

Журнал аналитической химии, 2020, T. 75, № 6, стр. 510-515
Особенности спектрофотометрического определения мономерных антоцианов
Л. А. Дейнека a, А. Н. Сидоров a, В. И. Дейнека a, *, Я. Ю. Кульченко a, И. П. Блинова a
a Белгородский государственный национальный исследовательский университет,
Институт фармации, химии и биологии
308015 Белгород, ул. Победы, 85, Россия
* E-mail: deineka@bsu.edu.ru
Поступила в редакцию 29.04.2019
После доработки 01.06.2019
Принята к публикации 28.12.2019
Аннотация
Показано, что дифференциальный спектрофотометрический метод определения мономерных антоцианов (в смеси с полимерными) непригоден для измерений в случае антоцианов, ацилированных замещенными коричными кислотами. Это объясняется конкуренцией между реакцией нуклеофильного присоединения молекулы воды (с высвобождением Н+) к флавилиевым ионам с образованием неокрашенной формы псевдооснования (A) и реакцией депротонирования флавилиевых ионов с образованием хиноноидных структур (Б), также имеющих окраску с некоторым батохромным сдвигом максимума абсорбции. Применимость дифференциального спектрофотометрического метода предложено оценивать путем сопоставления разностей спектров, полученных при различных рН (1, 2, 3, 4 и 4.5). Метод применим при преобладании направления А, когда указанные разности спектров неразличимы с учетом масштабирования. Метод неприменим при основном направлении Б, когда спектральные разности различаются. Во втором случае оправдан упрощенный метод определения антоцианов без учета полимерных антоцианов.
Антоцианы относятся к обширному классу флавонидов, выделяясь среди них необычно высокой растворимостью в воде [1, 2], существованием в виде нескольких рН-зависимых форм [3, 4], среди которых имеются окрашенные, с присущей всем флавоноидам высокой антиоксидантной активностью [1, 4, 5]. Перечисленные выше свойства определяют большой интерес к антоцианам как к природным красителям для пищевой и медицинской промышленности [1, 6]. Антоцианы могут быть выделены в кристаллическом состоянии [2, 7], но вследствие биосинтеза в растительных источниках в виде сложного набора соединений различного строения и высокой реакционной способности получение индивидуальных антоцианов со строго определенным составом проблематично. Из-за этого, например, молярные коэффициенты поглощения, найденные различными исследователями для одних и тех же антоцианов, могут заметно различаться [8]. Так, очевидно, что непригодность использованных в работе стандартных образцов антоцианов является причиной большого различия (около 200%) результатов количественного определении антоцианов в нескольких растительных объектах спектрофотометрическим и хроматографическим методами [9]. Однако если при анализе методом ВЭЖХ концентрацию антоцианов в градиуровочных растворах определять спектрофотометрически, то полученные результаты хорошо совпадают [10].
Решением проблемы может быть пересчет результатов спектрофотометрического метода на один из наиболее распространенных антоцианов, например на цианидин-3-глюкозид хлорид, используя наиболее достоверное значение молярного коэффициента поглощения [8]. Однако в экстрактах наряду с мономерными антоцианами (МА) могут присутствовать продукты их полимеризации – полимерные антоцианы (ПА) [8]. Абсорбцию ПА обычно стремятся исключить из конечной оптической плотности для расчета содержания МА, несмотря на то, что в экстракте могут появиться еще и пираноантоцианы [11].
Настоящая работа посвящена исследованию наиболее часто используемого для определения МА дифференциального спектрофотометрического метода (ДСФМ) [8], адаптированного в Российской Федерации в виде ГОСТ [12].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Экстракты антоцианов готовили настаиванием растительного материала в 0.1 М водном растворе соляной кислоты в течение суток и хранили в бытовом холодильнике. Для контроля видового состава экстракты очищали методом твердофазной экстракции на патронах ДИАПАК С18 (БиоХимМак СТ, Москва).
При спектрофотометрическом определении аликвоту экстракта (Va) разбавляли до метки в колбе емк. Vк, доводя рН до 1 (1 М HCl в воде), и записывали первый спектр (относительно этанола) с максимумом абсорбции A1(λ1) при длине волны λ1. К другой такой же аликвотной порции экстракта (Va) добавляли 1 М водный раствор NaOH, доводя рН до 4.5 и разбавляя до метки (Vк) ацетатным буферным раствором с рН 4.5, и записывали второй спектр, определяя второе значение оптической плотности A2(λ1) при той же длине волны.
Электронные спектры поглощения записывали на спектрофотометре Shimadzu UV 2550 в кварцевых кюветах. После получения базовой линии спектры записывали при обнулении при λ = = 700 нм (для исключения влияния рассеяния на коллоидных частицах экстракта). Оптическую плотность для расчета концентрации мономерных антоцианов в пробе находили прямым вычитанием двух изменений:
(1)
$A = {{А}_{{{\lambda (max)}}}}\left( {{\text{pH }}1} \right) - {{А}_{{\lambda ({\text{max}})}}}\left( {{\text{pH }}4.5} \right),$Концентрацию мономерных антоцианов в исходном экстракте рассчитывали с учетом использованных объемов:
где с – концентрация антоцианов в образце в пересчете на цианидин-3-глюкозид, М; l – длина оптического пути, см; 26900 – молярный коэффициент поглощения, л моль–1 см–1; Vк и Vа – объемы колбы и аликвоты соответственно, мл.При записи спектров при различных значениях рН использовали одни и те же объемы аликвот исходного раствора, рН доводили до нужного значения добавлением 0.1 или 0.01 М раствора NaOH и разбавляли растворы до метки. Численные характеристики полученных спектров с шагом 1 нм экспортировали из программы обработки УФ-спектров прибора Shimadzu в MSExcel для последующей обработки.
Для измерения рН растворов использовали рН-метр рН-150МИ с комбинированным стеклянным электродом ЭСК-10603.
Для контроля видового состава антоцианов в образцах применяли хроматографическую систему Agilent 1260 Infinity с диодно-матричным и масс-спектрометрическим детекторами; колонку (150 × 2.1 мм) Kromasil 100–5C18 при температуре термостата колонок 40°С и градиентный режим элюирования. Элюент А содержал 10 об. % муравьиной кислоты и 8 об. % ацетонитрила в воде. Элюент Б – 10 об. % муравьиной кислоты и 20 об. % ацетонитрила в воде. Режим элюирования: 0 мин – 100% А, 20 мин – 100% Б, 30 мин – 100% Б, 31 мин – 0% Б. Расход подвижной фазы 200 мкл/мин. Хроматограммы записывали при 515 нм (рис. 1). Масс-спектрометрические измерения выполняли в режиме ионизации электрораспылением со сканированием положительно заряженных ионов, задавая напряжение на фрагменторе 225 В для частичной фрагментации исходных “молекулярных” ионов.
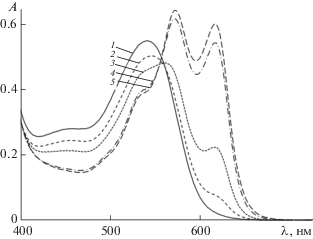
Рис. 1.
Электронные спектры поглощения экстракта цветков Clitoria ternatea при рН 1 (1), 2 (2), 3 (3), 4 (4) и 4.5 (5).
Хроматограммы записывали, хранили и обрабатывали, используя программное обеспечение Agilent ChemStation.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Дифференциальный спектрофотометрический метод определения антоцианов основан на том, что в сильнокислых растворах (рН ≤ 1) эти соединения существуют в окрашенной флавилиевой форме (схема 1, I ). Это позволяет выполнять первое измерение в максимуме оптической плотности, положение которого зависит от типа агликонов и характера их гликозилирования, включая ацилирование некоторыми органическими кислотами. Поправки на рассеяние света на коллоидных частицах (при 700 нм) осуществляются в спектрофотометре Shimadzu UV-2550 автоматически при активации соответствующей функции. В таком случае:
(4)
${{A}_{1}} = {{А}_{{{\lambda (max)}}}}\left( {{\text{pH }}1} \right) - {{А}_{{700{\text{ нм}}}}}\left( {{\text{pH }}1} \right).$
Схема 1. Схема превращения флавилиевого иона (I) в псевдооснование (II) и в халконные (IIIa и IIIb) формы.
Выполнение второго измерения основано на предположении о том, что при повышении рН окрашенная флавилиевая форма постепенно превращается в неокрашенную форму псевдооснования (схема 1 , II [3]) за счет нуклеофильной атаки молекулой воды положения 2 флавилиевого иона с последующим выбросом протона (реакция гидратации, hydration reaction). Считается, что при рН 4.5 флавилиевая форма полностью исчезает. В этом случае оптическая плотность раствора определяется только концентрацией ПА, что позволяет выполнить второе измерение:
(5)
${{A}_{2}} = {{А}_{{{\lambda (max)}}}}\left( {{\text{pH }}4.5} \right) - {{А}_{{{\text{700 нм}}}}}\left( {{\text{pH }}4.5} \right).$Предполагается, что разность результатов двух измерений (конечный показатель в ДСФМ) соответствует оптической плотности, определяемой только концентрацией МА, уравнение (2). Форма псевдооснования (или полуацетальная форма) находится в равновесии с цис-халконной формой (схема 1 , IIIa) (слабоокрашенной в коротковолновой области), которая, в свою очередь, находится в равновесии с также слабоокрашенной транс-халконной формой (схема 1 , IIIb). Несмотря на то, что повышение рН с 1.0 до 4.5 обычно приводит к существенной потере интенсивности окраски, описанный подход имеет несколько ограничений.
1) Равновесия (между формами II, IIIa и IIIb) не зависят от рН, поэтому изменение интенсивности окраски антоцианов можно контролировать при различных рН при заданной константе гидратации флавилиевого иона, Kh. Из данных работы [10] следует, что не существует такой Kh, при которой доля флавилиевой формы в растворе при рН 1 была бы равна 100%, а при рН 4.5 – 0%. Однако этот недостаток (потеря примерно 1.5% флавилиевой формы при вычитании оптических плотностей) можно устранить поправкой в молярном коэффициенте поглощения. Таким образом, отличие от нуля абсорбции антоцианов при рН 4.5 необязательно указывает на присутствие полимерных форм.
2) По данным [3], равновесие между формой псевдооснования и цис-халконной формой устанавливается быстро, а между цис- и транс-халконными формами – медленно [3]. В связи с этим перевод всех форм во флавилиевую форму требует определенного времени (в зависимости от исходного состояния). По нашим данным для достижения равновесия требуются примерно сутки. При выдержке в течение 6 ч раствора при рН 1 при комнатной температуре “потеря” антоцианов не превысит 1–2%. В этом отношении вызывает вопрос рекомендация официальной методики [8] выдерживать такой раствор перед измерением в течение от 15 мин до 1 ч, поскольку при более длительной выдержке наблюдается увеличение оптической плотности (!?).
3) Наиболее серьезное ограничение, которое ставит под вопрос возможность применения ДСФМ, заключается в следующем. При повышении рН, кроме превращения в форму псевдооснования (А), возможно иное направление течение реакции – депротонирование флавилиевых ионов с образованием хиноноидных структур (Б) [3] (схема 2 ) – незаряженных форм IVa и IVb и заряженной V. При этом число и устойчивость таких структур и вероятность их образования зависят от строения антоцианов [13]. Хиноноидные структуры также окрашены (с небольшим батохромным сдвигом максимума абсорбции [14] для незаряженных форм), поэтому измерение абсорбции при рН 4.5 бессмысленно, а применение ДСФМ может привести к существенной недооценке содержания МА.

Схема 2 . Схема превращения флавилиевого иона (I) в хиноноидные незаряженные (IVa и IVb) и в хиноноидную заряженную (V) формы.
Для оценки возможности применения ДСФМ можно воспользоваться предлагаемой ниже схемой: 1) записывают в численном виде электронный спектр поглощения экстракта при рН 1 с шагом 1 нм, получая функцию AрН 1(λ); 2) аналогично находят функции при рН 2, 3 и 4; 3) рассчитывают разностные спектры:
(6)
${{A}_{{1--2}}}\left( \lambda \right) = {{A}_{{{\text{рН 1}}}}}\left( \lambda \right) - {{A}_{{{\text{рН 2}}}}}\left( \lambda \right),$(7)
${{A}_{{1--3}}}\left( \lambda \right) = {{A}_{{{\text{рН 1}}}}}\left( \lambda \right) - {{A}_{{{\text{рН 3}}}}}\left( \lambda \right),$(8)
${{A}_{{1--4}}}\left( \lambda \right) = {{A}_{{{\text{рН 1}}}}}\left( \lambda \right) - {{A}_{{{\text{рН 4}}}}}\left( \lambda \right).$Если при изменении рН в ряду 1 → 2 → 3 → 4 происходит только уменьшение концентрации флавилиевой формы, то полученные разности (6)–(8) могут различаться только интенсивностью – разность любой пары не отличается от нулевой линии при подборе коэффициента g, например:
В этом случае применение ДСФМ оправдано. В противном случае применение этого метода следует исключить.
В ряде случаев невозможность применения ДСФМ очевидна уже при визуальном анализе набора спектров, записанных при различных рН. Так, в случае экстракта лепестков цветков Clitoria ternarea (голубой тайский императорский чай) при рН 4.5 оптическая плотность очень велика, а в максимуме превосходит оптическую плотность раствора флавилиевой формы (рис. 1). Следует отметить, что антоцианы этого растения уникальны – они построены на дельфинидине со сложным чередующимся гликозилированием (глюкозой) и ацилированием (пара-кумаровой кислотой) кольца В [15]. Дифференциальный спектрофотометрический метод неприменим также для определения антоцианов краснокочанной капусты (рис. 2), в антоцианах которой наблюдается двойное ацилирование цианидин-3-софорозид-5-глюкозида по гликозидному радикалу в положении 3 синаповой, феруловой или пара-кумаровой кислотами [16]. Результаты некоторых измерений представлены в табл. 1 с указанием возможности применения ДСФМ, установленной по спектральным разностям.
Рис. 2.
Электронные спектры поглощения экстракта краснокочанной капусты при рН 1 (1), 2 (2), 3 (3), 4 (4), 4.5 (5).
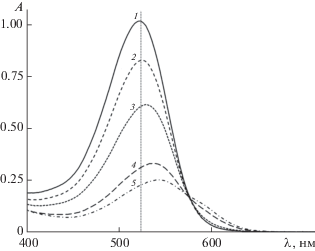
Таблица 1.
Применимость дифференциального спектрофотометрического метода к определению мономерных антоцианов в некоторых растительных объектах
| Растительный материал | Типы антоцианов | рН 1.0 (рН 4.5) | α*, мол. % |
Применимость | |
|---|---|---|---|---|---|
| λmax, нм | A | ||||
| Арония, плоды | Моногликозиды | 510 (518) |
0.914 (0.081) |
91.1 | Да |
| Боярышник, плоды | Моногликозиды | 512 (511) |
0.660 (0.068) |
89.7 | Да |
| Виноград, плоды | Моногликозиды + ацилированные КК** | 518 (523) |
0.719 (0.118) |
83.6 | Нет |
| Ирис, цветки. | Смесь, включая ацилированные КК | 523 (529) |
0.880 (0.555) |
36.9 | Нет |
| Капуста, листья | В основном ацилированные КК | 522 (537) |
0.802 (0.253) |
68.5 | Нет |
| Кукуруза, обертки | Смесь, включая ацилированные МК*** | 511 (514) |
0.335 (0.054) |
83.9 | Да |
| Паслен, плоды | Ацилированные КК | 526 (532) |
1.056 (0.129) |
87.8 | Нет |
| Роза, цветы | Дигликозиды | 514 (523) |
1.405 (0.084) |
94.0 | Да |
| Черная смородина, плоды | Моно- и дигликозиды | 515 (518) |
0.272 (0.037) |
86.4 | Да |
| Черемуха, плоды | Моно и дигликозиды | 513 (520) |
0.902 (0.142) |
84.3 | Да |
По нашим наблюдениям ДСФМ практически неприменим к анализу экстрактов, содержащих антоцианы, ацилированные производными коричной кислоты. В таких случаях оправдано использование упрощенной методики, не предполагающей измерение оптической плотности при рН 4.5.
Список литературы
Bueno J.M., Ramos-Escudero F., Sáez-Plaza P., Muñoz A.M., Navas M.J., Asuero A.G. Analysis and antioxidant capacity of anthocyanin pigments. Part I: General considerations concerning polyphenols and flavonoids // Crit. Rev. Anal. Chem. 2012. V. 42. P. 102.
Дейнека В.И., Дейнека Л.А., Сидоров А.Н., Костенко М.О., Блинова И.П. Оценка растворимости антоцианов с использованием насадок для твердофазной экстракции // Журн. физ. химии, 2016. Т. 90. № 4. С. 622. (Deineka V.I., Sidorov A.N., Deineka L.A., Kostenko M.O., Blinova I.P. Estimating the solubility of anthocyanins using cartridges for solid-phase extraction // Russ. J. Phys. Chem. A. 2016. V. 90. № 4. P. 861.)
Broiillard R., Lang J. The hemiacetal-cis-chalcone equilibrium of malvin, a natural anthocyanin // Can. J. Chem. 1990. V. 68. P. 755.
Bueno J.M., Sáez-Plaza P., Ramos-Escudero F., Jiménez A.M., Fett R., Asuero A.G. Analysis and antioxidant capacity of anthocyanin pigments. Part II: Chemical structure, color, and intake of anthocyanins // Crit. Rev. Anal. Chem. 2012. V. 42. P. 126.
Wang H., Cao G., Prior R.L. Oxygen radical absorbing capacity of anthocyanins // J. Agric. Food Chem. 1997. V. 45. P. 304.
Khoo H.E., Azlan A., Tang S.T., Lim S.M. Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits // Food Nutr. Res. 2017. V. 61. Article 1361779.
Lasagabaster A., Martin C., Gofii M. Preparation of spherically agglomerated crystals of the 3,5-diglucoside of cyanidin (cyanin) // J. Chem. Tech. Biotechnol. 1994. V. 60. P. 397.
Mónica Giusti M., Wrolstad R.E. Characterization and measurement of anthocyanins by UV-visible spectroscopy // Curr. Protoc. Food Anal. Chem. 2001. F1.2.1.
Lee J., Rennaker C., Wrolstad R.E. Correlation of two anthocyanin quantification methods: HPLC and spectrophotometric methods // Food Chem. 2008. V. 110. P. 782.
Дейнека Л.А., Блинова И.П., Кульченко Я.И., Озер П.С., Саенко И.И., Дейнека В.И. Сохранность и переход между формами антоцианов в растворах // Успехи современного естествознания. 2016. № 2. С. 16.
Rentzsch M., Schwarz M., Winterhalter P. Pyranoanthocyanins – an overview on structures, occurrence, and pathways of formation // Trends Food Sci. Technol. 2007. V. 18. P. 526.
ГОСТ 32709-2014 Продукция соковая. Методы определения антоцианинов. М.: Стандартинформ, 2014. 18 с.
Pina F. Thermodynamics and kinetics of flavylium salts malvin revisited // J. Chem. Soc. Faraday Trans. 1998. V. 94. P. 2109.
Levi M.A.B., Scarminio I.S., Poppi R.J., Trevisan M.G. Three-way chemometric method study and UV-vis absorbance for the study of simultaneous degradation of anthocyanins in flowers of the Hibiscus rosa-sinensys species // Talanta. 2004. V. 62. P. 299.
Terahara N., Saito N., Honda T., Tokis K., Osajima Y. Acylated anthocyanins of Clitoria ternatea flowers and their acyl moieties // Phytochemistry. 1990. V. 29. P. 949.
Lo Scalzo R., Genna A., Branca F., Chedin M., Chassaigne H. Anthocyanin composition of cauliflower (Brassica oleracea L. var. botrytis) and cabbage (B. oleracea L. var. capitata) and its stability in relation to thermal treatments // Food Chem. 2008. V. 107. P. 136.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии