, (в) – ЦГМФК. Масс-спектры (а) и (б) получены в режиме регистрации положительно заряженных ионов, (в) – в режиме регистрации отрицательно заряженных ионов.

Таблица 2.
Характеристики выбранных ионных реакций биомаркеров, используемых при обнаружении циклогексилметилфторфосфоната
| Метаболит | Выбранная ионная реакция (значения m/z) |
Тип ионной реакции | Брутто-формула иона-фрагмента |
|---|---|---|---|
| ЦГМФК | 177.07 → 94.9904 | Количественная оценка | [CH4O3P]– |
| ЦГМФК | 177.07 → 76.9798 | Достоверное подтверждение | [CH2O2P]– |
| АБХЭ | 956.41 → 778.3366 | Количественная оценка | [C33H48O13N9]+ |
| АБХЭ | 956.41 → 673.2940 | Достоверное подтверждение | [C30H41O10N8]+ |
| ТА | 342.15 → 260.0682 | Количественная оценка | [C10H15O5NP]+ |
| ТА | 342.15 → 214.0628 | Достоверное подтверждение | [C9H13O3NP]+ |
Оптимизация условий хроматографического разделения. В методе ВЭЖХ–МС/МС высокого разрешения назначение хроматографической системы и требования, предъявляемые к ней, несколько отличаются от таковых в традиционных вариантах ВЭЖХ. Масс-спектрометр высокого разрешения позволяет регистрировать строго специфический параметр каждого определяемого соединения – набор отношений m/z образующихся ионов. При работе в режиме мониторинга выбранных ионных реакций для большинства соединений не возникает необходимости полного хроматографического разделения компонентов смеси, поскольку набор выбранных ионных реакций для каждого соединения специфичен, и влияние сигнала от посторонних компонентов на сигнал аналита в этом случае ничтожно мало. Кроме того, подвижная фаза в варианте ВЭЖХ–МС может содержать только летучие компоненты.
Исследуемые три метаболита циклогексилметилфторфосфоната обладают кислотными свойствами, поэтому для увеличения доли незаряженных частиц данных веществ и уменьшения размывания хроматографических пиков использовали подвижную фазу, состоящую из 0.1%-ного раствора муравьиной кислоты в воде и 0.1%-ного раствора муравьиной кислоты в ацетонитриле. В случае детектирования АБХЭ и ТА присутствие муравьиной кислоты в подвижной фазе также способствовало увеличению доли положительно заряженных ионов в камере ионизации за счет протонирования аминогрупп данных биомаркеров. Величины времен удерживания, факторов удерживания и эффективности хроматографической колонки (число теоретических тарелок на метр, тт/м) для выбранных аналитов в выбранных условиях разделения представлены в табл. 3.
Таблица 3.
Хроматографические параметры разделения маркеров циклогексилметилфторфосфоната (to= = 1.9 мин, n = 3, P = 0.95)
| Метаболит | tR, мин | k | N, тт/м |
|---|---|---|---|
| ЦГМФК | 10.4 ± 0.2 | 4.5 | 135 000 |
| АБХЭ | 13.0 ± 0.2 | 5.8 | 230 000 |
| ТА | 11.5 ± 0.2 | 5.1 | 115 000 |
В качестве критериев установления присутствия выбранных маркеров циклогексилметилфторфосфоната в образцах плазмы использовали время удерживания и совпадение двух пар выбранных ионных реакций для каждого из аналитов. На рис. 3 представлены хроматограммы образца плазмы с расчетным содержанием 2 нг/мл циклогексилметилфторфосфоната, полученного после разбавления исходного образца плазмы и проведения описанных выше процедур пробоподготовки для выявления уникальных маркеров ЦГМФК, АБХЭ и ТА. На хроматограммах контрольных образцов плазмы, не содержащих циклогексилметилфторфосфоната, отсутствовали пики, мешающие определению выбранных биомаркеров. Приведены величины аналитического сигнала по наиболее интенсивной из выбранных ионных реакций. Вторую ионную реакцию для каждого аналита использовали для надежного подтверждения факта присутствия конкретного специфического маркера. Видно, что применение масс-спектрометрии высокого разрешения в выбранных условиях нивелирует мешающее влияние матрицы на аналитический сигнал целевого аналита, что, наряду с повышением чувствительности, обусловливает высокую специфичность определения.
Рис. 3.
Хроматограммы образца плазмы крови человека с содержанием 2 нг/мл циклогексилметилфторфосфоната, полученные в выбранных оптимальных условиях пробоподготовки и анализа методом ВЭЖХ–МС/МС при определении АБХЭ (а), ТА (б) и ЦГМФК (в) в режиме регистрации выбранных ионных реакций.

Метрологические характеристики и анализ реальных объектов. Последовательным разбавлением незаражeнной плазмой исходного образца с внесенными in vitro 8 нг/мл циклогексилметилфторфосфоната (образец ОЗХО) готовили образцы с концентрациями 0.05, 0.1, 0.2, 0.5, 2 и 5 нг/мл. Пробоподготовку и анализ каждой аликвоты проводили в соответствии с описанными выше процедурами. Предел обнаружения циклогексилметилфторфосфоната для каждого из маркеров рассчитывали при условии соблюдения отношения интенсивности сигнала пика аналита к величине шума S/N > 5. Матричный эффект рассчитывали, сравнивая площади пика контрольного образца плазмы с добавкой 50 нг/мл ЦГМФК или ТА после соответствующей процедуры пробоподготовки, с площадью пика модельного водного раствора, содержащего 50 нг/мл ЦГМФК или ТА соответственно. Метрологические характеристики разработанного подхода представлены в табл. 4. При концентрации циклогексилметилфторфосфоната в плазме крови менее 0.1 нг/мл для АБХЭ и ЦГМФК и менее 0.05 нг/мл в случае ТА резко уменьшаются интенсивности аналитических сигналов (по-видимому, вследствие термической деструкции данных аналитов в камере ионизации), поэтому, несмотря на высокие коэффициенты чувствительности градуировочной зависимости, предел обнаружения циклогексилметилфторфосфоната в плазме при детектировании в виде ТА ограничен значением 0.05 нг/мл, а в случае ЦГМФК и АБХЭ – 0.1 нг/мл. Можно полагать, что обнаружение в реальной плазме крови пострадавших совокупно трех биомаркеров циклогексилметилфторфосфоната (ЦГМФК, АБХЭ и ТА) однозначно указывает на факт интоксикации и обеспечивает достоверную идентификацию ФВНПД.
Таблица 4.
Метрологические характеристики определения циклогексил-метилфторфосфоната в плазме крови в виде ЦГМФК, АБХЭ и ТА (n = 3, P = 0.95)
| Метаболит | Линейный диапазон, нг/мл | Уравнение градуировочного графика | Коэффициент корреляции | Матричный эффект, % | cmin, нг/мл |
|---|---|---|---|---|---|
| ЦГМФК | 0.2–8 | Si = 161 000ci | 0.9995 | 93 ± 7 | 0.1 |
| АБХЭ | 0.2–8 | Si = 87 500ci | 0.9952 | –* | 0.1 |
| ТА | 0.1–8 | Si = 805 400ci | 0.9992 | 90 ± 10 | 0.05 |
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 18-33-20068 мол_а_вед.
Список литературы
Convention on the prohibition of the development, production, stockpiling and use of chemical weapons and their destruction. www.opcw.org. 1997. (17.03.2019).
Pan H., Hu XZ., Jacobowitz D.M., Chen C., McDonough J., Van Shura K., Lyman M., Marini A.M. Alpha-linolenic acid is a potent neuroprotective agent against soman-induced neuropathology // Neurotoxicology. 2012. V. 33. № 5. P. 1219.
Perkins M.W., Pierre Z., Sabnekar P., Sciuto A.M., Song J., Soojhawon I., Oguntayo S., Doctor B.P., Nambiar M.P. Aerosolized delivery of oxime MMB-4 in combination with atropine sulfate protects against soman exposure in guinea pigs // Inhal. Toxicol. 2012. V. 24. № 9. P. 539.
Black R.M., Clarke R.J., Read R.W., Reid M.T.J. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to the analysis of chemical warfare samples, found to contain residues of the nerve agent sarin, sulphur mustard and their degradation products // J. Chromatogr. A. 1994. V. 662. № 2. P. 301.
Black R.M., Read R.W. Rapid screening procedures for the hydrolysis products of chemical warfare agents using positive and negative ion liquid chromatography–mass spectrometry with atmospheric pressure chemical ionisation // J. Chromatogr. A. 1999. V. 862. № 2. P. 169.
Driskell W.J., Shih M., Needham L.L., Barr D.B. Quantitation of organophosphorous nerve agent metabolites in human urine using isotope dilution gas chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2002. V. 26. № 1. P. 6.
Matsuda Y., Nagao M., Takatori T., Niijima H., Nakajima M., Iwase H., Kobayashi M., Iwadate K. Detection of the sarin hydrolysis product in formalin-fixed brain tissues of victims of the Tokyo subway terrorist attack // Toxicol. Appl. Pharmacol. 1998. V. 150. № 2. P. 310.
Minami M., Hui D., Katsumata M., Inagaki H., Boulet C.A. Method for the analysis of the methylphosphonic acid metabolites of sarin and its ethanol-substituted analogue in urine as applied to the victims of the Tokyo sarin disaster // J. Chromatogr. B. 1997. V. 695. № 2. P. 237.
Shih M.L., Smith J.R., McMonagle J.D., Dolzine T.W., Gresham V.C. Detection of metabolites of toxic alkylmethylphosphonates in biological samples // Bio. Mass Spectrom. 1991. V. 20. № 11. P. 717.
Barr J.R., Driskell W.J., Aston L.S., Martinez R.A. Quantitation of metabolites of the nerve agents sarin, soman, cyclohexylsatin, VX, and Russian VX in human urine using isotope-dilution gas chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. № 5. P. 372.
Ciner F.L., McCord C.E., Plunkett R.W., Martin M.F., Croley T.R. Isotope dilution LC-MS-MS for the detection of nerve agent exposure in urine // J. Chromatogr. B. 2007. V. 846. № 1–2. P. 42.
Kanamori-Katoaoka M., Seto Y. Simultaneous and rapid analysis of nerve gases and proteinous toxins by liquid chromatography-mass spectrometry // Jpn. J. Forensic Toxicol. 2005. V. 23. P. 21.
Liu Q., Hu X., Xie J. Determination of nerve agent degradation products in environmental samples by liquid chromatography-time-of-flight mass spectrometry with electrospray ionization // Anal. Chim. Acta. 2004. V. 512. № 1. P. 93.
Hayes T.L., Kenny D.V., Hernon-Kenny L. Feasibility of direct analysis of saliva and urine for phosphonic acids and thiodiglycolrelated species associated with exposure to chemical warfare agents using LC−MS/MS // J. Med. Chem. Def. 2004. V. 2. P. 1.
D'Agostino P.A., Chenier C.L., Hancock J.R. Packed capillary liquid chromatography-electrospray mass spectrometry of snow contaminated with sarin // J. Chromatogr. A. 2002. V. 950. № 1–2. P. 149.
Evans R.A., Jakubowski E.M., Muse W.T., Matson K., Hulet S.W., Mioduszewski R.J., Thomson S.A., Totura A.L., Renner J.A., Crouse C.L. Quantification of sarin and cyclosarin metabolites isopropyl methylphosphonic acid and cyclohexyl methylphosphonic acid in minipig plasma using isotope-dilution and liquid chromatography-time-of-flight mass spectrometry // J. Anal. Toxicol. 2008. V. 32. № 1. P. 78.
Riches J., Morton I.; Read R.W., Black R.M. The trace analysis of alkyl alkylphosphonic acids in urine using gas chromatography–ion trap negative ion tandem mass spectrometry // J. Chromatogr. B. 2005. V. 816. № 1–2. P. 251.
Mawhinney D.B., Hamelin E.I., Fraser R., Silva S.S., Pavlopoulos A.J., Kobelski R.J.J. The determination of organophosphonate nerve agent metabolites in human urine by hydrophilic interaction liquid chromatography tandem mass spectrometry // J. Chromatogr. B. 2007. V. 852. № 1–2. P. 235.
Sporty J.L., Lemire S.W., Jakubowski E.M., Renner J.A., Evans R.A., Williams R.F., Schmidt J.G., van der Schans M.J., Noort D., Johnson R.C. Immunomagnetic separation and quantification of butyrylcholinesterase nerve agent adducts in human serum // Anal. Chem. 2010. V. 82. № 15. P. 6593.
Fidder A., Hulst A.G., Noort D., de Ruiter R., van der Schans M.J., Benschop H.P., Langenberg J.P. Retrospective detection of exposure to organophosphorus anti-cholinesterases: mass spectrometric analysis of phosphylated human butyrylcholinesterase // Chem. Res. Toxicol. 2002. V. 15. № 4. P. 582.
Knaack J.S., Zhou Y., Abney C.W., Jacob J.T., Prezioso S.M., Hardy K., Lemire S.W., Thomas J, Johnson R.C. A High-throughput diagnostic method for measuring human exposure to organophosphorus nerve agents // Anal. Chem. 2012. V. 84. № 21. P. 9470.
Noort D., Fidder A., van der Schans M.J., Hulst A.G. Verification of exposure to organophosphates: generic mass spectrometric method for detection of human butyrylcholinesterase adducts // Anal. Chem. 2006. V. 78. № 18. P. 6640.
Adams T.K., Capacio B.R., Smith J.R., Whalley C.E., Korte W.D. The application of the fluoride reactivation process to the detection of sarin and soman nerve agent exposures in biological samples // Drug Chem. Toxicol. 2005. V. 27. № 1. P. 77.
Williams N.H., Harrison J.M., Read R.W., Black R.M. Phosphylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents // Arch. Toxicol. 2007. V. 81. № 9. P. 627.
Black R.M., Harrison J.M., Read R.W. The interaction of sarin and soman with plasma proteins: the identification of a novel phosphonylation site // Arch. Toxicol. 1999. V. 73. № 2. P. 123.
Peeples E.S., Schopfer L.M., Duysen E.G., Spaulding R., Voelker T., Thompson C.M., Lockridge O. Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry // Toxicol. Sci. 2005. V. 83. № 2. P. 303.
Bao Y., Liu Q., Chen J., Lin Y., Wu B., Xie J. Quantification of nerve agent adducts with albumin in rat plasma using liquid chromatography-isotope dilution tandem mass spectrometry // J. Chromatogr. A. 2012. V. 1229. P. 164.
Crow B.S., Pantazides B.G., Quiñones-González J., Garton J.W., Carter M.D., Perez J.W., Watson C.M., Tomcik D.J., Crenshaw M.D., Brewer B.N., Riches J.R., Stubbs S.J., Read R.W., Evans R.A., Thomas J.D., Blake T.A., Johnson R.C. Simultaneous measurement of tabun, sarin, soman, cyclosarin, VR, VX, and VM adducts to tyrosine in blood products by isotope dilution UHPLC−MS/MS // Anal. Chem. 2014. V. 86. № 20. P. 10397.
Williams N.H., Harrison J.M., Read R.W., Black R.M. Phosphylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents // Arch. Toxicol. 2007. V. 81. № 9. P. 627.
Корягина Н.Л., Савельева Е.И., Каракашев Г.В., Бабаков В.Н., Дубровский Я.А., Уколова Е.С., Хлебникова Н.С., Мурашко Е.А., Конева В.Ю., Уколов А.И., Копейкин В.А., Радилов А.С. Определение конъюгированных с белками метаболитов фосфорорганических отравляющих веществ в плазме крови // Журн. аналит. химии. 2016. Т. 71. № 8. С. 883. (Koryagina N.L., Savel’eva E.I., Karakashev G.V., Babakov V.N., Dubrovskii Ya.A., Ukolova E.S., Khlebnikova N.S., Murashko E.A., Koneva V. Yu., Ukolov A.I., Kopeikin V.A., Radilov A.S. Determination of protein adducts of organophosphorus nerve agents in blood plasma // J. Analyt. Chem. 2016. V. 71. № 8. P. 849.)
Jiang W., Murashko E.A., Dubrovskii Y.A., Podolskaya E.P., Babakov V.N., Mikler J., Nachon F., Masson P., Schopfer L.M., Lockridge O. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of titanium oxide-enriched peptides for detection of aged organophosphorus adducts on human butyrylcholinesterase // Anal. Biochem. 2013. V. 439. № 2. P. 132.
Mathews T.P., Carter M.D., Johnson D., Isenberg S.L., Graham L.A., Thomas J.D., Johnson R.C. High-confidence qualitative identification of organophosphorus nerve agent adducts to human butyrylcholinesterase // Anal. Chem. 2017. V. 89. № 3. P. 1955
Hamelin E.I., Schulze N.D., Shaner R.L., Coleman R.M., Lawrence R.J., Crow B.S., Jakubowski E.M., Johnson R.C. Quantitation of five organophosphorus nerve agent metabolites in serum using hydrophilic interaction liquid chromatography and tandem mass spectrometry // Anal. Bioanal. Chem. 2014. V. 406. № 21. P. 5195.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии