

Журнал аналитической химии, 2020, T. 75, № 6, стр. 502-509
Мицеллярно-экстракционное концентрирование и цветометрическое определение некоторых фенолов
С. Ю. Доронин a, *, Е. С. Жестовская b, Э. И. Цыгулёва a
a Саратовский государственный университет им. Н.Г. Чернышевского, Институт химии
410012 Саратов, ул. Астраханская, 18/3, Россия
b Научный центр “Сигнал”
107014 Москва, ул. Большая Оленья, 8, Россия
* E-mail: doroninsu@mail.ru
Поступила в редакцию 27.02.2019
После доработки 04.07.2019
Принята к публикации 06.12.2019
Аннотация
Рассмотрено извлечение и концентрирование фенола и его гидроксипроизводных с применением индивидуальных и смешанных мицелл поверхностно-активных веществ неионного и катионного типов с цифровой регистрацией аналитического сигнала. Данный подход основан на цветной реакции фенолов с диазотированным 4-нитроанилином и мицелярной экстракции продуктов их взаимодействия (азосоединения). Оптимизированны параметры мицеллярной экстракции (рН, концентрации реагентов, NaOH и этанола) для эффективного извлечения азосоединений исследованных фенолов. Предложенный способ позволяет определять фенол и резорцин на уровне ПДК с погрешностью, не превышающей 10%. Для качественной оценки присутствия фенолов наряду с цветометрическими (R, G, B) применены геометрические (площадь – S, периметр – P) параметры соответствующих лепестковых диаграмм. Градуировочные зависимости линейны в пределах 1 × 10–7–3 × 10–5 М для фенола; 1 × 10–7–2 × 10–5 М для резорцина и флороглюцина. Разработанный цветометрический способ апробирован для определения резорцина в лекарственном препарате “Резорцинол”.
Фенол и его производные обладают канцерогенными, тератогенными, кумулятивными свойствами (например, ПДК в воде фенола и резорцина составляют 0.001 мг/л [1]) и относятся к основным органическим загрязнителям водных объектов. В обзорах [2, 3] показано, что фенолы можно определять на уровне ПДК и ниже без предварительного концентрирования с применением хроматографических, люминесцентных и электрохимических методов анализа. Тем не менее, разработка простых в исполнении, экономичных, чувствительных и селективных методов, в том числе тест-методов, с предварительным концентрированием указанных аналитов является актуальной аналитической задачей. Применяют жидкостно–жидкостную, твердофазную, сверхкритическую флюидную, парофазную экстракцию, экстракционное вымораживание, а также сорбционные и мембранные способы концентрирования [2]. Эта стадия пробоподготовки часто приводит к увеличению стоимости анализа и связана, как правило, с применением токсичных и летучих органических растворителей. В последнее время альтернативой органическим растворителям стали разбавленные водные растворы нелетучих малотоксичных поверхностно-автивных веществ (ПАВ), которые применяют как экстрагенты для концентрирования веществ по методологии на основе ''точки помутнения'' (clound point, CP) [2–4]. Она основана на разделении гомогенных растворов ПАВ (их смесей) при нагревании, изменении рН, добавлении различных высаливателей на две изотропные фазы: одна из них, обогащенная ПАВ (surfactant-rich phase; micellar-rich phase), концентрирует вещества, до фазового разделения распределенные по всему объему раствора; другая фаза, обедненная ПАВ, или водная фаза (micellar-dilute phase, micelle-poor, surfactant depleted, aqueous phase) содержит ПАВ с концентрацией до критической концентрации мицеллообразования (ККМ) и остаточные количества экстрагируемого вещества. Обогащенная ПАВ фаза обычно является экстрагентом [4]. Эту методологию применяют для концентрирования аналитов как неорганической, так и органической природы с высокими значениями коэффициентов извлечения [4, 5].
Одним из важных факторов, влияющих на характер и количественные характеристики СР-экстракции, является выбор типа ПАВ. Так, в ряде работ фенолы определяют спектрофотометрически по собственному поглощению с использованием в качестве экстрагентов неионных ПАВ [6–8]. В этом случае применяют только такие ПАВ, которые не содержат в своей структуре ароматических фрагментов, мешающих определению фенолов в УФ-области спектра. Рекомендации по выбору ПАВ для извлечения фенола и близких к нему по растворимости веществ даны в работе [9]. Оценивалось влияние степени полимеризации в полиэтоксилатной группе (т.е. число полиэтоксилатных групп), числа алкильных групп и степень разветвления цепи.
Применение СР-экстракции для извлечения и концентрирования фенолов сочетается со спектрофотометрией, ВЭЖХ, капиллярным электрофорезом, мицеллярно-электрокинетической жидкостной хроматографией [3]. Наряду с указанными методами в последнее время все большее распространение получает цветометрия [10], в которой аналитическим сигналом является интенсивность параметров цветности электронного изображения, полученного с использованием цифровых устройств, таких как фотоаппарат, сканер и др., обработанного при помощи различных графических редакторов. Данный способ позволяет быстро, объективно и автоматизированно оценивать цветометрические характеристики исследуемых окрашенных твердых или жидких образцов [11–13].
Выше отмечено, что применение для СР-методологии ПАВ, содержащих в своей структуре ароматический фрагмент, затрудняет определение фенолов, имеющих максимумы поглощения в УФ-диапазоне. Решением данной проблемы может быть применение цветных реакций фенола и его замещенных с органическими реагентами, что позволяет сместить максимум поглощения в видимую часть спектра, устранив поглощение ПАВ. Данный подход лежит в основе цветометрического определения фенолов. Для этой цели применяют их взаимодействие с 4-аминоантипирином [14]. Эта реакция имеет ряд недостатков: использование большого количества реагентов (например, хлороформа – летучего токсичного растворителя); зависимость степени извлечения фенола от pH водного и водно-солевого раствора, а также от природы высаливателя. Данные о применении СР-методологии для предварительного концентрирования фенолов и последующего цветометрического их определения в виде азопроизводных – продуктов азосочетания с 4-динитрофенилдиазонием в литературе отсутствуют.
Цель настоящего исследования – разработка цветометрического способа определения фенола, резорцина и флороглюцина в водных средах после предварительного их СР-концентрирования комбинированными системами на основе неионных и катионных ПАВ в виде дериватизатов с 4-динитрофенилдиазонием.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Электронные спектры поглощения исследуемых растворов регистрировали на спектрофотометре Shimadzu UV-2550 с программным обеспечением UVProbe-2.34 в кюветах с толщиной поглощяющего слоя l = 1 см. Электронные изображения получали фотоаппаратом Canon EOS 50 D (EFS 17–85 mm, ultrasonic) с последующей их обработкой в графическом редакторе Adobe Photoshop CS5. Для разделения фаз применяли высокоскоростную центрифугу с охлаждением HERMLE Z383K (Германия).
Реактивы. В работе применяли: фенол (≥99%, Sigma, Германия); резорцин (99%, Sigma, Германия); флороглюцин (99%, Sigma, Германия); 4‑нитроанилин (4-НА) (≥99%, Sigma, Германия); нитрит натрия ч. (ХимМед); натрия гидроксид (99.99%, Sigma, Германия); карбонат натрия (≥99%, Sigma, Германия); неионные ПАВ: ОП-10 (ХИМПЭК, Россия), Тритон Х-114 (Sigma, Германия), Бридж-35 (Sigma, Германия), цетилтриметиламмония хлорид (ЦТМАХ, 25% в H2O, Sigma, Германия); этанол “для хроматографии” (Chromasolv®, Merck, Германия); воду деионизированную с удельным сопротивлением 18.2 мОм/см, полученную с помощью установки NANO Pure (Thermo Scientific, США).
Исходные стандартные 0.01 М растворы фенолов готовили растворением точных навесок препаратов в воде. Диазотирующий агент (4-НА + + NaNO2) с концентрацией реактантов 0.001 М готовили в 1 М HCl.
Способы CP-концентрирования фенолов. Для определения следовых количеств фенолов применяли реакцию с диазотированным 4-нитроанилином в щелочной среде и Тритон X-114 в качестве экстрагента. Поскольку реакция образования азосоединений, полученных реакцией исследуемых фенолов с 4-нитрофенилдиазонием, протекает в щелочной среде, их предварительную СР-экстракцию осуществляли двумя способами. Для этого применяли два типа высаливателей: NaOH и Na2CO3.
Способ 1: в пробирку помещали 100 мкл 4-НА, 100 мкл NaNO2 и 500 мкл HCl. Затем добавляли порцию 3 × 10–6–2 × 10–5 М раствора исследуемого фенола, 700 мкл этилового спирта, 1.75 мл 20%-ного раствора ПАВ и 1.4 мл 10 М раствора NaOH. Доводили объем до 5 мл водой и перемешивали. После разделения фаз отбирали 500 мкл мицеллярно-насыщенной фазы ПАВ, разбавляли ее 1 М раствором NaOH или водой и фотометрировали. Аналогично готовили раствор сравнения, не содержащий фенолов.
Способ 2: в пробирку помещали 100 мкл 4-НА, 100 мкл NaNO2 и 500 мкл HCl. Затем добавляли порцию 3 × 10–6–2 × 10–5 М раствора фенола, 1 мл 1.2 М раствора Na2CO3, аликвоту 20%-ного раствора ПАВ. Доводили объем до 5 мл водой, перемешивали и в зависимости от применяемого ПАВ центрифугировали при 4500 об/мин в течение 10 мин (Тритон Х-114) или нагревали на водяной бане (ОП-10, Бридж-35). В обоих вариантах водную фазу отделяли декантацией, а мицеллярную фазу разбавляли 50%-ным (по объему) этанолом.
Для цветометрического определения концентрации фенолов в водных средах получали цифровые изображения окрашенных обогащенных ПАВ фаз (они образуются через 20 мин после смешивания растворов по способам 1 и 2), полученных в специализированном снабженном лампами дневного света боксе, внутренняя поверхность которого обработана черной матовой краской. Для получения изображений применяли зеркальный фотоаппарат Nikon D 5100 (параметры фотоаппарата: ISO – 160, выдержка – 1/250, приближение – 18 мм, F – 3.5; объектив AF-S Nikkor 18–55 мм; качество изображений – RAW). Цветометрические параметры определяли путем математической обработки цифровых изображений с использованием графического редактора Adobe Photoshop CS6. Для этого часть изображения предварительно усредняли до одного пикселя с помощью фильтра “Blur” (Average), и определяли значения координат цветовых параметров R, G, B, L и K.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Дериватизация исследованных фенолов. Поскольку в качестве аналитов выбрали неокрашенные фенолы, реакцией диазотирования 4‑нитроанилина (наиболее реакционноспособный ариламин, образующий устойчивую соль хлорид 4-нитрофенилдиазония) и последующего азосочетания их переводили в окрашенные аналитические формы соответствующих азосоединений (схема 1):

R1 = R2 = H (фенол); R1 = OH, R2 = H (резорцин); R1 = R2 = OH (флороглюцин)
Схема 1. Реакции диазотирования 4-нитроанилина и азосочетания 4-нитрофенилдиазония с исследуемыми фенолами.
СР-концентрирование азосоединений фенолов мицеллярными фазами неионных ПАВ. Образующиеся азосоединения экстрагировали в мицеллярно-насыщенные фазы различных неионных (ОП-10, Тритон Х-114, Бридж-35) ПАВ. Для оптимизации условий CP-экстракции фенолов варьировали рН, концентрации реактантов, NaOH и этанола. Следует отметить, что кислотность среды, создаваемая добавлением различных концентраций растворов NaOH или Na2CO3, влияет не только на возможность проведения мицеллярной экстракции фазами ПАВ, но и на получение соответствующей окрашенной аналитической формы (схема 2 ):
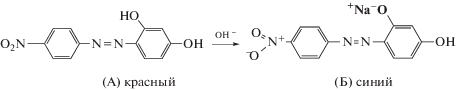
Схема 2 . Образование анионной формы азосоединения в щелочной среде.
Так, например, изучено состояние продукта азосочетания диазотированного 4-НА с резорцином (Магнезон I, форма А) в водно-мицеллярных средах при различных pH. Как видно из рис. 1, спектр Магнезона I в водной среде характеризуется максимумом средней интенсивности при 450 нм (форма А), в спектре 4-НА регистрируется максимум поглощения при 380 нм. В кислой среде набдюдается гипсохромный сдвиг максимума поглощения полосы 450 нм, что связано с образованием его протонированной формы. В щелочной среде спектр Магнезона I представлен двумя максимумами поглощения при 450 и 555 нм, что обусловлено переходом формы А в форму Б. При увеличении концентрации NaOH форма Б преобладает в растворе (рис. 1). Для продукта азосочетания и диазотирования 4-НА с фенолом и флороглюцином аналогичных эффектов в спектрах поглощения не наблюдали.
Рис. 1.
Спектры поглощения Магнезона I (1–4) и 4‑НА (5) при различных pH: смагнезона I = 2 × 10–5 М, раствор сравнения – дистиллированная вода, l = 1 см, 1 – 0.1 М НСl, 2 – 0.01 М НСl. 3 – 2.8 М NaOH, 4 – 1 М NaOH, 5 – 2 × 10–5 М 4-НА.

Спектрофотометрически изучено влияние концентрации 4-НА на образование формы Б в диапазоне концентраций 3 × 10–6–2 × 10–4 М (рис. 2а). Установлено, что оптическая плотность увеличивается до концентрации 4-НА 2 × 10–5 М, поэтому дальнейшие исследования проводили при этой его концентрации.
Рис. 2.
Зависимость оптической плотности системы: резорцин–4-НА–N${\text{O}}_{2}^{ - }$–ОП-10 от концентрации 4‑НА: срезорцина = 2 × 10–5 М, сNaOH = 2.8 М, сОП-10 = = 7% (а); влияние концентрации Тритона Х-114 (1), Бридж-35 (2) и ОП-10 (3) различных ПАВ на степень извлечения резорцина: срезорцина = 2 × 10–5 М, сNaOH = 2.8 М (б).

Ранее нами на примере систем неионное ПАВ–электролит (неэлектролит) показано [15], что наибольшее влияние на процесс фазового разделения растворов неионных ПАВ при 20 ± 5°С оказывают концентрации щелочи (NaOH) и органических растворителей (этанол). Для системы НПАВ–NaOH–C2H5OH фазовое разделение достигалось при концентрации NaOH 2.2–3 М и этанола 2–30 об. %. В связи с этим фазовое разделение в системе фенол–неионное ПАВ–NaOH–C2H5OH исследовали при оптимальных концентрациях щелочи и этанола 2.8 М и 15 об. % соответственно. Кроме того, поскольку реакция с фенолом протекает в слабощелочной среде, наряду с NaOH применяли водный раствор Na2CO3. В этом случае для достижения фазового разделения системы необходимо либо дополнительное центрифугирование (Тритон Х-114, Бридж-35), либо нагревание (ОП-10, Тритон Х-114, Бридж-35).
Степень извлечения аналитов рассматривали на примере трех неионных ПАВ в диапазоне их концентраций 2–12% (рис. 2б). Тритон Х-114 и его технический аналог ОП-10 являются представителями оксиэтилированных производных алкилфенолов c общей формулой СnH2n + 1C6H4O(C2H4O)mH, Бридж-35 – полиоксиэтилированный лауриловый эфир (C12H25(C2H4O)23OH). Как видно из рис. 2б, для всех исследованных НПАВ при их концентрациях ≥7 мас. % степень извлечения резорцина практически одинакова. Наилучший эффект достигается при использовании Тритона Х-114, содержащего в своей структуре разветвленный алкильный радикал, для которого степень извлечения резорцина мало зависит от концентрации в интервале от 3 до 12 мас. %. Некоторые характеристики растворов исследуемых ПАВ представлены в табл. 1.
Таблица 1.
Некоторые характеристики исследуемых растворов неионных ПАВ
| ПАВ | Содержание основного вещества, % | ККМ, мМ | Высаливатель | |||
|---|---|---|---|---|---|---|
| Na2CO3 | NaOH | |||||
| tпом*, °С | локализация мицеллярной фазы | tпом, °С | локализация мицеллярной фазы | |||
| ОП-10 | 80 | 0.23 | 80–90 | Внизу | 20–25 | Вверху |
| Тритон Х-114 | 98 | 0.20 | 70–80 | 20–25 | ||
| Бридж-35 | 98 | 0.075 | >100 | 20–25 | ||
Установлено, что степень извлечения аналитических форм фенолов также зависит от их природы. Так, например, 2%-ного раствора Тритона Х-114 вполне достаточно для практически полного извлечения 2 × 10–5 М продукта взаимодействия фенола с 4-НА, в то время как азоформа А той же концентрации частично остается в водной фазе. Увеличение числа OH-групп в исследуемом фенольном аналите приводит к снижению его степени извлечения в мицеллярную фазу неонного ПАВ.
Исследовано влияние катионного (ЦТМАХ) ПАВ на изменение оптической плотности системы резорцин–4-НА–N${\text{O}}_{2}^{ - }$–ОП-10–ЦТМАХ. Установлено, что присутствие ЦТМАХ усиливает аналитический сигнал. При 20 ± 5°С наибольший эффект достигается добавлением 4 × 10–4–1 × 10–5 М ЦТМАХ в систему; концентрация 7 × × 10–4 М выбрана в качестве оптимальной (табл. 2). Как видно из табл. 2, степень извлечения каждого исследуемого фенола максимальна при экстракции в мицеллярные фазы Тритона Х-114. Присутствие в системе ЦТМАХ, с одной стороны, снижает степень извлечения аналитов, а с другой, позволяет стабилизировать ее во времени, улучшая при этом сходимость результатов (постоянство оптической плотности) определения фенолов.
Таблица 2.
Степени извлечения (%) исследованных фенолов в системе диазотированный 4-НА–фенольный аналит–ПАВ (n = 3, P = 0.95)
| ПАВ | Фенол | Резорцин | Флороглюцин | |||
|---|---|---|---|---|---|---|
| способ 1 | способ 2 | способ 1 | способ 2 | способ 1 | способ 2 | |
| ОП-10 | – | 90 ± 3 | 89 ± 3 | 90 ± 3 | 83 ± 3 | 75 ± 3 |
| ОП-10 + ЦТМАХ | – | 85 ± 2 | 74 ± 1 | 84 ± 2 | 75 ± 1 | 68 ± 1 |
| Тритон Х-114 | – | 93 ± 2 | 90 ± 2 | 90 ± 2 | 84 ± 2 | 76 ± 2 |
| Тритон Х-114 + ЦТМАХ | – | 89 ± 1 | 74 ± 1 | 84 ± 1 | 79 ± 1 | 69 ± 1 |
| Бридж-35 | – | 91 ± 2 | 89 ± 2 | 90 ± 2 | 84 ± 2 | 76 ± 2 |
| Бридж-35 + ЦТМАХ | – | 88 ± 1 | 75 ± 1 | 85 ± 1 | 79 ± 1 | 70 ± 1 |
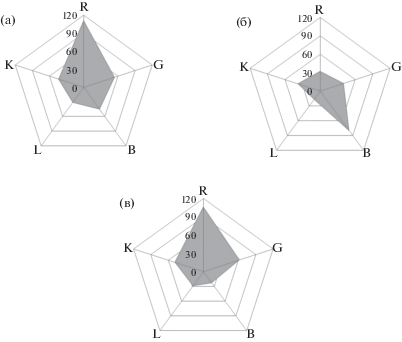
Цветометрическое определение фенолов. Цифровая технология [16–18] регистрации интенсивности электронных изображений реализована нами для цветометрического определения фенола, резорцина и флороглюцина в модельных растворах после проведения реакции дериватизации аналитов с диазотированным 4-НА и последующим концентрированием азосоединений в мицеллярные фазы неионных ПАВ. Применения трех цветометрических параметров не всегда достаточно для разработки селективного метода определения соответствующего фенола. Для усовершенствования этого подхода применяют многокомпонентные системы для построения лепестковых диаграмм (ЛД) с различным количеством осей как индивидуальных отпечатков соответствующего аналита. Нами построены лепестковые диаграммы с применением пяти выбранных цветометрических параметров: R – красный, G – зеленый, B – синий, L – светлота, K – черный (рис. 3). Как видно из рис. 3, каждый фенол имеет свой индивидуальный профиль, который характеризуется соответствующей формой ЛД. Следует отметить, что при низких концентрациях (1 × 10–7–8 × 10–7 М) такие профили для фенола, резорцина и флороглюцина становятся малоразличимыми, что не позволяет достоверно их идентификацию.
Рис. 3.
Лепестковые диаграммы исследованных фенолов (6 × 10–6 М): (а) – фенол, (б) – резорцин, (в) – флороглюцин.
Для количественной оценки содержания фенолов применяли как цветометрические (оптимальный параметр цветности – G-канал), так и геометрические параметры полученных ЛД: площадь (S) и периметр (P). Градуировочные зависимости линейны в пределах 1 × 10–7–3 × 10–5 М для фенола; 1 × 10–7–2 × 10–5 М для резорцина и флороглюцина. Корреляционные уравнения в координатах интенсивность G-канала (IG)–pc(фенола) и площадь ЛД (S)–рc(фенола), а также величины достоверностей апроксимации для указанных аналитов составили соответственно: IG = 52.6рс – 216 (R2 = 0.982), S = 8180рс – 34000 (R2 = 0.977); IG = = 54.3рс – 235 (R2 = 0.985), S = 9380рс – 43360 (R2 = 0.985); IG = 38.4рс – 141 (R2 = 0.970), S = = 6130рс – 24650 (R2 = 0.973).
Правильность результатов определения фенолов с предварительным СР-концентрированием оценивали методом введено–найдено по параметру G (табл. 3). Предложенный способ позволяет определять содержание фенолов в диапазоне 1 × 10–7–1 × 10–4 М, при этом погрешность цветометрического определения не превышает 10–12%.
Таблица 3.
Результаты цветометрического определения фенолов в модельных растворах (№№ 1–3) и в препарате “Резорцинол” (№ 4) методом введено–найдено (n = 3, Р = 0.95)
| № | Аналит | Введено, М | Найдено, (Xср ± ΔX), М | sr |
|---|---|---|---|---|
| 1 | Фенол | 6.0 × 10–7 | (6.0 ± 0.23) × 10–7 | 0.07 |
| 1.5 × 10–6 | (1.3 ± 0.27) × 10–6 | 0.08 | ||
| 1.5 × 10–5 | (1.5 ± 0.13) × 10–5 | 0.04 | ||
| 2 | Резорцин | 6.0 × 10–7 | (5.7 ± 0.17) × 10–7 | 0.05 |
| 1.5 × 10–6 | (1.5 ± 0.33) × 10–6 | 0.10 | ||
| 1.5 × 10–5 | (1.4 ± 0.20) × 10–5 | 0.06 | ||
| 3 | Флороглюцин | 6.0 × 10–7 | (5.5 ± 0.17) × 10–7 | 0.05 |
| 1.5 × 10–6 | (1.3 ± 0.27) × 10–6 | 0.08 | ||
| 1.5 × 10–5 | (1.3 ± 0.23) × 10–5 | 0.07 | ||
| 4 | Резорцин* | 1.0 × 10–3 | (1.4 ± 0.07) × 10–2 | 0.02 |
| 2.0 × 10–3 | (1.6 ± 0.20) × 10–2 | 0.06 | ||
| 4.0 × 10–3 | (1.9 ± 0.17) × 10–2 | 0.05 |
Разработанный цветометрический способ определения фенолов апробирован для определения резорцина в лекарственном препарате Резорцинол. Препарат предварительно разбавляли этанолом в 1000 раз и определяли концентрацию резорцина по уравнению IG = 54.3рс – 235. Полученный результат (1.5 ± 0.2) × 10–2 М не содержит систематической погрешности и хорошо согласуется с концентрацией резорцина, заявленной производителем – 1.5 × 10–2 М.
* * *
Таким образом, методология CP-концентрирования дериватизатов фенола и его аналогов с использованием простых и комбинированных систем на основе неионных и катионных ПАВ показала, что возможно их раздельное цветометрическое определение с пониженными пределами обнаружения на уровне концентраций 10–7–10–5 М. Такой подход не только позволяет определять исследуемые соединения на уровне ПДК и ниже (ПДК в воде фенола составляет 1.1 × 10–6 М, резорцина – 0.9 × 10–6 М), но и контролировать их содержание в фармацевтических препаратах. Предложенный способ экспрессен, экономически и экологически обоснован, однако вопрос повышения селективности цветометрического определения с предварительным СР-концентрированием близких по свойствам фенолов остается актуальным для дальнейших исследований.
Список литературы
СанПиН 2.1.4.1074-01. Питьевая вода. Гигиенические требования к качеству воды централизованных систем питьевого водоснабжения. Контроль качества. М., 2002. 62 с.
Жестовская Е.С., Доронин С.Ю. Мицеллярная экстракция в “точке помутнения” – как способ концентрирования фенолов // Бутлеровские сообщения. 2016. Т. 45. № 2. С. 66.
Ferrera Z.S., Sanz C.P., Santana C.M., Rodriguez J.J.S. The use of micellar systems in the extraction and preconcentration of organic pollutants in environmental samples // Trends Anal. Chem. 2004. V. 23. P.469.
Чернова Р.К., Доронин С.Ю. Определение органических аналитов в растворах ПАВ: ионные и мицеллярные эффекты. Саратов: Изд-во Сарат. ун-та, 2017. 200 с.
Carabias R.M, Rodrıguez G.E., Moreno C.B. Surfactant cloud point extraction and preconcentration of organic compounds prior to chromatography and capillary electrophoresis // J. Chromatog. A. 2000. V. 902. P. 251.
Ghasemi E1, Kaykhaii M. Developing a new micro cloud point extraction method for simultaneous preconcentration and spectrophotometric determination of uranium and vanadium in brine // Anal. Sci. 2015. V. 31. № 5. P. 407.
Zain N.N.M., Abu Bakar N.K., Mohamad S., Saleh N.Md. Optimization of a greener method for removal phenol species by cloud point extraction and spectrophotometry // Spectrochim. Acta A. 2014. V. 118. P. 1121.
Wang Z., Zhao F., Li D. Determination of solubilization of phenol at coacervate phase of cloud point extraction. Colloids and surfaces // Physicochem. Eng. Aspects. 2003. V. 216. P. 207.
Taechangama P., Scamehornb J.F., Osuwana S., Rirksomboona T. Effect of nonionic surfactant molecular structure on cloud point extraction of phenol from wastewater // Physicochem. Eng. Aspects. 2009. V. 347. P. 200.
Иванов В.М., Моногарова О.В., Осколок К.В. Возможности и перспективы развития цветометрического метода в аналитической химии // Журн. аналит. химии. 2015. Т. 70. № 10. С. 1011.
Байдичева О.В., Рудакова Л.В., Рудаков О.Б. Применение цифровых технологий в цветных тестах биологически активных веществ // Бутлеровские сообщения. 2008. Т. 13. № 2. С. 50.
Рудаков О.Б., Хорохордина Е.А., Грошев Е.Н., Чан Хай Данг, Селиванова Е.Б. Цифровой цветометрический контроль качества строительных материалов // Научный Вестник ВГАСУ. 2013. Т. 7. № 2. С. 104.
Апяри В.В., Горбунов М.В., Исаченко А.И., Дмитриенко С.Г., Золотов Ю.А. Иcпользование бытовых цветорегистрирующих устройств в количественном химическом анализе // Журн. аналит. химии. 2017. Т. 72. № 11. С. 1165.
Чурилина Е.В., Суханов П.Т., Коренман Я.И., Ильин А.Н. Двухфазные системы на основе поли-n-винилкапролактама для экстракционного концентрирования фенолов из водных растворов // Вестник ВГУИТ. 2012. № 1. С. 112.
Доронин С.Ю., Чернова Р.К., Бурмистрова А.А. Экстракционное концентрирование органических аналитических форм системами на основе ПАВ // Бутлеровские сообщения. 2011. Т. 52. № 6. С. 94.
Zarei A.R., Gholamian F., Chalavi S. Micelle-mediated extraction and cloud point pre-concentration for the spectrophotometric determination of phenol in water samples // South Afr. J. Chem. 2011. V. 64. P. 158.
Зяблов А.М., Жиброва Ю.А., Селеменев В.Ф. Усовершенствование экстракционно-инструментальных методик определения парацетамола c применением ВЭЖХ, ТСХ, фотоколориметрии и цифровой цветометрии // Сорбционные и хроматографические процессы. 2006. Т. 6. № 6. С. 1424.
Рудаков О.Б., Хорохордина Е.А., Чан Хай Данг / Матер. IV Всерос. симп. “Разделение и концентрирование в аналитической химии и радиохимии”. Краснодар, 24 сентября–04 октября 2014 г. С. 108.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии